
Abstract
Oral-Facial-Digital 1 (OFD1) Syndrome is an X-linked developmental disorder caused by mutations in the gene Ofd1. OFD1 syndrome involves malformation of the face, oral cavity, and digits and may be characterized by cystic kidneys and mental retardation. Deletion or missense mutations in Ofd1 also result in loss of primary cilia, a microtubule-based cellular projection that mediates multiple signaling pathways. Ofd1 mutant mice display pleiotropic developmental phenotypes, including neural, skeletal, and cardiac defects. To address how loss of Ofd1 and loss of primary cilia affect early differentiation decisions, we analyzed embryoid bodies (EBs) derived from Ofd1 mutant embryonic stem (ES) cells. Ofd1 mutant EBs do not form primary cilia and display defects in Hedgehog and Wnt signaling. Additionally, we show that ES cells lacking Ofd1 display an increased capacity to differentiate into neurons. Nevertheless, neurons derived from Ofd1 mutant ES cells fail to differentiate into V3 interneurons, a cell type dependent on ciliary function and Hedgehog signaling. Thus, loss of Ofd1 affects ES cell interpretation of developmental cues and reveals that EBs model some aspects of ciliopathies, providing insights into the developmental origins of OFD1 syndrome and functions of cilia.
Introduction
C
Hh signal transduction is mediated through the primary cilium, and localization of several Hh pathway components to primary cilia is necessary for their function. Smoothened, a 7-pass transmembrane protein, moves into the primary cilium in the presence of Sonic Hh (Shh) [7]. Gli proteins, the effectors of the Hh pathway, also localize to the primary cilium, and this localization is essential for formation of both activator and repressor forms [8]. Smoothened translocation to the cilium triggers the switch from formation of Gli repressors to Gli activators [8 –11]. Gli3, for example, is processed to a truncated repressor form in the absence of Hh ligand [12 –15]. This processing is inhibited by pathway activation in a cilium-dependent manner [8,10,11]. Gli proteins are presumed to shuttle from the cilium to the nucleus to control transcription of Hh target genes. Thus, the cilium coordinates multiple steps of the Hh pathway to regulate Hh pathway transcriptional activity.
As cilia play diverse roles in development and signaling, ciliary dysfunction manifests as human genetic syndromes known as ciliopathies, which include Meckel, Joubert, Senior-Loken, Bardet-Biedl, and Oral-Facial-Digital 1 (OFD1) syndromes [2 –4,16 –20]. OFD1 syndrome is characterized by polydactyly and deformity of the oral cavity and face and is caused by mutations in the gene Ofd1 [20,21]. Ofd1 encodes a protein that localizes to the distal end of centrioles where it functions as a cap to regulate centriole length [22,23]. As OFD1 is X-linked, males lacking OFD1 do not form cilia, resulting in prenatal lethality [22]. The developmental phenotypes displayed in Ofd1 mutant mice resemble those seen in humans with OFD1. Likewise Ofd1 mutant mice share many developmental abnormalities with other mouse mutants lacking cilia [24 –27]. We recently described a mouse embryonic stem (ES) cell line that contains a gene trap insertion into the gene Ofd1 (Ofd1Gt ) [23,28]. The Ofd1Gt ES cell line is male and thus lacks both Ofd1 and primary cilia.
ES cells are derived from the inner cell mass of the blastocyst and can differentiate into all cell types of the body [29 –31]. In addition to representing a potential for stem cell-based therapies, ES cells are a tool for investigating cell fate decisions and the mechanisms of development [31,32]. Mouse ES cells are able to maintain their pluripotency in culture by activation of the Janus kinase/signal transducers and activators or transcription (JAK/STAT) and bone morphogenic protein (BMP) pathways [33]. Upon differentiation in suspension culture, ES cells form aggregates called embryoid bodies (EBs) [34,35]. EBs form 3 layers comprised of endoderm, mesoderm, and ectoderm and have the potential to form nearly all cell types of the embryo. [31,34,35].
Here, we used the Ofd1Gt ES cell line to address the role of Ofd1 and primary cilia in ES cell differentiation. We found that Ofd1Gt EBs have Hh signaling defects and exaggerated β-catenin-dependent pathway activation in response to Wnt3a. Further, differentiated Ofd1Gt ES cells displayed increased neural differentiation. Examination of mouse mutant embryos lacking cilia demonstrated that cilia do not limit neural differentiation in vivo. Nevertheless, Ofd1Gt EBs do not form V3 interneurons similarly to mouse mutants with abrogated ciliogenesis. V3 interneurons require high levels of Hh signaling in the ventral neural tube for induction, thus indicating that the role of cilia in EB differentiation recapitulates the role of cilia in Hh-mediated neuronal patterning.
Materials and Methods
Tissue culture
Embryonic stem cells and EBs were grown as described previously [23,28]. For Hh pathway activation, wild-type and Ofd1Gt (RRF427; Bay Genomics) EBs were cultured for 7 days and incubated with recombinant Shh (1 μM; R&D Systems) or SAG (0.1 μM; Axxora) for 8, 18, or 24 h. Similarly, Wnt pathway activation was performed by incubating EBs for 2–4 days and adding recombinant Wnt3a (0.1 μg/mL; R&D Systems) for 4 h. BMP4 (0.1 μM; R&D Systems) and Noggin (1 μM; jCBS) were added to EBs for 48 h after being cultured for 3–6 days.
Lentiviral infection
ES cells were trypsinized and incubated for 1 h with concentrated lentivirus for either pSico-Smad1-puro or pSico-puro empty vector (gifts of Drs. Rik Derynck and Michael McManus). The cells were plated overnight and placed under selection the following morning using 2 μM puromycin. Resistant cells were selected for 5 days and knockdown was assayed. Two different Smad RNAi lentiviruses were used and gave similar results.
Immunofluorescence
For wholemount EBs
EBs were grown for 10–12 days in low suspension culture and subsequently plated onto chamber slides coated with poly-lysine and Matrigel (BD) for 48 h. EBs were washed and fixed with 4% paraformaldehyde (PFA) for 20 min and washed 3 times with phosphate buffered saline with 0.1% Triton X-100 (PBT). Subsequently, the EBs were exposed to primary block (2% bovine serum albumin and 1% serum in PBT) for 1 h. Primary antibody was added overnight in primary block at 4°C. The next day, EBs were washed and secondary block (2% bovine serum albumin and 10% serum in PBT) was added for 1 h. The EBs were then incubated with secondary antibodies for 1–2 h followed by washes with PBT. Nuclei were stained with 4′,6-diamidino-2-phenylindole in primary block for 20 min and washed again with PBT 3 times. Slides were mounted and left overnight to dry.
For embryo and EB sections
Embryos and EBs were fixed for 1 h in 4% PFA, washed, and imbedded in OCT tissue-freezing medium (OCT Tissue-Tex). The blocks were sectioned on a Microm HM 550 (Thermo-Fisher) at 12 μm thickness. The slides were washed 3 times with PBS and stained using the protocol above, except for the cilia staining protocol, which included a 2 min 100% methanol fixation.
For blastocysts
Blastocysts were flushed from the uterus at E3.5 and immunofluorescently analyzed similarly to the wholemount EBs.
All images were taken on a Nikon C1si Spectral confocal microscope.
Antibodies
The following antibodies were used for immunofluorescence and immunoblotting at the following dilutions: mouse anti-γ-tubulin (1:500; Abcam GTU488); mouse anti-Tuj1 (1:500; Covance MMS435); rabbit anti-Tuj1 (1:2,000; Covance PRB435); mouse anti-Nestin (1:300; BD Pharmingen 556309); mouse anti-Islet1/2 (1:50; DSHB); mouse anti-Nkx2.2 (1:20; DSHB); rabbit anti-β-actin (1:5,000; Abcam); rabbit anti-phospho-Smad 1/5 (1:1,000; Cell Signaling); rabbit anti-phospho-Smad 1/5/8 (1:1,000; Chemicon); rabbit anti-Rootletin (1:20,000; gift from Tiansen Li); mouse anti-acetylated tubulin (1:500; Sigma); rabbit anti-Ofd1 (1:5,000); mouse anti-α-tubulin (1:5,000). The mouse anti-Neurogenin2 antibody (1:100) was a gift from Dr. D. J. Anderson. Rabbit anti-Arl13b (1:500) was a gift from Dr. Tamara Caspary. Mouse anti-Gli3 and guinea pig anti-Gli2 were gifts from Drs. Suzie Scales (Genentech) and Jonathan Eggenschwiler, respectively, and were used at (1:4,000).
Quantitative real-time polymerase chain reaction
RNA was extracted from EBs at indicated time points using Qiagen RNeasy Plus Mini kit. RNA was then subjected to first-strand cDNA synthesis using iScript (Biorad or Fermentas). Expression levels were then analyzed in triplicate using a 7300 real-time polymerase chain reaction machine (Applied Biosystems) and normalized to β-actin. The following primers were used: β-actin F: CACAGCTTCTTTGCAGCTCCTT β-actin R: CGTCATCCATGGCGAACTG Nestin F: TTAAGGCCAGAACCCCCAC Nestin R: CTCTGCATTTTTAGGATAGGGAGC Hnf4 F: CAGACGTCCTCCTTTTCTTGTGATA Hnf4 R: TGTTTGGTGTGAAGGTCATGATTA T-brachyury F: CTGGGAGCTCAGTTCTTTCGA T-brachyury R: GAGGACGTGGCAGCTGAGA Keratin 18 F: CGCTTGCTGGAGGATGGA Keratin 18 R: CTTCTGCACAGTTTGCATGGA Sox1 F: TGAAGGAACACCCGGATTACA Sox1 R: GCCAGCGAGTACTTGTCCTTCT Gli1 F: CTTCACCCTGCCATGAAACT Gli1 R: TCCAGCTGAGTGTTGTCCAG Patched F: TGATTGTGGAAGCCACAGAAAA Patched R: TGTCTGGAGTCCGGATGGA CyclinD1 F: CCAAGTTCCCTAGCAAGCTG CyclinD1 R: CTTTCATGTCACAGGGCAGA C-myc F: CAACGTCTTGGAACGTCAGA C-myc R: TCGTCTGCTTGAATGGACAG Follistatin F: ACGTGTGAGAACGTGGACTG Follistatin R: CATTCGTTGCGGTAGGTTTT Axin2 F: CTCCCCACCTTGAATGAAGA Axin2 R: ACTGGGTCGCTTCTCTTGAA Ngn2 F: GCTGTGGGAATTTCACCTGT Ngn2 R: AAATTTCCACGCTTGCATTC Tuj1 F: TGAGGCCTCCTCTCACAAGT Tuj1 R: CGCACGACATCTAGGACTGA Sox3 F1: CGTAACTGTCGGGGTTTTGT Sox3 F2: CACAACTCCGAGATCAGCAA Sox3 R1: AACCTAGGAATCCGGGAAGA Sox3 R2: GTCCTTCTTGAGCAGCGTCT
Western immunoblot
Cells were lysed in RIPA lysis buffer with protease and phosphatase inhibitor, and protein concentration was measured. For immunoprecipitate, 0.5 μg of rabbit anti-Ofd1 antibody was added to 400 μg protein lysate overnight and bound to rProtein G agarose (Invitrogen). Samples were run on a 9% polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in 5% milk and incubated with primary antibody overnight at 4°C. The next day the membrane was washed and probed with secondary for 1 h. After 3 washes, the membrane was incubated with chemiluminescent substrate for 1 min and exposed for 1–30 min.
Fluorescence-activated cell sorting
EBs were grown for 10–18 days and dissociated using collagenase/dispase (1 mg/mL) for 10 min followed by 0.25% trypsin with Dnase I (1 mg/mL) for 10 min. Cells were resuspended in EB media, washed with PBS, and fixed in 2% PFA for 15 min permeabilized with 1% saponin for 15 min. Cells were resuspended in primary block (3% goat serum in PBS) for 15 min and incubated with primary for 2 h, followed by washing and incubation with secondary antibody for 1 h. Samples were washed several more times and transferred to flow cytometry tubes. Analysis was performed using a fluorescence-activated cell sorting (FACS) Calibur Flow Cytometer (BD) and FlowJo software (Treestar).
In situs
Protocol used for in situs has been described previously [36].
Cell proliferation by bromodeoxyuridine incorporation
ES cells were plated in 24-well low-suspension plates, grown for 7 days, and analyzed using the Cell Proliferation ELISA bromodeoxyuridine kit (Roche). Absorbance measured in triplicate.
Results
To understand how Ofd1 and primary cilia contribute to cell fate decisions and to establish whether ES cells may be an appropriate model system to address ciliary function, we examined whether differentiated ES cells possess primary cilia. Wild-type ES cells were grown in nonadherent suspension culture in the absence of leukemia inhibitory factor (LIF) to form EBs. When probed for Arl13b, a ciliary component, and γ-tubulin, a centrosomal component, wild-type EBs displayed abundant cilia associated with centrosomes (Fig. 1A and Supplementary Fig. S1A; Supplementary Data are available online at
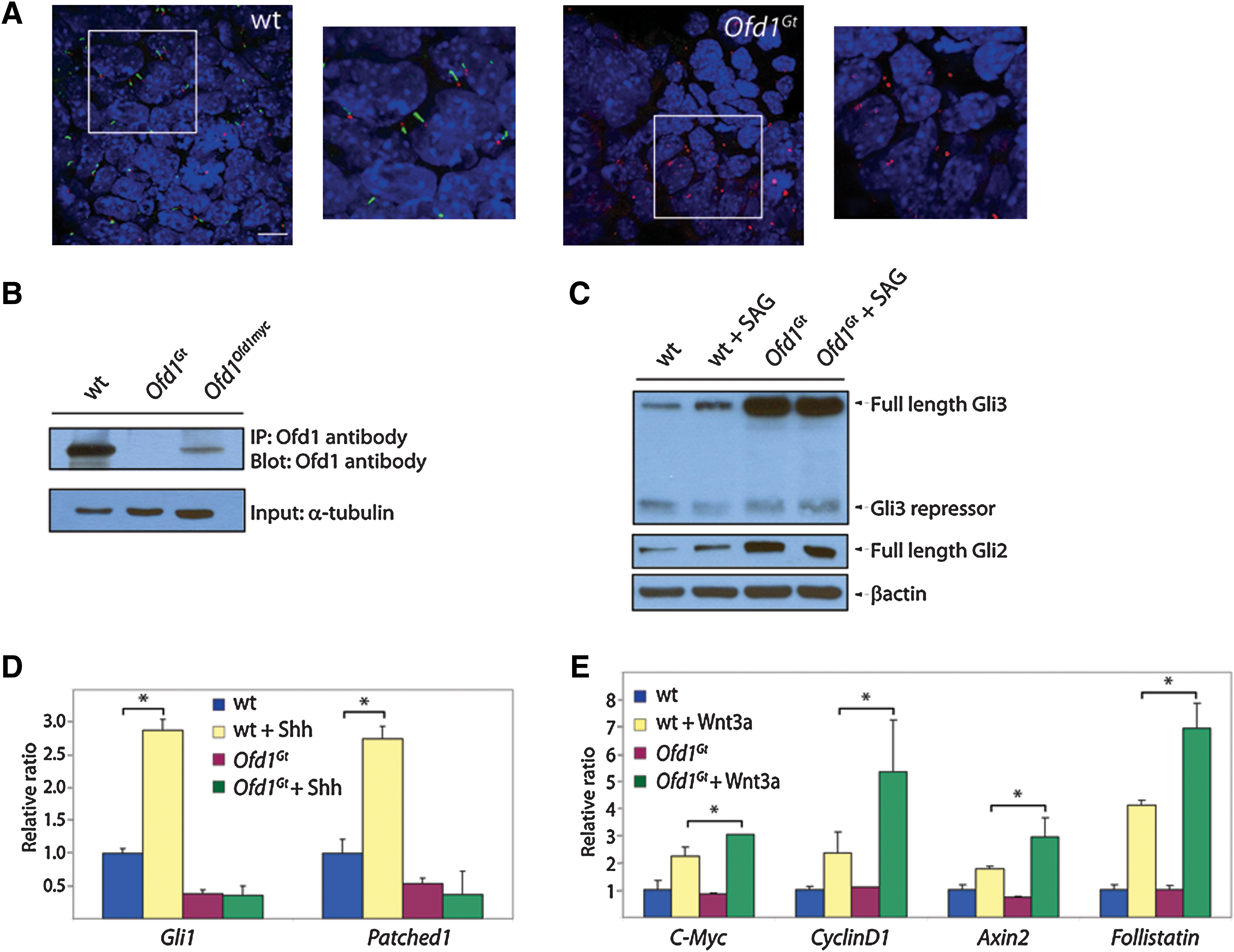
Ofd1Gt
EBs lack cilia and display altered Hh and Wnt signaling.
Given that wild-type EBs possess cilia, we addressed whether Ofd1Gt EBs, which do not make Ofd1 protein, lack primary cilia like their ES cell counterparts (Fig. 1B) [23]. Indeed, primary cilia were completely absent in the Ofd1Gt EBs, although centrosomes were present (Fig. 1A and Supplementary Fig. S1B).
Previous studies have found that mutations that disrupt primary cilia abrogate vertebrate Hh signaling and can alter Wnt signaling [10,24,37]. To ascertain whether Ofd1Gt EBs also display signaling abnormalities, we analyzed Gli processing and gene induction in the presence of either SAG, a small molecule activator of the Hh pathway, or Wnt3a [38]. Efficiency of Gli3 processing was assessed by measuring levels of truncated Gli3 compared with full-length Gli3. SAG induction in wild-type EBs caused increased full-length Gli3 and reciprocally decreased truncated Gli3, indicating that pathway activation inhibits Gli3 processing to the repressor form in EBs (Fig. 1C and Supplementary Fig. S1C). In contrast, addition of SAG failed to change the amount of full-length or truncated Gli3 in Ofd1Gt EBs (Fig. 1C and Supplementary Fig. S1C). Interestingly, Ofd1Gt EBs displayed higher levels of full-length Gli3 and Gli2 protein compared with wild-type EBs (Fig. 1C and Supplementary Fig. S1C). Together, these results indicate that Ofd1Gt EBs display defects in Gli processing and degradation.
To determine whether Ofd1Gt EBs generate a transcriptional response to Hh pathway stimulation, we assayed downstream target genes, Gli1 and Patched1 (Ptch1), in the presence of Hh ligand. Wild-type EBs exhibited a nearly 3-fold increase in Gli1 and Ptch expression upon induction with Shh, whereas Gli1 and Ptch levels in the Ofd1Gt EBs remained the same (Fig. 1D). Notably, the increased full-length Gli3 and Gli2 protein levels observed in Ofd1Gt EBs did not correlate with increased activity, indicating that this stabilized protein is unable to activate the Hh transcriptional program. These data indicate that Ofd1Gt EBs, like mouse mutants lacking cilia, cannot activate genes in response to Shh.
At least some genes essential for cilium formation are required to restrain canonical Wnt signaling in mice and mouse embryonic fibroblasts [37,39]. Therefore, we assessed whether Ofd1Gt EBs display increased responsiveness to Wnt3a. We exposed wild-type and Ofd1Gt EBs to recombinant Wnt3a and assayed for transcriptional activation of Wnt target genes, C-myc, CyclinD1, Axin2, and Follistatin. Addition of Wnt3a in wild-type EBs induced a response in each gene, but stimulation in Ofd1Gt EBs with equal concentrations of Wnt3a induced a greater increase in gene expression (Fig. 1E). Thus, Ofd1Gt EBs overactivate canonical Wnt target genes in the presence of Wnt3a, similar to Kif3a mutant cells [37].
As Ofd1, cilia, and Hh and Wnt signaling are all critical for mammalian development, we examined how Ofd1 influences ES cell differentiation [40 –42]. We analyzed expression of a panel of differentiation markers in wild-type and Ofd1Gt EBs. Whereas markers of epithelium (K18), trophectoderm (Eomes), and endoderm (Hnf4) were expressed at similar levels in wild-type and Ofd1Gt EBs, Ofd1Gt EBs expressed less T-Brachyury, an early mesodermal marker, than wild-type EBs (Fig. 2A). The most pronounced difference, however, was the increased expression of Sox1, a marker of neural precursors, in Ofd1Gt EBs compared with wild-type EBs (Fig. 2A). Expression analysis of several other neural markers, including Sox3, Nestin, Tuj1, and Ngn2, also revealed a dramatic increase in Ofd1Gt EBs, revealing a 5–35-fold change between Ofd1Gt and wild-type EBs (Fig. 2B). These data suggest that in the absence of Ofd1, ES cells have an increased capacity to differentiate toward the neural lineage.
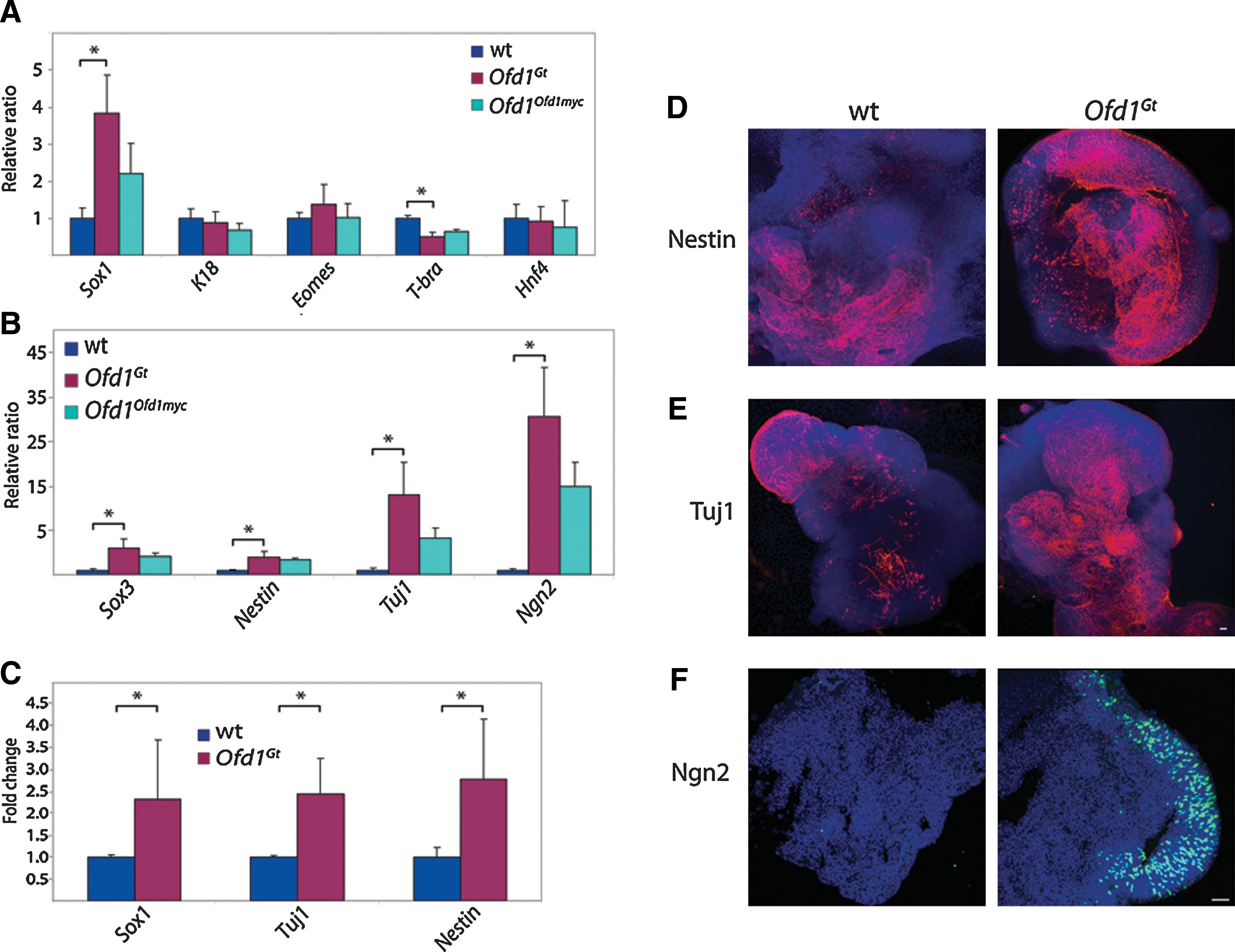
Ofd1Gt
EBs exhibit increased neural differentiation.
To test whether increased neural induction was associated with loss of Ofd1 protein or whether it was due to Ofd1-independent differences in the mutant cell line, we made use of a carboxy-terminal Myc-tagged Ofd1 allele reintroduced into the endogenous locus (Ofd1Ofd1myc ) [23,28]. The Ofd1Ofd1myc ES cells display lower levels of Ofd1 protein than wild-type and approximately half the number of cilia (Fig. 1B and Supplementary Fig. S1B). Correlatively, Ofd1Ofd1myc EBs express levels of neural markers intermediate to wild-type and Ofd1Gt EBs (Fig. 2A, B). Thus, by re-expressing Ofd1 at lower levels than wild type, we were able to partially rescue the phenotypes seen in the Ofd1Gt EBs, suggesting that the increased neural differentiation observed in Ofd1Gt EBs is due to loss of Ofd1 protein and primary cilia.
To distinguish whether increased neural marker expression observed in Ofd1Gt EBs is a result of the presence of more neural cells or higher expression of neural genes within a normal number of neural cells, we analyzed wild-type and Ofd1Gt EBs by flow cytometry and immunofluorescence. We found that Ofd1Gt EBs contained a higher percentage of neural cells as assayed by FACS for Sox1, Tuj1, and Nestin expressing cells (Fig. 2C). Likewise, we observed expanded immunofluorescent staining of Nestin, Tuj1, and Ngn2 in Ofd1Gt EBs compared with wild-type EBs (Fig. 2D–F). Of note, Ngn2 staining in wild-type EBs at day 12 of differentiation was dramatically lower than Ofd1Gt EBs, whereas at later time points, wild-type EBs showed Ngn2 staining more comparable to, although still less than, Ofd1Gt EBs (Fig. 2F and Supplementary Fig. S1D). This suggests that Ngn2 may be induced at an earlier time point in Ofd1Gt EBs than in wild-type EBs, indicating that loss of Ofd1 changes the timing of neural differentiation.
There are several ways in which increased neural induction could occur in Ofd1Gt EBs, including increased proliferation rates, decreased levels of apoptosis, or misregulation of cell fate decisions. Although the population-doubling rate of wild-type and Ofd1Gt ES cells is indistinguishable, Ofd1 may impinge upon proliferation rates of differentiated cells [23]. Consequently, we tested proliferation in wild-type and Ofd1Gt EBs by bromodeoxyuridine incorporation and phospho-histone H3 staining during differentiation. Both assays revealed that wild-type and Ofd1Gt EBs have similar rates of cell division throughout differentiation (Fig. 3A–C). In addition, we tested levels of apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, which was comparable between wild-type and Ofd1Gt EBs (data not shown). On the basis of these data, it seems most likely that Ofd1Gt EBs form neurons at the expense of other cell types.
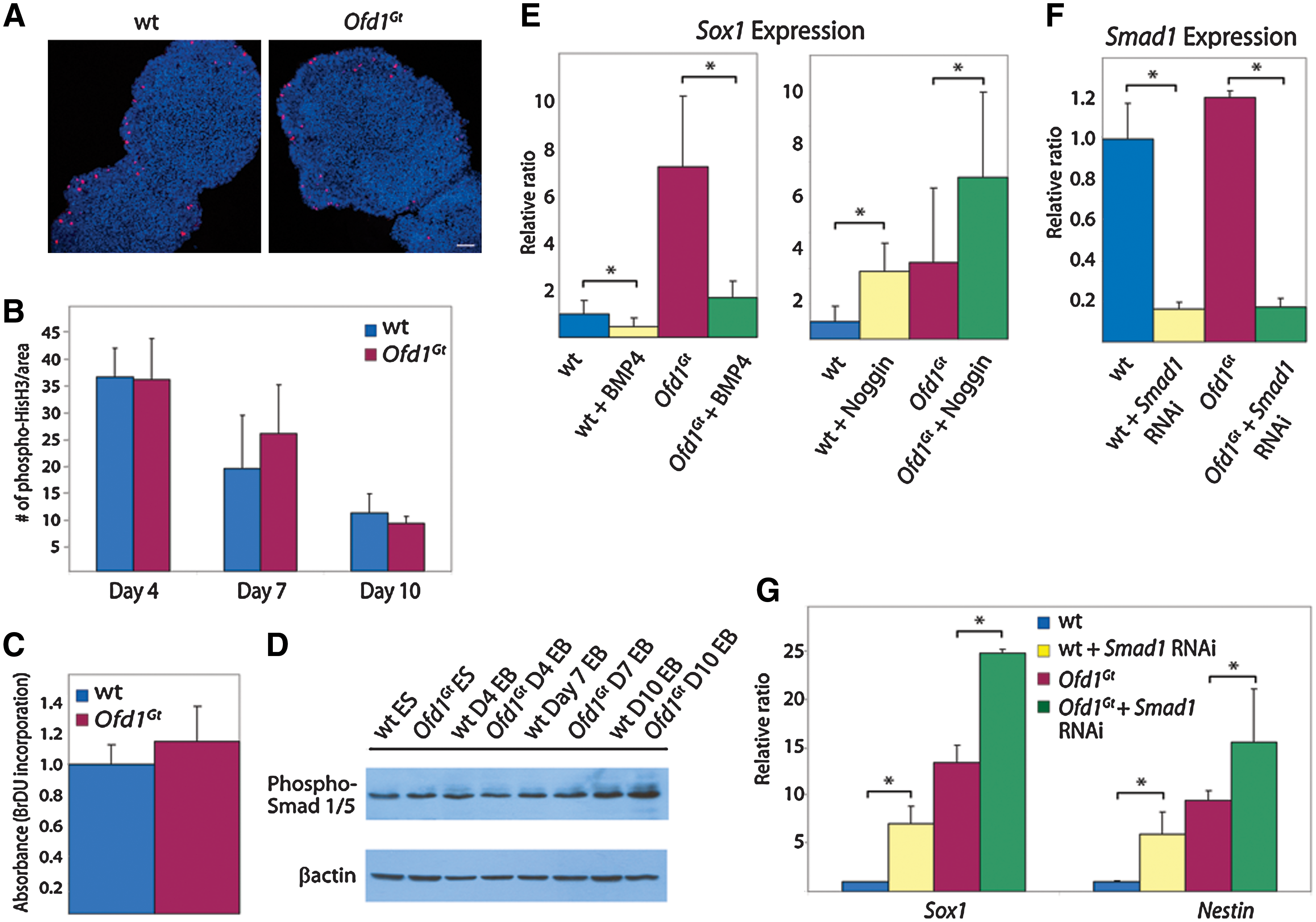
Proliferation rates and BMP signaling are unaltered in Ofd1Gt
EBs.
Mammalian neural induction involves the interplay of several signaling pathways, including the BMP, Wnt, Nodal, fibroblast growth factor, and Hh pathways [43,44]. In particular, inhibition of the BMP pathway is a key determinant of neural fate in many vertebrates [43,45 –47]. Defects in BMP pathway signaling can lead to early and increased neural induction; conversely, increased BMP signaling results in loss of forebrain development [48,49]. Thus, we tested BMP activity and the effects of BMP activation and inhibition on neural induction in wild-type and Ofd1Gt EBs. The BMP pathway is mediated through phosphorylation of Smad proteins 1/5/8, which then bind to Smad4, relocate to the nucleus, and initiate transcription of target genes [50]. To determine whether pathway activity is altered in Ofd1Gt EBs, levels of phosphorylated Smad1/5 were measured at time points throughout differentiation. Phosphorylated Smad levels were indistinguishable between wild-type and Ofd1Gt EBs, suggesting that BMP activity is unaffected by loss of Ofd1 (Fig. 3D).
To further test whether increased neural induction in Ofd1Gt EBs is due to altered responses to BMPs or BMP antagonists, we added either BMP4 or Noggin, a BMP inhibitor, to differentiating EBs and assessed neural induction. In response to BMP4, both wild-type and Ofd1Gt EBs displayed decreased Sox1 expression (Fig. 3E). Conversely, addition of Noggin resulted in increased Sox1 expression in wild-type and Ofd1Gt EBs (Fig. 3E).
These results were substantiated by analyzing neural induction after interruption of the BMP pathway downstream of ligand interaction. Smad1 is a mediator of the BMP pathway as it is phosphorylated upon BMP pathway activation and translocates to the nucleus with Smad4 to activate downstream BMP target genes. Wild-type and Ofd1Gt ES cells were transduced with a Smad1 RNAi lentivirus and subsequently differentiated. Both wild-type and Ofd1Gt -transduced EBs exhibited >80% knockdown of Smad1 RNA levels and increased Sox1 and Nestin expression compared with control EBs (Fig. 3F, G). On the basis of these data, BMP activity and response do not appear to depend upon Ofd1 as wild-type and Ofd1Gt EBs react to pathway activation and inhibition similarly.
Ofd1Gt EBs have defects in Hh and Wnt signaling that are similar to mouse mutants lacking Kif3a, a kinesin essential for ciliary formation [27,37]. Thus, we wanted to determine if there was a general role for cilia in restriction of neurogenesis. Although Kif3a has functions beyond cilia formation, many phenotypes in Ift88, Kif3a, and Ofd1 null embryos are attributed to loss of cilia [22,25,27,37,39]. Therefore, we analyzed Ift88 and Kif3a mutant embryos to determine whether neural induction is increased. Neural marker expression was assessed in E9.5 Kif3a and Ift88 mutants and compared with their wild-type littermates. Overall, there was a 30%–50% decrease in Ngn2, Tuj1, and Nestin expression in the Kif3a and Ift88 mutants (Fig. 4A). In situs of E7.5 and E8.5 embryos probed for brain markers Sox2, Krox20, and Engrailed indicated that there was no appreciable difference in neural specification between wild-type and mutant embryos (Supplementary Fig. S2A–C). To examine neural differentiation in greater detail, E9.5 wild-type and Ift88 mutant embryo neural tubes were examined for Nestin and Tuj1 (Fig. 4B). Stage-matched mutant embryonic neural tubes were comparable to wild type, indicating that neural induction is unperturbed in mice lacking cilia. Thus, it appears either that cilia do not serve as a constraint of neurogenesis in vivo as they do in EBs or that increased neural differentiation is specific to loss of Ofd1.

Neural induction in mouse embryos lacking cilia and Ofd1 mutant missense EBs.
Loss of cilia causes decreased processing of Gli3 into Gli3 repressor in both Ift88 and Kif3a mutant embryos, consistent with what is seen in the Ofd1Gt EBs [10,51]. To determine whether reduced levels of Gli3 repressor affect neural induction, we examined neural markers in Gli3xt/xt mutant embryos. Similar to the Ift88 and Kif3a mutants, a 30%–50% decrease in neural marker expression was observed in Gli3xt/xt mutants as compared with their wild-type littermates at E9.5 (Fig. 4A). These results exclude Gli3 from mediating Ofd1-dependent effects on neural differentiation, and further indicate that the stabilized full-length Gli3 seen in the Ofd1Gt EBs does not function in neurogenesis.
To further dissect the differentiation potential of Ofd1Gt EBs, we tested whether Ofd1Gt EBs could form ventral neural subtypes. The ventral neural tube is patterned by Shh produced in the notochord and floor plate [52]. Mice lacking primary cilia have dorsalized neural tubes, including a loss of the most ventral neurons, V3 interneurons, and a decrease in motor neuron formation [11,16]. To determine whether this same phenotype occurs in vitro, wild-type and Ofd1Gt EBs were assayed for Nkx2.2, a marker of V3 interneurons. Whereas wild-type EBs showed widespread Nkx2.2 expression, Ofd1Gt EBs had very little or low Nkx2.2 expression (Fig. 4C). In contrast, Ofd1Gt EBs form motor neurons at levels similar to wild-type EBs as assessed by Islet1/2 levels (Supplementary Fig. S2D). Thus, neurons that require the highest levels of Hh signaling, V3 interneurons, do not form in the absence of cilia, whereas motor neurons, which require intermediate levels, are formed in Ofd1Gt EBs.
One reason for the disparity between the roles that cilia play in neural induction in EBs and embryos may be differences in the prevalence of cilia in EBs and mouse embryos during early development. ES cells can be ciliated and become more highly ciliated upon differentiation, whereas blastocysts, from which ES cells are derived, have not been reported to possess cilia. The first evidence of ciliation in the mouse embryo is in the embryonic node at E7.5 [53]. As differences in the prevalence of cilia could affect how ES cells and inner cell mass cells respond to signaling pathways before and during neural induction, we stained wild-type blastocysts to see if cilia were present. Most blastocysts displayed acetylated tubulin staining of mid-bodies and mitotic spindles (Fig. 4D). Moreover, we observed several blastocysts that possessed long, acetylated tubulin-containing projections, but these projections did not extend from the basal body as indicated by co-staining with Rootletin, a marker of the ciliary rootlet (Fig. 4D). As primary cilia derive from basal bodies, our interpretation is that these acetylated tubulin-positive projections are not cilia, but a distinct type of microtubular cellular structure.
Finally, to assess the effects of human disease-associated OFD1 mutations on neurogenesis, we examined whether 4 ES cell lines containing distinct OFD1-associated missense mutations in highly conserved residues or domains display defects in neural induction [23]. Although Ofd1 missense lines, G139S and KDD359-361FSY, retain some Ofd1 protein that localizes correctly to the centrioles, all missense mutant lines show a complete or partial loss of cilia [23]. Upon differentiation, all Ofd1 missense mutant EBs displayed increased expression levels of neural markers Sox1, Nestin, Sox3, and Tuj1 compared with wild-type EBs (Fig. 4E). ES cells with mutations G139S and KDD359-361FSY are ciliated at ∼20% and 35% of wild-type levels, whereas mutations S75F and A80T prohibit Ofd1 centriolar localization and cilia formation [23]. Despite differences in ciliation frequency, neural induction was relatively similar among all 4 missense mutant lines (Fig. 4E). Centrioles in the Ofd1 missense mutant cells are structurally abnormal and lack distal appendages [23]. Thus, it is possible that the centriolar role of Ofd1 may contribute independently to its ciliogenic functions and its ability to restrain neural induction.
Discussion
Primary cilia are essential for the development of diverse tissues and organs. We have found that Ofd1 is essential for EB ciliogenesis, restrains EB neurogenesis, and is essential for V3 interneuron differentiation. These phenotypes may be attributable to the demonstrated misregulation of Hh and Wnt signaling in Ofd1Gt EBs. However, mouse embryos lacking Ift88 or Kif3a, other proteins essential for ciliogenesis, do not show increased neural induction, raising the possibility that the neural induction defect is caused by altered centriolar structure.
To help elucidate the signaling mechanisms causing increased neural induction, we investigated the involvement of the BMP, Hh, and Wnt pathways, known regulators of neural induction and differentiation. Inhibition of the BMP pathway has been shown to be important for neural induction in diverse vertebrates, and in some species, BMP inhibition is sufficient to induce neural tissue [43,45,48,49,54]. Mammals require BMP signaling for proper neural induction, but also require inputs from additional pathways. When induced with BMP agonists or antagonists, wild-type and Ofd1Gt EBs displayed comparable changes in neural induction, suggesting that the cilium is not essential for interpretation of BMP signals. Likewise, levels of phosphorylated Smads, the downstream mediators of BMP signaling, were similar in wild-type and Ofd1Gt EBs, indicating that BMP pathway activity is not dependent on Ofd1 or the primary cilium. Further, disruption of the intracellular BMP signal transduction pathway using Smad RNAi resulted in comparable increases in neural marker expression. These experiments suggest that loss of cilia has no effect on BMP signaling, and misregulation of other pathways likely results in increased neurogenesis in Ofd1Gt EBs.
Both Hh and Wnt signaling pathways are important for neural differentiation and specification [43,52,55]. Previous studies have shown that cells and mouse embryos lacking primary cilia have defective Hh signaling and can display overactive Wnt responsiveness [37]. Consistent with this data, we found that Ofd1Gt EBs also have an overactive canonical response to Wnt3a. Additionally, we show that Ofd1Gt EBs cannot activate downstream Hh target genes upon treatment with Shh and show abrogated processing of Gli3 to the truncated repressor form. Thus, Ofd1Gt EBs recapitulate the known biochemical and transcriptional Hh signaling defects displayed by mouse embryos lacking cilia.
Despite similarities in signaling profiles between Ofd1Gt EBs and embryos lacking cilia, we observed increased neural induction in Ofd1Gt EBs, but not in Ift88 or Kif3a mutants. One possible reason for the divergence in neural induction observed in embryos without cilia and unciliated EBs may be differences in the temporal regulation of ciliogenesis. Approximately 15% of undifferentiated ES cells are ciliated, whereas blastocysts do not appear to be. Early discrepancies in ciliation frequency could influence how signaling is mediated and consequently how cell fates are determined. Another fundamental difference between EB differentiation and embryo development is the spatial disorganization of EB tissues. During development, embryonic architecture limits the exposure of prospective neural tissue to other tissues, signals, and cell–cell contacts. These barriers are less present in the chaotic EB environment.
Variation of growth conditions could also account for neurogenesis differences between Ofd1Gt EBs and Ift88 and Kif3a mutant embryos. EBs are cultured in vitro in the presence of signaling factors than normally regulate embryo development. Nonetheless, this formula of factors may be quite distinct than the milieu found in the developing embryo.
Although Hh and Wnt signaling are abnormal in the Ofd1Gt EBs, the effects of these two pathways on neurogenesis either alone or cooperatively is unclear. Wnt signaling is important for maintenance of neural precursors and specification of the dorsal spinal cord, whereas Shh signaling from the notochord induces neural proliferation and specifies subclasses of ventral interneurons [15,16,48,54 –59]. There are, however, instances in which the Hh and Wnt pathways coordinately regulate neural development. For example, Gli3 repressor inhibits canonical Wnt signaling by binding β-catenin during spinal cord patterning [56]. Additionally, glycogen synthase kinase 3 (GSK3), a component of both the Hh and Wnt pathways, is required for normal proliferation of neural progenitors [55]. Future studies are needed to dissect the molecular interactions of these two pathways during ES cell differentiation.
Another area of future study is to explore whether defects in primary cilia or defective centrioles may affect ES cell differentiation in distinct ways. Ofd1 null mice show phenotypes resembling those of other mutant embryos lacking cilia, such as Ift88 mutants, suggesting that Ofd1 functions primarily in cilium assembly. Indeed, Ofd1 missense mutant cells lacking cilia, either partially or completely, all displayed increased neural induction. All Ofd1 missense mutant lines also have abnormal centriolar structure, even in cases in which the mutant Ofd1 localizes properly to the distal centriole [23]. Thus, it is difficult to discern whether increased neural differentiation is a result of loss of cilia or defective centrioles. To distinguish these 2 possibilities, neural differentiation will need to be assayed in ES cells that lack cilia but have normal centriolar structure.
The neural tube is patterned by signaling molecule gradients initiated in the notochord and roof plate. V3 interneurons are the most ventral subtype, requiring the highest levels of Hh signaling for specification. Indeed, mouse mutants without cilia lack V3 interneurons and form few motor neurons due to the absence of Hh signaling. Consistent with this finding and the loss of Hh responsiveness in Ofd1Gt EBs, we observed few, if any, V3 interneurons in the Ofd1Gt EBs. In contrast, Ofd1Gt EBs do form motor neurons, which are induced more dorsally in the neural tube than are V3 interneurons. Given that inhibition of Gli3 repressor participates in motor neuron development, it is likely that formation of motor neurons in Ofd1Gt EBs is caused by decreased Gli3 processing [15,60 –62]. Hence, the defects in Ofd1Gt EB neural specification recapitulate defects in neural tube patterning observed in mouse mutants with dysfunctional cilia.
This work demonstrates that ES cell differentiation is a useful way to study the developmental mechanisms underlying ciliopathies. Although we differentiated ES cells spontaneously in suspension culture, other protocols that direct differentiation to specific cell types such as pancreatic cells, cardiomyocytes, and motor neurons may be utilized to study the role of primary cilia in the development of specific cell types. For example, directed differentiation of Ofd1Gt ES cells to cardiomyocytes or neural stem cells may allow one to study how primary cilia affect stem cell maintenance or the ability to respond to stress or damage [5]. This system also potentially alleviates the need to derive and immortalize cell lines, which may facilitate correlating cell biological and embryological findings. Taken together, these results examining the role of Ofd1 and primary cilia in neural differentiation demonstrate the utility of the ES cell system to study the intricate role of cilia in developmental signaling and patterning.
Footnotes
Acknowledgments
The authors thank the UCSF Nikon Imagining Center at UCSF as well as the UCSF Lentiviral RNAi and FACS core. We also thank Holly Ingram for use of the 7300 Real-Time polymerase chain reaction system and members of the Reiter lab for valuable discussions. The Nkx2.2 and Islet1/2 antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development (NICHD) and maintained by The University of Iowa, Department of Biology, Iowa City, IA. This work was funded by grants from the California Institute for Regenerative Medicine, the National Institutes of Health (RO1AR054396), the Burroughs Wellcome Fund, the Packard Foundation, the Sandler Family Supporting Foundation, and the Helmsley Charitable Trust to J.F.R., and also by fellowships from Genentech and the California Institute for Regenerative Medicine to J.H.
Author Disclosure Statement
All authors state that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.