
Abstract
Silent mating type information regulation 2 homolog 1 (SIRT1) plays a critical role in reactive oxygen species-triggered apoptosis in mouse embryonic stem (mES) cells. Here, we investigated a possible role for the PTEN/Akt/JNK pathway in the SIRT1-mediated apoptosis pathway in mES cells. Akt was activated by removal of anti-oxidant 2-mercaptoethanol in SIRT1 −/− mES cells. Since PTEN is a negative regulator of Akt and its activity can be modulated by acetylation, we investigated if SIRT1 deacetylated PTEN to downregulate Akt to trigger apoptosis in anti-oxidant-free culture conditions. PTEN was hyperacetylated and excluded from the nucleus in SIRT1 −/− mES cells, consistent with enhanced Akt activity. SIRT1 deficiency enhanced the acetylation/phosphorylation level of FOXO1 and subsequently inhibited the nuclear localization of FOXO1. Cellular acetylation levels were enhanced by DNA-damaging agent, not by removal of anti-oxidant. c-Jun NH2-terminal kinase (JNK) was activated by removal of anti-oxidant in SIRT1-dependent manner. Although p53 acetylation was stronger in SIRT1 −/− mES cells, DNA-damaging stress activated phosphorylation and enhanced cellular levels of p53 irrespective of SIRT1, whereas removal of anti-oxidant slightly activated p53 only with SIRT1. Expression levels of Bim and Puma were increased in anti-oxidant-free culture conditions in an SIRT1-dependent manner and treatment with JNK inhibitor blocked induction of Bim expression. DNA-damaging agent activated caspase3 regardless of SIRT1. Our data support an important role for SIRT1 in preparing the PTEN/JNK/FOXO1 pathway to respond to cellular reactive oxygen species.
Introduction
E
Silent mating type information regulation 2 homolog 1 (SIRT1) is an evolutionally conserved NAD+-dependent class III histone deacetylase [6]. SIRT1 is predominantly localized in the nucleus, but is involved in cellular functions in the cytoplasm. SIRT1 plays a role in cellular functions such as longevity, apoptosis, and stress resistance by deacetylating important proteins, including p53, NF-κB, Ku70, PTEN, and FOXO transcription factors [7,8].
Phosphatidylinositide 3-kinase (PI3K) and its downstream protein kinase Akt play important roles in cell survival and proliferation by inhibition of apoptotic pathways by phosphorylating downstream targets such as BCL2-associated agonist of cell death (BAD), caspase 9, IκB kinase (IKK), and FOXO [9]. Although Akt predominantly localizes in the cytoplasm and is activated by PI3K near the plasma membrane, Akt can be activated and can function in the nucleus. Components in PI3K signaling pathways are found in the nucleus, including PIP2, PIP3, PI3K, and PDK1 [9,10]. Nuclear Akt promotes cell survival by its kinase activity or by interacting with nuclear proteins such as FOXO and B26 [11].
PTEN is a negative regulator of the PI3K/Akt pathway by dephosphorylating PIP3 (phosphatidylinositol 3, 4, 5 phosphate) to PIP2 (phosphatidylinositol 4, 5 phosphate) [9,10,12]. PTEN activity can also be regulated by post-translational regulation, including phosphorylation, acetylation, oxidation, and control of its localization [9,10]. Acetylation of PTEN modulates its phosphatase activity and interaction with (postsynaptic density 95, PSD-85; discs large, Dlg; zonula occludens-1, ZO-1 (PDZ)-domain containing proteins [13,14]. Although PTEN is localized mainly to the cytoplasm, nuclear localization of PTEN has been reported. Nuclear PTEN regulates chromosome stability, DNA repair, cell cycle arrest, and apoptosis by controlling cyclin D, CENP-C, RAD51, p53, and AKT. PTEN can enter nucleus by several mechanisms such as passive diffusion, major vault protein-mediated nuclear transport, PI3K-S6K signaling, Ran-dependent mechanism, and monoubiquitination [10,14].
Members of the class O of forkhead box transcription factors (FOXO) regulate cell cycle, apoptosis, DNA repair, and ROS resistance by activating or repressing Bim, FasL, p27, Gadd45a, and manganese superoxide dismutase (MnSOD) [15,16]. Transcriptional activity and cellular localization of FOXO are tightly regulated by post-translational modifications. Akt and c-Jun NH2-terminal kinase (JNK) have opposing effects on FOXO activity and localization. Akt facilitates nuclear export of FOXO to block expression of apoptotic regulators [15,17], whereas JNK promotes nuclear import of FOXO to enhance expression of FOXO-regulated genes [18 –20]. ROS activates JNK by inhibiting MAP kinase phosphatase (MKPs) [21] and activating apoptosis signal-regulating kinase 1 (ASK1) [22]. FOXO transcriptional activity is also regulated by acetylation [23,24]. Although Sirt1 has been generally accepted to deacetylate and activate FOXO, the function of SIRT1 in FOXO regulation remains unclear.
BH3-only proteins trigger apoptosis by activating BCL2-associated X protein (BAX) or BCL2-antagonist/killer (BAK) [25,26]. Bim and Puma are potent proapoptotic BH3-only proteins and are critical for apoptosis of various cell types. Expression of Bim and Puma is controlled by FOXO-mediated transcriptional regulation [27,28].
We reported that SIRT1 is essential for ROS-mediated apoptosis in mES cells by facilitating mitochondrial localization of p53 [8]. We have now investigated how SIRT1 regulates the PTEN/JNK/FOXO pathway to modulate Bim and Puma expression in mES cells.
Materials and Methods
mES cells culture
Parental mES cell line R1 [29] and SIRT1
−/− mES cell line derived from R1 cells [30] were maintained on mitomycin C (MMC)-treated mouse embryonic fibroblasts in knock-out Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA;
Immunoblotting and immunoprecipitation
Total lysates were prepared using ice-cold Mg2+ lysis/wash buffer (Millipore) containing protease inhibitor mixture (Complete; Roche, Indianapolis, IN;
Antibodies
The following antibodies were used in western blot analyses at 1:1,000 dilution: anti-Bim (2819), anti-Bax (2772), anti-cleaved caspase 3 (9661), anti-phospho-p53 (Ser15; mouse Ser18) (16G8), anti-phospho-p53 (Ser392; mouse Ser389) (9281), anti-acetyl-p53 (Lys379) (2570), anti-acetylated lysine (9441), anti-PTEN (138G6), anti-Akt (9272), anti-phospho-Akt (Ser473) (9271), anti-phospho-JNK (Thr183/Tyr185) (9251), anti-p70S6kinase (49D7), anti-Foxo1 (C29H4), and anti-phospho-Foxo1(T24)/Foxo3a(T32) (9464) (Cell Signaling Technology, Beverly, MA;
Apoptosis assays
Apoptosis was measured by Annexin V/7AAD staining (BD Biosciences, San Jose, CA;
Statistical analysis
Each experiment was performed 3 to 5 times. Statistical significance was determined by unpaired Student's t-test. Values of P < 0.05 were considered significant.
Results
SIRT1 deficiency activates Akt in response to removal of 2-ME
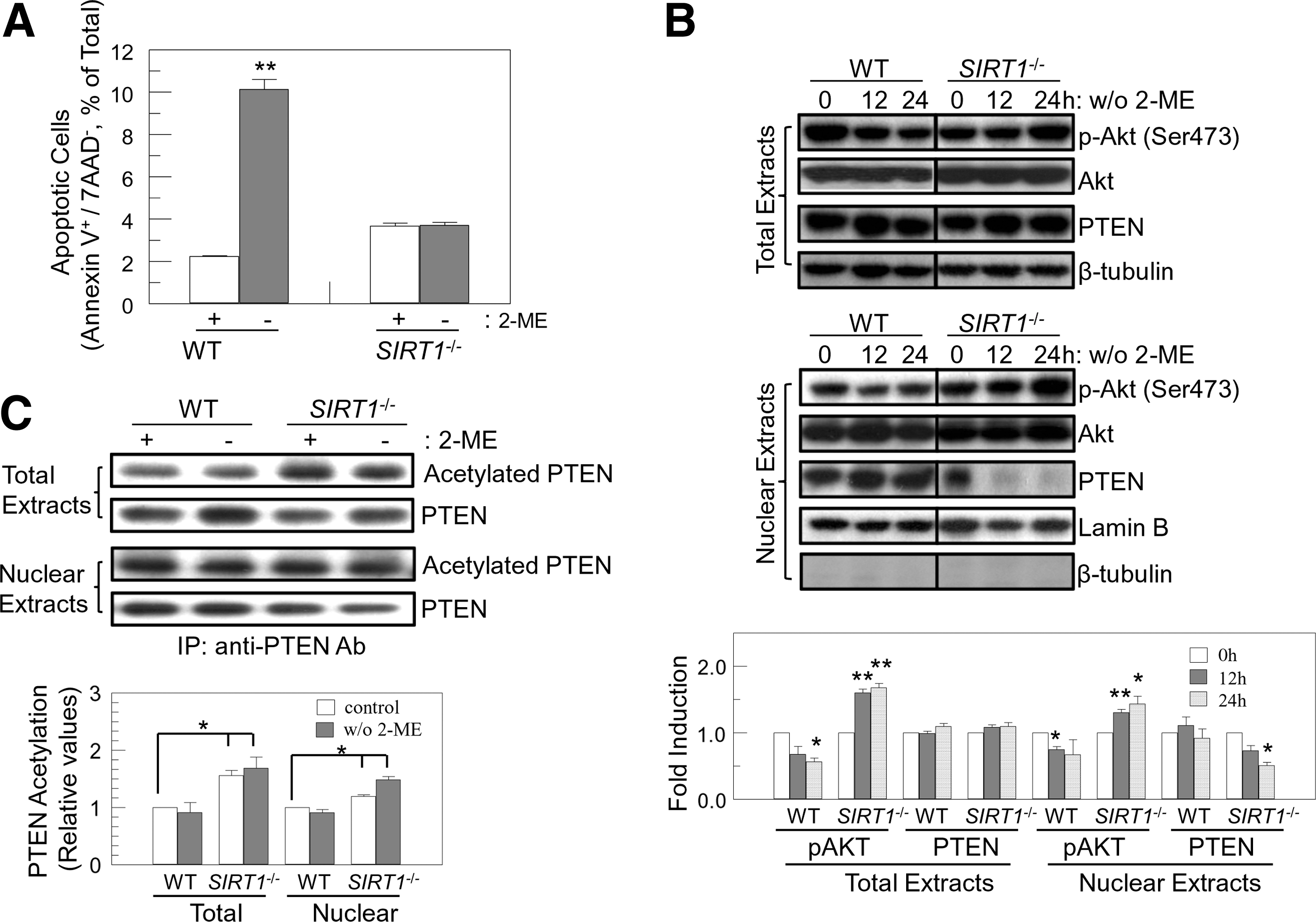
Endogenous ROS, generated by removal of anti-oxidants, triggers apoptosis through an SIRT1-mediated pathway (Fig. 1A) [8]. Since the PI3K/Akt pathway plays an important role in protecting cells from stresses, including ROS, we investigated whether the PI3K/Akt pathway was involved in SIRT1-mediated apoptosis in mES cells. To determine the role of PI3K/Akt in ROS-induced apoptosis, we assessed levels of Akt phosphorylation after 2-ME removal in the culture medium. Withdrawal of 2-ME suppressed Akt phosphorylation in wild-type (WT) mES cells, and enhanced phosphorylation of Akt in SIRT1 −/− cells (Fig. 1B, upper panel), which is consistent with apoptosis (Fig. 1A). Akt downstream molecules such as FOXO transcription factors are nuclear proteins [9]. Thus, we examined nuclear Akt activity in anti-oxidant-free conditions. Nuclear Akt manifested the same results as total cellular Akt: downregulation in WT and upregulation in SIRT1 −/− cells (Fig. 1B, lower panel). p-Akt levels in nuclear fraction were calculated after normalization of the amount of loaded proteins. As the amount of loaded protein at 12 h in SIRT1 −/− is lower than control based on the Lamin B protein levels, p-Akt at 12 h in SIRT1 −/− was enhanced compared to control.
AKT is activated by 2-ME withdrawal from SIRT1
−/− mES cells.
As PTEN, a lipid and protein phosphatase, acts as a negative regulator of Akt by dephosphorylating PIP3 [9,12], we speculated that change of PTEN activity after 2-ME withdrawal might regulate Akt. PTEN protein levels were not changed after 2-ME withdrawal (Fig. 1B, upper panel). However, PTEN was mainly sequestered in the cytoplasm in SIRT1 −/− cells after removal of 2-ME, consistent with increased Akt phosphorylation (Fig. 1B, lower panel). This suggests that exclusion of PTEN from the nucleus allows activation of Akt to protect ES cells from ROS-induced stress in SIRT1 −/− cells.
Recently, acetylation of PTEN has been reported to regulate its phosphatase activity and interaction with PDZ-containing proteins [33,34]. We evaluated whether SIRT1 deacetylated PTEN to regulate Akt activity in response to endogenous ROS. SIRT1 deficiency enhanced basal acetylation levels of PTEN and sustained acetylation levels after 2-ME withdrawal in SIRT1 −/− cells (Fig. 1C). Nuclear PTEN was also hyperacetylated in SIRT1 −/− cells (Fig. 1C). This suggests that SIRT1 deacetylates PTEN to prevent Akt activation after removal of 2-ME, which in turn initiates apoptosis in response to ROS.
Regulation of Foxo1 activity by SIRT1 in mES cells
As SIRT1 and Akt regulate FOXO1 transcription factor [7,15,16], we investigated whether the SIRT1-Akt-FOXO1 pathway was responsible for apoptosis in response to endogenous ROS. SIRT1-mediated deacetylation of FOXO activates its transcriptional activity and promotes its nuclear retention [15,16]. We examined acetylation levels of FOXO1 in WT and SIRT1 −/− mES cells. As we expected, basal acetylation levels of FOXO1 were significantly higher in SIRT1 −/− cells (Fig. 2A). Acetylation facilitates Akt-mediated phosphorylation (Thr24) of FOXO1, which in turn regulates transcriptional activity of FOXO1 [15,16]. Consistent with enhanced acetylation, FOXO1 was hyperphosphorylated (Thr24) in SIRT1 −/− cells. FOXO1 phosphorylation was suppressed in WT cells (Fig. 2A). Akt phosphorylates and inhibits FOXO by promoting interaction of FOXO with 14-3-3 proteins and nuclear export [15,16]. We therefore investigated whether SIRT1 deficiency inhibited nuclear import of FOXO1. As expected, nuclear localization of FOXO1 was increased by endogenous ROS in WT cells, but not in SIRT1 −/− cells (Fig. 2B). This suggests that SIRT1 deficiency inhibited FOXO1 by direct acetylation and phosphorylation via the PTEN/Akt pathway to make mES cells less sensitive to endogenous ROS.
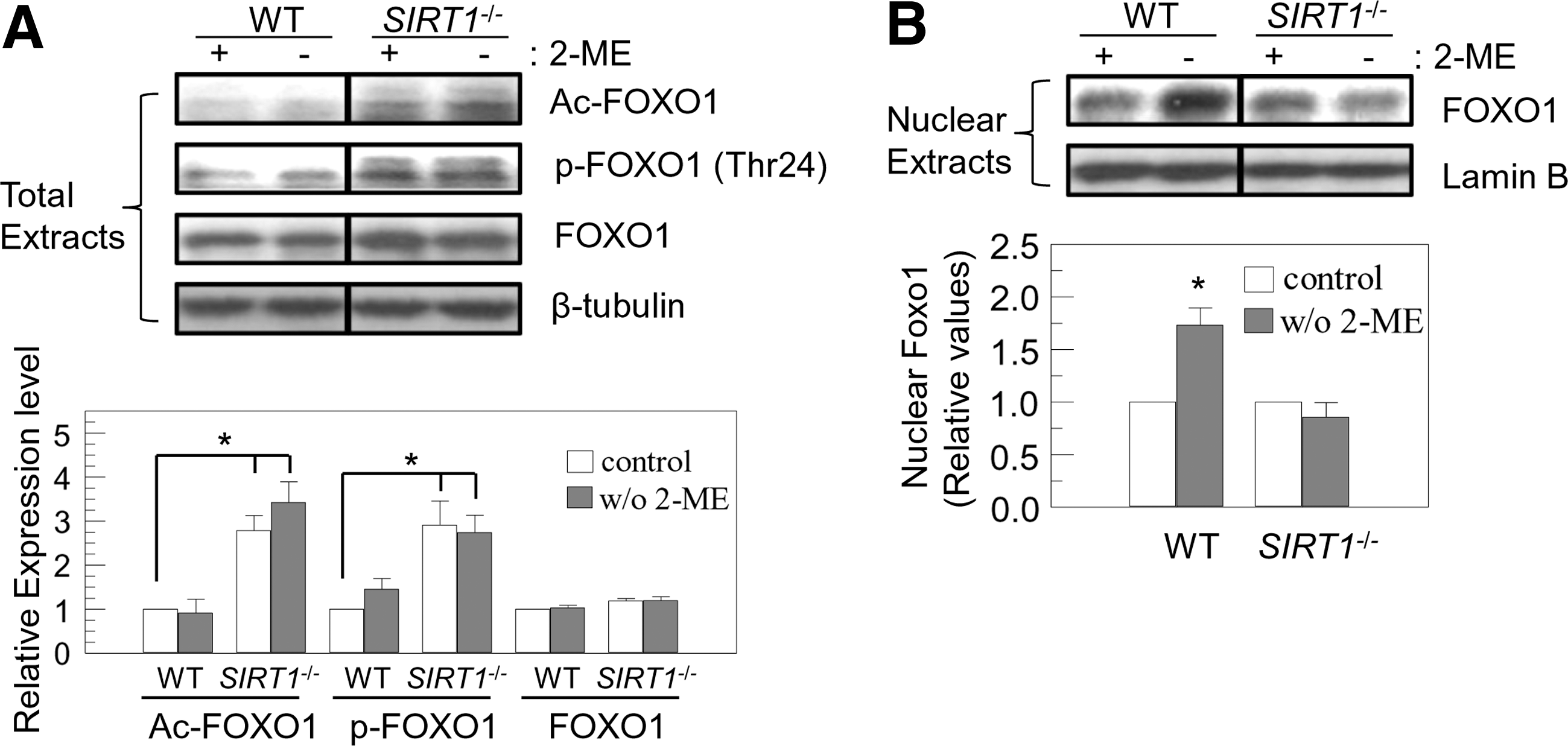
Nuclear import of FOXO1 is induced by removal of 2-ME in an SIRT1-dependent manner. WT and SIRT1
−/− mES cells were cultured with or without 2-ME (100 μM, 12 h). Total extracts
Differential regulation of JNK and p53 by ROS and DNA-damaging stress
Acetylation of FOXO1 and PTEN was not changed after 2-ME withdrawal, even though acetylation of FOXO1 and PTEN was enhanced in SIRT1 −/− cells (Figs. 1C and 2A). We therefore investigated cellular acetylation level upon 2-ME removal to address whether SIRT1 was activated to initiate ROS-induced apoptosis. As expected, cellular acetylation was markedly enhanced in SIRT1 −/− cells (Fig. 3A). Although treatment with DNA-cross linking agent, MMC, increased cellular acetylation both in SIRT1 −/− and to a less extent in WT mES cells, removal of 2-ME did not affect cellular acetylation in mES cells (Fig. 3A). This suggests that SIRT1 is not an ROS-sensitive protein, even though SIRT1 is required for the proper function of proteins involved in the ROS-mediated apoptosis. We next investigated which protein is activated by ROS to induce activation of PTEN/Akt/FOXO1 pathway. ROS activates FOXO by JNK, which can induce nuclear import of FOXO1 by inhibiting 14-3-3 binding to FOXO1 [16]. Therefore, we assessed JNK activation in response to removal of anti-oxidant 2-ME in mES cells. Phosphorylated JNK proteins were measured after culturing mES cells in 2-ME-free culture media. 2-ME-free culture media enhanced phosphorylated JNK protein levels in WT, but not in SIRT1 −/−, mES cells. However, MMC treatment did not affect JNK phosphorylation (Fig. 3B). Basal JNK phosphorylation was highly maintained in SIRT1 −/− cells (Fig. 3B). These data demonstrate that ROS and DNA-damaging stresses activate different pathways to initiate apoptosis. p53 responds to a broad range of stresses to control cellular fates and responses including apoptosis, cell cycle arrest, or senescence [35]. Removal of 2-ME is associated with mitochondrial p53 induction of apoptosis [8]. DNA-damaging stresses also activate the p53 pathway to induce apoptosis [36] or differentiation into somatic cells by repressing Nanog expression [37] in mES cells. p53 activity is tightly controlled by multiple post-translational modifications. Acetylation of p53 is involved in transcriptional activation of p53 [35]. Therefore, we evaluated stability and modifications of p53 before and after stresses of ROS and DNA cross-linking treatment. Basal levels of p53 acetylation were as expected higher in SIRT1 −/− cells. Although SIRT1 deficiency did not affect p53 protein level and phosphorylation on serine 18 of p53 before stresses, p53 phosphorylation on serine 389 was increased (Fig. 4). p53 protein level and phosphorylation at serine 389 were enhanced by removal of 2-ME in WT, but not in SIRT1 −/− cells (Fig. 4). In contrast, protein levels and phosphorylation at serine 18 and 389 of p53 were similarly induced both in WT and SIRT1 −/− cells by MMC treatment, even though acetylation on Lysine 379 was much higher in SIRT1 −/− cells (Fig. 4). These data suggest that intracellular ROS activates JNK and p53 to initiate apoptosis in an SIRT1-dependent manner. Meanwhile, direct DNA damage stress by DNA-cross linking agent activated p53 irrespective of the presence or absence of SIRT1. This suggests that ROS triggered by removal of 2-ME induced apoptosis through activation of JNK, and in part p53.
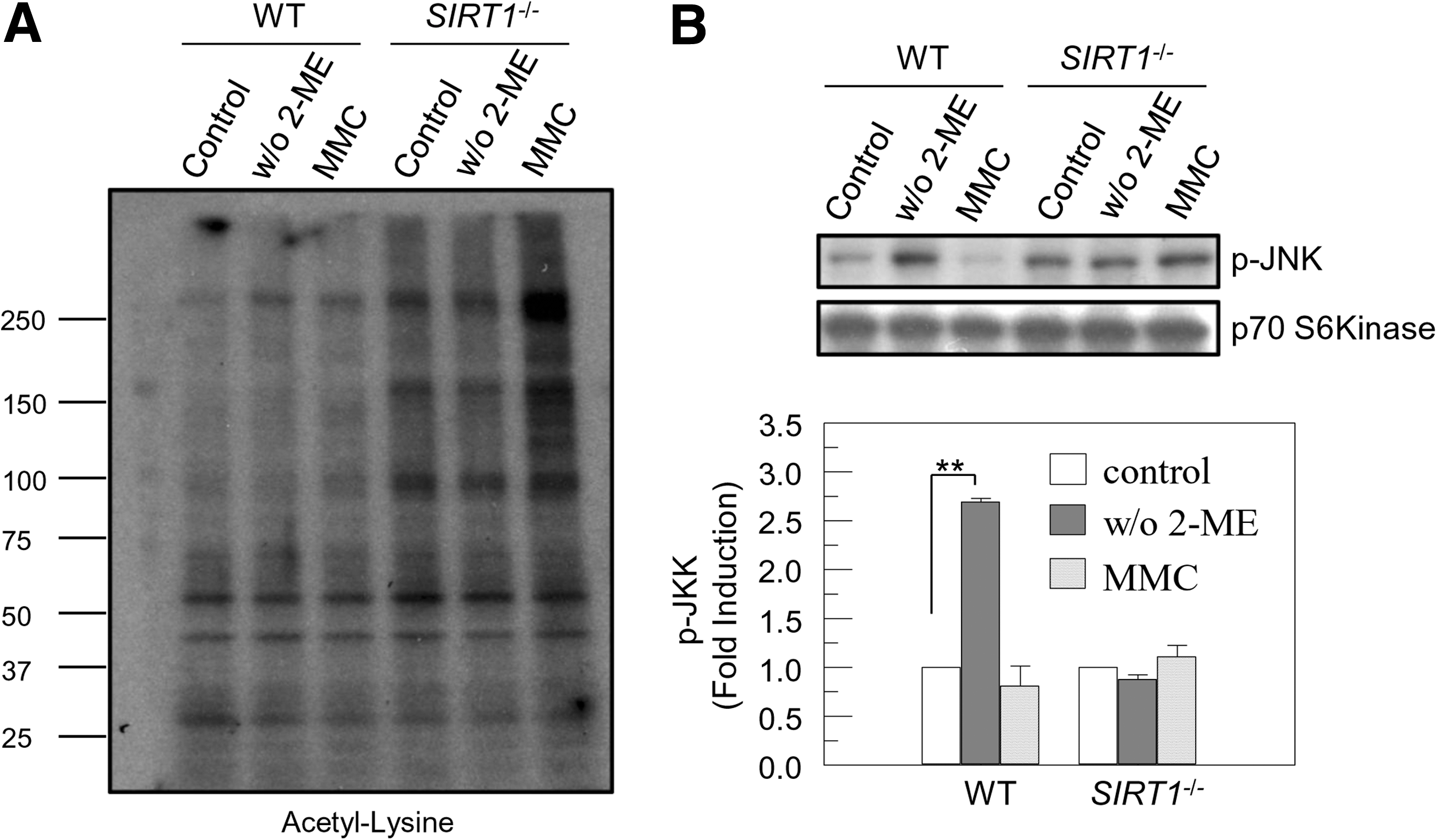
Removal of 2-ME enhances JNK phosphorylation, not cellular acetylation. WT and SIRT1
−/− mES cells were cultured with or without 2-ME (100 μM) for 24 h or treated with 2-ME and MMC (1 μg/mL) for 6 h, and then total extracts were prepared.
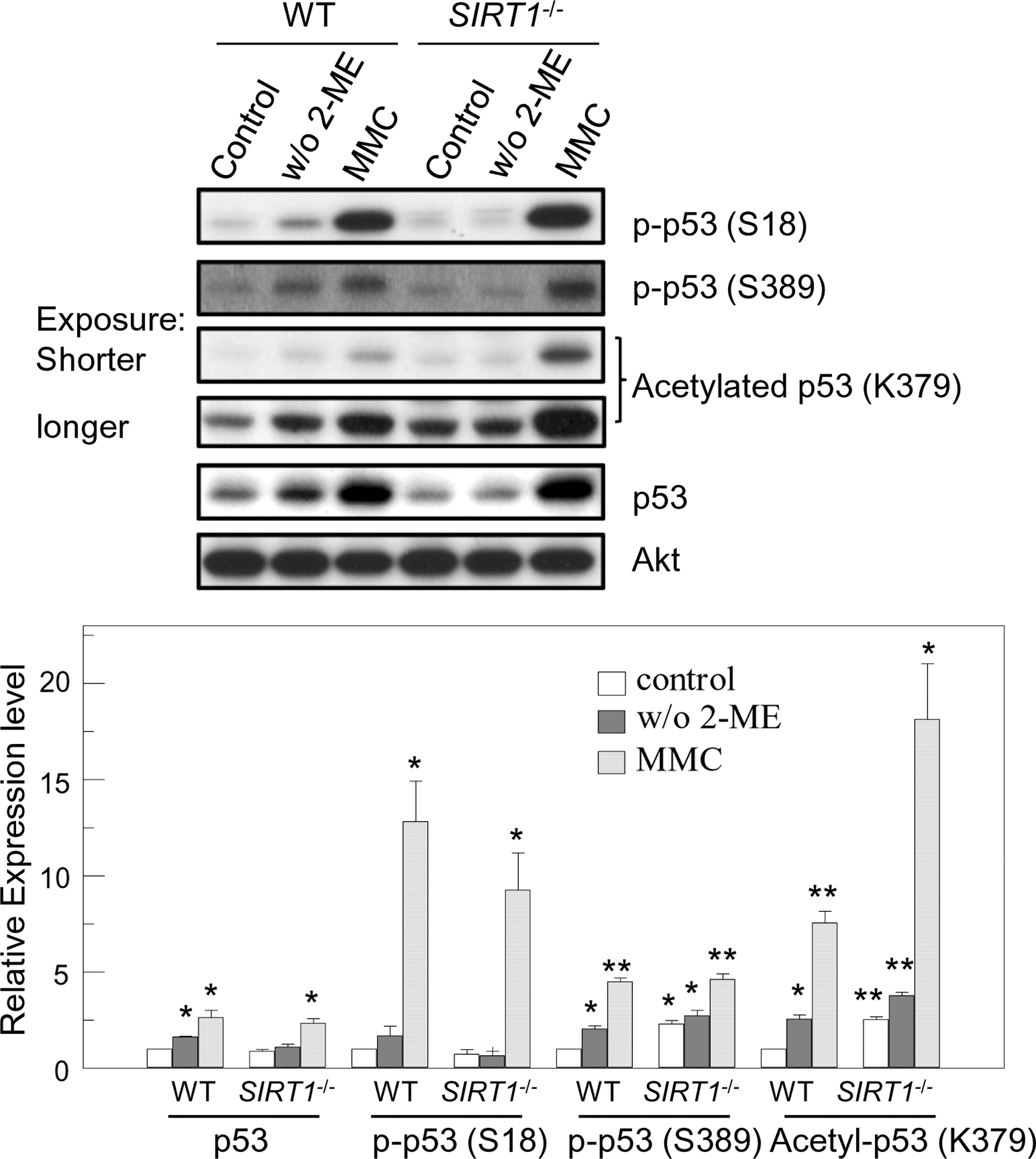
Differential regulation of p53 by ROS and DNA-damaging stress. WT and SIRT1 −/− mES cells were cultured with or without 2-ME (100 μM) for 24 h or treated with 2-ME and MMC (1 μg/mL) for 6 h, and then total extracts were prepared. Total p53 protein, and phospho and acetylated p53 were detected by immunoblotting. Relative expression levels (the value of control WT mES cells is set as 1) are shown. Values represent mean ± SEM (n = 3). *P < 0.01; **P < 0.01. ROS, reactive oxygen species.
Requirement of SIRT1 for induction of BH3-only proapoptotic Bcl-2 family proteins in context of 2-ME removal
BH3-only proteins initiate the mitochondrial apoptotic pathway in response to a variety of stresses by modulating multi-domain Bcl-2 family proteins [25,26]. Dephosphorylation and activation of FOXO induces expression of the proapoptotic BH-3 only proteins, Bim and Puma, to trigger apoptosis in response to ROS [27,38]. Puma is also induced by p53 activation [35]. To address whether BH3-only proteins were induced in ROS-triggered apoptosis in mES cells, we assessed expression levels of BH3-only proteins after removal of 2-ME. Expression of all 3 major isoforms of Bim proteins, extra long (EL), long (L), and short (S), was significantly increased 24 h after culture in 2-ME-free media in WT cells, but not by treatment of cells with DNA-damaging agent, MMC (Fig. 5A). In SIRT1 −/− cells, Bim protein level was not enhanced by MMC treatment or 2-ME withdrawal. Puma α was also induced in WT mES cells by MMC treatment as well as 2-ME withdrawal. In SIRT1 −/− mES cells, Puma α was induced by MMC treatment, but not by 2-ME withdrawal (Fig. 5A). Bax expression was unaffected by 2-ME withdrawal or MMC treatment (Fig. 5A). Thus, we found that Bim and Puma expression is induced by endogenous ROS in an SIRT1-dependent manner and Puma is induced by MMC treatment independent of SIRT1. Caspase 3, a key mediator of apoptosis in mammalian cells, was activated by both 2-ME withdrawal and MMC treatment in WT cells, but only by MMC treatment in SIRT1 −/− cells (Fig. 5A). Notably, basal Bim and Puma expressions were suppressed in SIRT1 −/− cells [35.3% ± 9.7% (Bim), 35.8% ± 10% (Puma); % of WT cells; mean ± SEM; n = 3; P < 0.05].
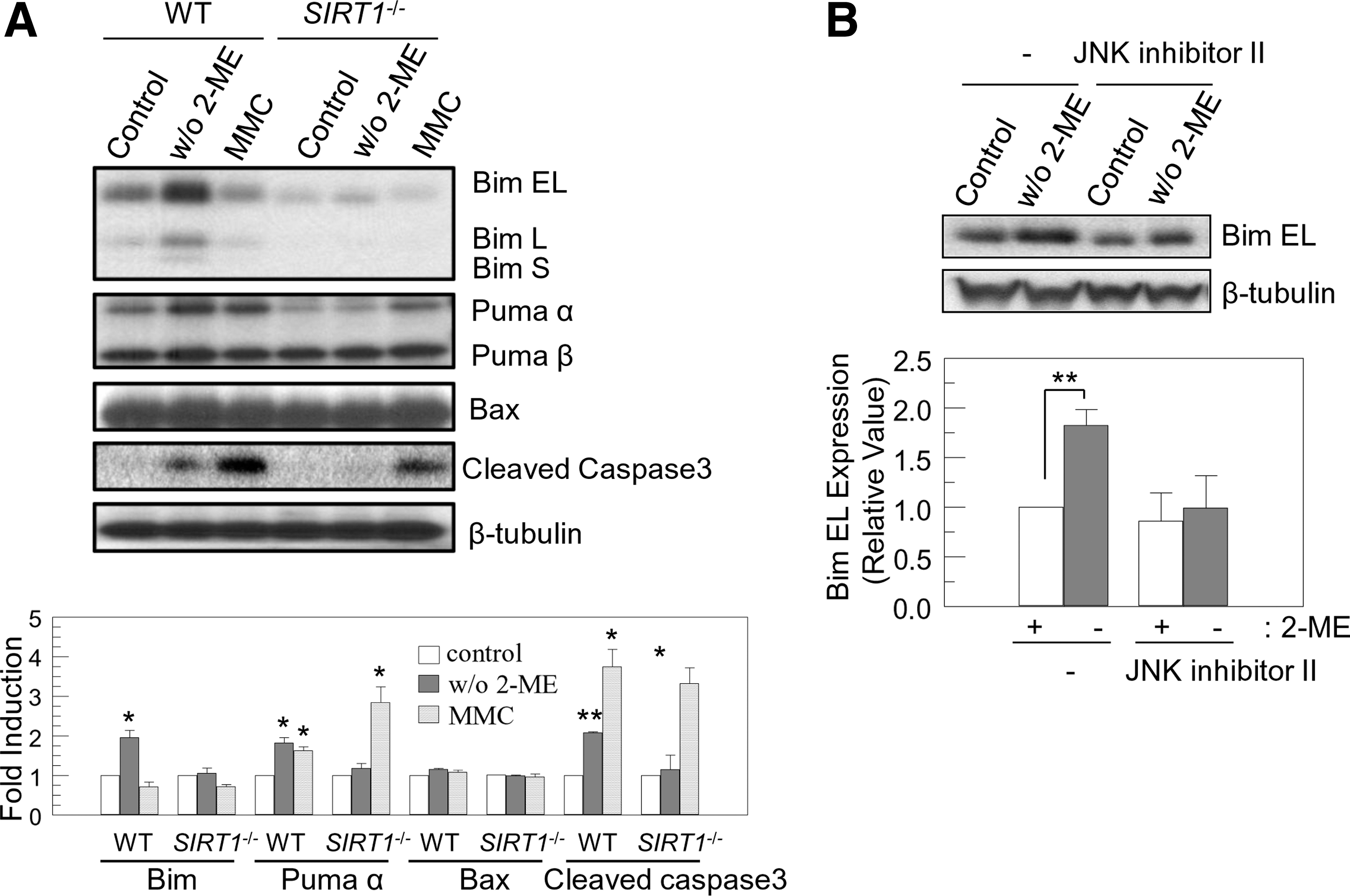
Removal of 2-ME induces expression of Bim and Puma to activate Caspase3 through JNK.
JNK activity has been shown to induce Bim expression in neurons [19] and to control ROS-mediated apoptosis [21]. Therefore, we assessed JNK activation in response to withdrawal of anti-oxidant 2-ME in mES cells. To determine whether JNK mediated endogenous ROS-triggered Bim induction, we pretreated WT cells with the JNK inhibitor SP600125 in the absence or presence of 2-ME. JNK inhibitor decreased both ROS-triggered and basal Bim EL expression (Fig. 5B). In SIRT1 −/− cells, a significant amount of JNK was phosphorylated without stimulation (Fig. 3B). Since JNK can function as an anti-apoptotic as well as pro-apoptotic regulator [39], we tested whether the hyperactivated JNK protected SIRT1 −/− mES cells from reactive oxygen stress. SIRT1 −/− cells were pretreated with JNK inhibitor SP600125 (10 μM) for 24 h, and then further cultured with or without 2-ME (100 μM) for 24 h. Pretreatment of JNK inhibitor induced apoptosis in SIRT1 −/− cells and slightly induced 2-ME-withdrawal-induced apoptosis [3.7% ± 0.15% vs. 12.7% ± 0.12% (control vs. JNK inhibitor, P < 0.01); 12.7% ± 0.12% vs. 15.2% ± 0.18% (JNK inhibitor: with vs. without 2-ME, P < 0.05); AnnexinV-positive/7AAD-negative population; mean ± SEM; n = 3]. However, treatment with JNK inhibitor did not affect Bim expression (data not shown). JNK showed anti-apoptotic function in an SIRT1-deficient cellular context. These data suggest that ROS-induced expression of Bim and Puma by activating JNK requires SIRT1 activity. In contrast, DNA-damaging stress activates p53, which results in Puma induction in an SIRT1-independent manner.
The PTEN/Akt pathway also regulates BAD and IKK to modulate apoptosis [9], but BAD phosphorylation and the NF-kB pathway were not affected by removal of 2-ME in mES cells (data not shown).
Together, these data suggest that ROS triggers apoptosis by activation of p53 and JNK, which require SIRT1 for proper regulation of the PTEN/Akt/FOXO pathway that induces Bim and Puma (Fig. 6).
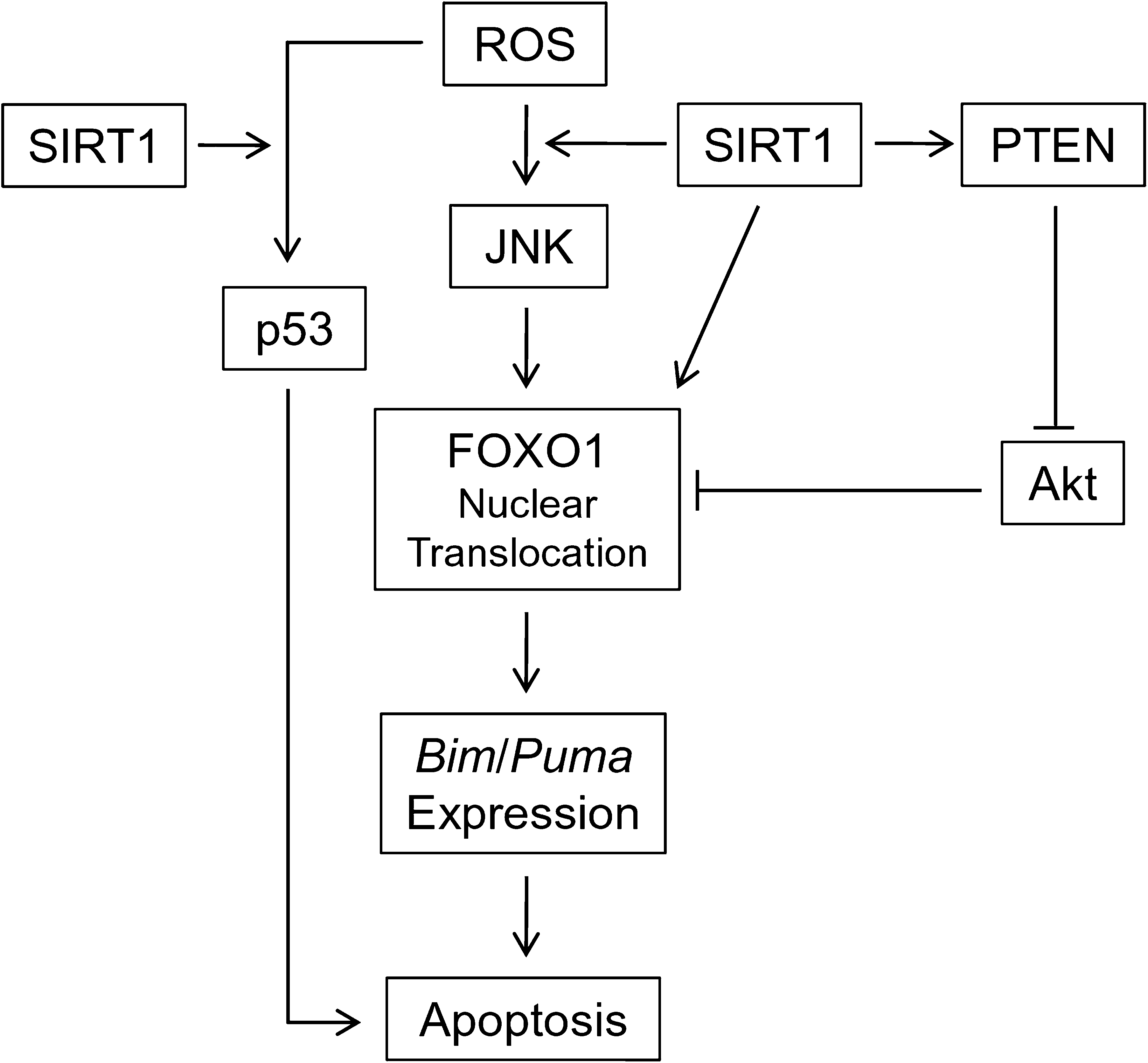
Schematic diagram of SIRT1 function in ROS-induced apoptosis in mES cells. SIRT1 is required for ROS-induced JNK activation, which results in activation and nuclear translocation of FOXO1 to induce Bim/Puma expression. SIRT1 also positively regulates FOXO1 by direct deacetylation and via inactivation of Akt. Together with p53, Bim/Puma initiates mitochondrial-mediated apoptosis.
Discussion
We report here that enhanced ROS due to removal of 2-ME induces expression of Bim and Puma through JNK/FOXO signaling in an SIRT1-dependent manner. Hyperacetylation of PTEN/FOXO1 makes SIRT1 −/− cells resistant to ROS. The function of SIRT1 in longevity and cellular protection from stresses are controversial. SIRT1 has been reported to extend life span of model organisms and protect cells from extracellular stresses [7]. However, there are also several reports showing proapoptotic function of SIRT1 [20].
Our data showed that PTEN was hyperacetylated and sequestered in the cytoplasm after removal of 2-ME, but Akt was activated by removal of 2-ME in SIRT1 −/− cells. As PTEN activity can be abrogated by oxidation and formation of a disulfide bond between Cys124 and Cys71 [13,14] and acetylation of PTEN on Lys125 and 128 also inhibits its lipid phosphatase activity to downregulate PI3K/AKT pathway [34], ROS stimulates Akt by inhibiting PTEN phosphatase activity in SIRT1 −/− cells. Activated PI3K/Akt pathway promotes nuclear export of PTEN [40]. Nuclear PTEN is enhanced upon apoptotic stresses and C-terminal deleted PTEN favors nuclear localization. Given that the deleted C-terminal tail is required for PTEN binding to PDZ-domain scaffolding proteins [41] and Lys402 acetylation in the C-terminal tail has been reported to enhance interaction with PDZ domains [33], nuclear export of PTEN is promoted to protect cells from intracellular ROS by activating nuclear Akt and blocking the nuclear PTEN-mediated apoptotic pathway in SIRT1 −/− cells [10,14].
Our data propose that JNK mediates activation and nuclear import of FOXO1 to initiate apoptosis in response to ROS. FOXO1 showed increased phosphorylation on Thr24 in SIRT1 −/− cells, which is a negative phosphorylation site by Akt. FOXO1 is also regulated by acetylation/deacetylation, which is controlled by SIRT1. The precise function of SIRT1 in the regulation of FOXO transcriptional activity is controversial: inhibition, activation, or dependent on cellular context [15,16]. These controversial results are due to the complexity of cellular acetylation. In general, SIRT1 is believed to activate FOXO activity, but SIRT1 can also deacetylate histone, histone acetyltransferases, or other transcriptional co-factors for FOXO transcriptional activity. Therefore, the effects of SIRT1 on final outcome of cellular response to various stresses might be different on different cells. SIRT1 deacetylates FOXO1, which in turn increases FOXO1 DNA binding, and inhibits negative phosphorylation by Akt. Activated FOXO1 induces expression of p27 (cell cycle arrest), MnSOD (ROS resistance), GAD45 (DNA repair), and Bim (apoptosis) [15,16]. Thus, SIRT1 can protect cells from ROS under mild stress, but SIRT1 promotes apoptosis by enhanced expression of Bim under severe stress. We did not observe any changes in acetylation/phosphorylation of FOXO1 after removal of 2-ME. JNK promotes nuclear import of FOXO1 by phosphorylating 14-3-3, which results in releasing FOXO1 from 14-3-3 [15,16]. ROS activates JNK in WT cells, not in SIRT1 −/− cells. ROS activates JNK by activating ASK1 and inhibiting MKPs. Hsp90-Akt has been reported to bind to and phosphorylate ASK1 to keep ASK1 in an inactive state to protect cells from apoptosis [42]. Since Akt was activated by removal of 2-ME in SIRT1 −/− cells, activated Hsp90-Akt might block activation of ASK1-JNK in SIRT1 −/− cells.
JNK was constitutively activated in SIRT1 −/− cells and JNK inhibition induced apoptosis in SIRT1 −/− cells. Although JNK is generally considered to be a pro-apoptotic regulator [16], it can also function as a anti-apoptotic regulator [39]. JNK-induced AP-1 activity increases expression of MnSOD to respond to ROS or collaborates with NF-kB, Akt or p53 to enhance anti-apoptotic genes [43,44]. Activated Akt might work together with JNK to keep SIRT1 −/− cells from undergoing apoptosis. This anti-apoptotic JNK activity may be responsible for keeping basal expression of Bim and Puma at a low level in SIRT1 −/− cells.
We showed differential regulation of ROS and DNA-damaging stress by SIRT1. ROS activates PTEN/JNK/FOXO1 and p53 pathways in an SIRT1-dependent manner, whereas DNA-damaging stress activates p53 pathway exclusively, irrespective of SIRT1. p53 functions as a cellular gatekeeper to coordinate diverse cellular responses such as cell cycle arrest, DNA repair, senescence, autophagy, and apoptosis in response to stresses [35]. While acetylated p53 level is enhanced in SIRT1 −/− cells before and after treatment of MMC, protein and phosphorylation on Ser18 and 389 of p53 was similarly induced after treatment of MMC in both WT and SIRT1 −/− cells, which lead to similar induction of Puma and activation of caspase3 in both cells. Although p53 acetylation has been shown to correlate with p53 activation and stabilization, p53 mutants, with substitution of 6 or 7 Lys residues with Arg, show no differences compared with WT p53. However, mutations in 8 Lys residues completely block p53-mediated cell cycle arrest and pro-apoptotic genes, even though mutant p53 shows normal p53 protein stability, mdm2 induction, and DNA-binding activity [35]. Partial acetylation is enough for p53 to regulate cellular responses to stresses. Nuclear PTEN and JNK promote p53 stabilization and apoptosis [35,45]. Our results showed that JNK was activated and nuclear PTEN was sustained by removal of 2-ME in WT cells, suggesting a relationship between SIRT1 and p53 through JNK and PTEN.
Lysine acetylation is one of the major post-translational modifications, and has been found in a broad range of proteins, preferentially large macromolecular complexes involved in controlling many cell signaling processes. High-resolution mass spectrometry identified 3,600 lysine acetylation sites on 1,750 human proteins [46]. SIRT1 deficiency enhanced acetylation levels of broad range of proteins (Fig. 3A). Differential status of cellular acetylations in cellular signal pathways other than PTEN/JNK/FOXO1 might be involved in responses to ROS in mES cells.
Our data provide novel findings that SIRT1 regulates acetylation of PTEN to control its activity and localization in response to intracellular ROS initiation of apoptosis. However, DNA-damaging stress activates p53 irrespective of its acetylation. Physiologically low levels of ROS regulate intracellular signal pathways to modulate cellular processes, whereas higher amounts of ROS induce cell death pathways [4]. ROS are generated during metabolic processes. Oxidative respiration in mitochondria is a major source of ROS. Lineage-specific differentiation is usually accompanied by metabolic changes, which makes differences in the cellular redox system. A low-level ROS pulse is required specifically for cardio and vascular differentiation from mES cells [5]. ROS also enhances spontaneous differentiation of human ES cells into mesendodermal cell lineage [47]. Jnk1 −/− Jnk2 −/− mES cells show severe defects in mesodermal differentiation [48]. Since SIRT1 −/− mES cells inhibited apoptosis in response to cellular ROS, and ROS is an important signal for lineage differentiation of ES cells, further investigation of SIRT1 target molecules during ES cell differentiation should be important for understanding lineage-specific differentiation.
Footnotes
Acknowledgments
The authors are grateful to Michael W. McBurney (Ottawa Health Research Institute) for providing SIRT1 −/− mES cells. These studies were supported by Public Health Service grants R01 HL56416 and R01 HL67384 from the National Institutes of Health to H.E.B.
Author Disclosure Statement
The authors have no conflicts of interest to disclose.