
Abstract

Loss of Protein Homeostasis Has Been Hypothesized to Play a Critical Role in Aging
Accumulation of damaged proteins is considered to be a major mechanism by which homeostasis declines during aging. Protein homeostasis is defined as the set of transcriptional, translational, and posttranslational processes, including folding, trafficking, localization, and degradation, that maintain the correct amount, distribution, and structures of proteins within a cell. Mounting evidence suggests that maladaptive alterations in proteostasis, especially alterations in mechanisms that maintain protein quality control, such as chaperone function, unfolded protein response (UPR), ubiquitin/proteasome proteolysis, lysosomal function, macroautophagy, and chaperone-mediated autophagy (in mammals) contribute to aging (Fig. 1). 1 Morever, dysfunctional proteostasis plays a significant role in numerous degenerative pathologies, including Alzheimer, Huntington, and Parkinson diseases.
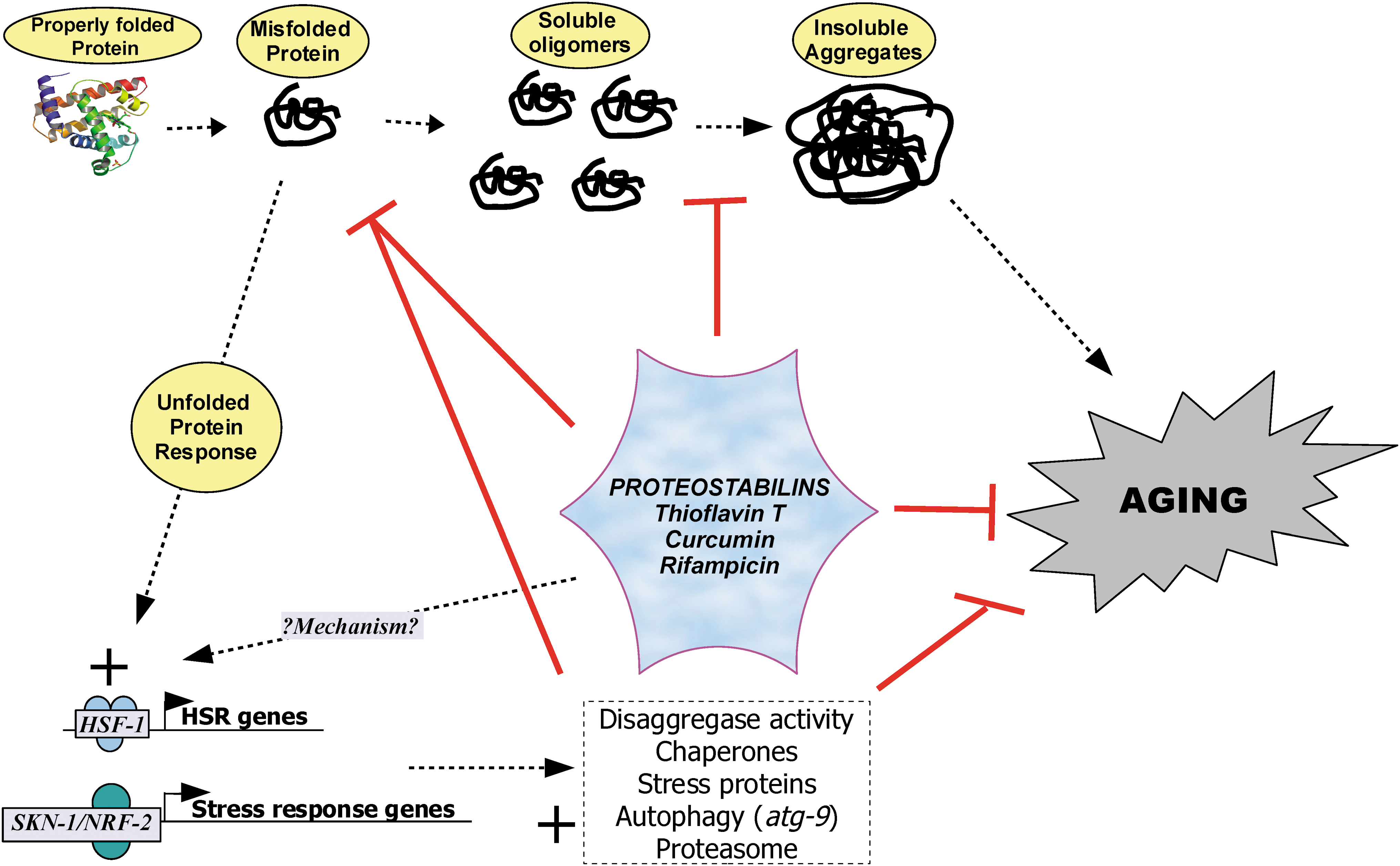
Pathways of proteostasis. Protein misfolding and aggregation contribute to cellular dysfunction and loss of homeostasis. Alavez et al.5 have demonstrated that thioflavin T and other small molecules (“proteostabilins”) stabilize misfolded proteins to reduce aggregation and activate various protein quality control pathways. HSR, Heat shock response. (Color image available online at
One of the key targets of protein quality control is elimination of misfolded or modified proteins that oligomerize or aggregate. For example, in the nematode worm Caenorhabditis elegans, there is a pool of metastable proteins that begin to accumulate in misfolded conformations during early adulthood and lead to increasing loss of protein quality control with age. 2 Furthermore, several hundred mostly beta-sheet–enriched proteins that become more insoluble with age have been identified in adult C. elegans. 3 Homologs of these proteins are overrepresented in aggregates from humans with neurodegenerative diseases, 3 suggesting that these proteins represent an important set of age-sensitive molecules. Their aggregation state may be a useful biomarker of compromised proteostasis associated with aging. Studies in rats demonstrating reduced chaperone levels with age in liver endoplasmic reticulum are consistent with the reduced protein quality control observed in worms, although more work is needed to understand the role of proteostasis in mammalian aging. 4
Alavez and Colleagues Report a Fascinating Study Demonstrating Extension of the Life Span of C. elegans by Protein-Stabilizing Compounds That Inhibit Protein Aggregation
To test the hypothesis that inhibition of protein misfolding and aggregation may help ameliorate dysfunctional proteostasis associated with aging, Alavez et al. 5 treated adult C. elegans with the flavanoid thioflavin T (ThT), a compound known to bind and inhibit aggregation of amyloids, such as those involved in Alzheimer disease. Treatment with 50 or 100 μM ThT thoughout adult life extended median life span by 60% and maximum life span by 43%–78%. In addition, improved health was observed throughout adulthood, including reduced loss of movement associated with aging. Protein aggregate inhibitors curcumin and rifampicin also increased life span, but to a lesser extent (≈45%). Combination of curcumin and ThT did not confer additional life span increase, suggesting that they act through a common mechanism, presumably stabilization and/or inhibition of protein aggregation.
To test directly whether ThT could suppress toxic effects of protein aggregation, worms were engineered to express temperature-sensitive aggregating proteins human β-amyloid(3-42) or polyglutamine (polyQ) in muscle. At 25°C, the worms become paralyzed, due to the toxic effects of the aggregates. ThT suppressed paralysis, most likely by direct interaction with and stabilization of the pathological proteins. To show that this effect is general, ThT was used to suppress multiple worm temperature-sensitive mutations: gas-1 of mitochondrial complex 1, levamisole resistance in the α-subunit of the nicotinic receptor, unc-52 (perlecan), and unc-54 (myosin II heavy chain).
Dietary restriction (DR) is well known to increase life span in numerous species. ThT did not cause DR, as evidenced by a lack of a significant decrease in the pharyngeal pumping rate. However, the 100 μM dose of ThT was toxic to dietary-restricted worms, indicating that the combined effects of these treatments may be maladaptive in a dose-dependent manner. This effect suggests that a cautionary note should be sounded for humans attempting to combine antiaging regimens and compounds without data on their potential interactions. A lower dose (10 μM) of ThT extended the life span of DR-fed worms by an additional 24%, indicating that at least some of the mechanisms by which ThT extends life span are independent of DR.
To test involvement of protein quality control pathways, RNA interference (RNAi) knockdown and/or mutation of key proteostasis genes were studied. Thus, the chaperone Hsp16.42, heat shock/protein quality control regulator HSF-1, and cell stress regulator SKN-1 (Nrf2-like) (Fig. 1) are required for ThT to extend life span and to inhibit amyloid-mediated paralysis. These results indicate that protein quality control pathways also play a role in the ability of ThT to extend life span. Consistent with these results, ThT also increased expression of chaperones Hsp-6, Hsp16.2, and Hsp-70. However, the relative importance of protein quality control pathways versus a direct effect on protein stability is yet to be determined.
The Big Picture for Aging: Are These Results Significant for Humans?
The results of the Alavez et al. paper lead to several important questions.
How much of aging can be attributed to dysfunction of proteostasis?
The short answer is that we do not know. The strongest consensus at this time would probably be that dysfunction in proteostasis is one of several mechanisms by which organisms age. Furthermore, this mechanism may play a greater role in postmitotic rather than in dividing cells. Quiescent cells do not undergo cell proliferation, which can dilute out misfolded or aggregated proteins.
Can upregulation of protein quality control pathways prevent aging in worms?
Yes. Flies and worms that overexpress Hsp-70 and small heat shock proteins (sHSPs), such as Hsp16 and Hsp22, 6 –11 have extended life spans. Furthermore, mutations that decrease insulin signaling pathways or act as dietary restriction mimetics are associated with increased expression of Hsp-16. 12
Does proteostasis play a similar role in humans or other mammals?
Organisms that are mostly postmitotic as adults, such as C. elegans or Drosophila melanogaster, might be expected to be especially sensitive to alterations in protein quality control pathways. Consistent with this hypothesis is the observation that to date there are no reports of increased life span in transgenic mice that are engineered to express higher levels of proteostasis regulators or effectors. However, rapamycin, an inhibitor of the target of rapamycin (TOR) pathway, has been shown to extend life span in mice. 13 Rapamycin increases autophagy, so there remains the possibility that autophagy plays a critical role in the rapamycin-mediated increase in longevity. On the other hand, an enhanced heat shock response may act to increase tumorigenesis and thereby shorten life span by buffering misfolded or mutant proteins found in cancer cells.
There are numerous reports of loss of proteostasis with increased age in mammals. In old rodents, endoplasmic reticulum (ER) chaperones, calnexin, protein disulfide isomerase (PDI), and binding immunoglobulin protein (BiP) have been observed to decrease with age in some organs and tissues. 14 –18 Furthermore, the UPR has been reported to be aberrant in aged animals. 17,18 On the other hand, preliminary attempts to correlate 20S/26S proteasome activity with life span among endotherm vertebrate species has been unsuccessful, 19 although a full exploration of comparative vertebrate protein quality control should be carried out.
More work is clearly needed to determine whether enhanced protein quality control can extend human health span or life span.
Medical Implications of This Work
The compounds investigated by Alavez et al. appear to extend longevity in C. elegans through two basic mechanisms: (1) Biophysical stabilization of misfolded proteins and inhibition protein aggregation, and (2) activation of protein quality control pathways though the heat shock response (HSR) regulator HSF-1 and cell stress regulator SKN1 (Nrf2-like). We define compounds acting via both mechanisms as “proteostabilins.”
As discussed above, the capacity of proteostabilins to increase life span or health span for humans remains to be established. However, approaches to stabilize protein conformation are expected to benefit diseases caused by: (1) Gain-of-function changes resulting in toxic protein aggregates, exemplified by Alzheimer and Huntington diseases, or (2) loss-of-function mutations resulting in misfolded proteins such as Gaucher disease and cystic fibrosis (CF). Compounds can be classified according to their mechanism of action, including pharmacological (chemical) chaperoning to maintain proper folding by direct interaction, kinetic stabilizers to shift equilibrium of a multimeric protein toward the most stable nonpathogenic form, and regulators of proteostasis that alter HPR, UPR, or lysosomal/autophagic pathways. 20 A number of agents with these properties have been under active investigation for a number of diseases (Table 1).
HSR, Heat shock response; ER, endoplasmic reticulum; CTFR, cystic fibrosis transmembrane conductance regulator.
Currently Available Drugs That Maintain Protein Stability, Prevent Protein Aggregation, or Stimulate Protein Stress Response Pathways
Extending life span by agents that help maintain proteostasis or act as proteostabilins remains hypothetical for mammals, including humans. However, there is evidence that such compounds may be useful to treat or prevent proteostatic diseases, many of which are associated with aging. Although no proteostatic therapeutic is Food and Drug Administration (FDA)–approved, there are a multitude of compounds being investigated for possible therapies of a variety of diseases where protein stability plays a critical role.
Direct protein stabilization
Agents that work through protein stabilization in the ER include 4-phenylbutyrate (4-PBA) and taurine-conjugated ursodeoxycholic acid (Table 1), which have been shown to increase glucose tolerance, normalize hyperglycemia, increase insulin sensitivity, and resolve fatty liver disease in a mouse model of type II diabetes (leptin deficient, ob/ob) by relieving ER stress. 21 4-PBA also suppresses a destabilizing mutation in nephrin linked to congenital nephrotic syndrome in cultured human cells by restoring normal intracellular trafficking. 22 4-PBA partially restores the proteostasis of several misfolded soluble and transmembrane proteins, including the cystic fibrosis transmembrane conductance regulator (CFTR), for which there is an ongoing clinical trial. Identification of compounds that act to kinetically stabilize transthyretin to prevent amyloidoses linked to malignant transthyretin aggregation is the subject of active research. Hundreds of such agents such as 3,5-dibromo-4-hydroxyphenyl–based stilbene and dihydrostilbene (Table 1) block aggregation of transthyretin in cell culture. 23
Modulation of proteostasis by modulation of HSR, UPR, and autophagy
Numerous compounds stimulate proteostasis by modulating protein quality control pathways. For example, celastrol (Table 1) increases HSF-1 levels in mammals to stimulate HSR and demonstrates protective activity in a murine model of Alzheimer disease. 24 Diltiazem and verapamil (Table 1), FDA-approved for hypertension, increase transcription of numerous cytoplasmic and ER chaperones by inhibiting L-type Ca2+ channels in the plasma membrane. The resulting enhanced folding, trafficking, and activity of lysosomal enzymes helps suppress the pathologies associated with lysosomal storage diseases in cell culture models. 25 α1-Antitrypsin deficiency results from α1-antitrypsin trapped in ER segments that are subject to autophagy. Carbamazepine (Table 1), an autophagy stimulator, has been shown to boost the capacity of the autophagic pathway to catabolize the aggregates of α1-antitrypsin. 26
Proteostabilins
The compounds investigated by Alavez et al. appear to work by at least two mechanisms. As proteostabilins, they are expected to show similar activity in other systems. Curcumin has been shown to inhibit amyloid formation and toxicity 27 as well as be protective in a mouse model of Parkinson disease. 28 A bioavailable nanoparticle preparation of curcumin had antiamyloid activity in culture and a mouse model. 29 Curcumin has also been shown to modestly extend life span in C. elegans, 5 D. melanogaster, 30 and mice. 31 At least two other nutriceutical-based agents may act as proteostabilins. Polyphenols in coffee extracts suppress amyloid toxicity in a C. elegans model at least partially through the cell stress regulator SKN-1 (Nrf2). 32 Coffee itself has been shown to have anti-Alzheimer disease activity in mice. 33,34 Epigallocatechin gallate (EGCG) (Table 1), a major component of green tea, has similar antiamyloid/anti-Alzheimer activities, including the ability to disaggregate amyloid fibrils and protect cultured cells. 35,36 EGCG has potential therapeutic value for Parkinson disease as well. 35 It has also been shown to increase life span signficantly in C. elegans, upregulating SKN-1 (Nrf2) and Daf-16, 37,38 as well as modestly increasing life span in mice. 31
Conclusion
The mechanisms underlying proteostasis present attractive targets for pharmaceutical “proteostabilins” to extend health span and life span. Such efforts may be aided by further study of the specific roles played by proteostatic pathways in aging. Drugs to correct proteostatic dysfunction in diseases such as Alzheimer disease are already under investigation or development. In the meantime, coffee or green tea may prove useful in staving off some neurodegenerative diseases.