
Abstract
Members of the homeobox (Hox) gene family are known to mediate expansion of hematopoietic stem cells (HSCs) and progenitors. The absence of oncogenic properties promoted HOXB4 as prime candidate in the quest to expand HSCs for clinical purposes. Despite its potential to expand HSCs, studies with mutant mice showed that Hoxb4 is not essential for HSC generation and function under physiological conditions. Expression studies and the existence of functional redundancy in particular between paralog Hox genes suggest that HOXA4 might have potent properties to expand HSCs. Here we measured the ability of HOXA4 to promote ex vivo expansion of HSCs and progenitors using retrovirus-mediated overexpression. Our results provide evidence that HOXA4-transduced HSCs and primitive progenitors expand in culture conditions and demonstrate that the potential of expanded HOXA4 HSCs to give rise to mature myeloid and lymphoid progeny in normal proportions remained intact. Interestingly, constitutive overexpression of HOXA4 resulted in an unbalanced expansion of lymphoid/myeloid progenitors in bone marrow chimeras favorable to B-cell progenitors responsive to interleukin-7. This expansion was specific for these progenitors and not for the more primitive Whitlock-Witte-initiating cells. These data indicate that early stages of B-cell development associated with proliferation are in particular sensitive to HOXA4. Thus, this study supports the potential use of HOXA4 to expand both HSCs and B-cell progenitor populations for therapeutic strategies.
Introduction
H
Redundancy within the Hox network for controlling developmental programs in the embryo that emerged from studies with compound mutants [17,18] might also apply to their regulatory functions in hematopoiesis, as shown by lack of strong hematopoietic phenotypes in single Hox gene mutants. Surveys of Hox gene expression in HSC-enriched populations by us and others showed dominancy of the Hoxa cluster genes in primitive hematopoiesis [4,15,19,20]. Quantitative reverse transcription–polymerase chain reaction (Q-RT-PCR) analysis showed one log higher expression levels for Hoxa4 than Hoxb4 in d14.5 fetal liver populations enriched for HSCs. The fact that during this phase of development HSCs undergo their major expansion [21] suggests that Hoxa4 might be an important determinant of HSC self-renewal expansion divisions under physiological conditions. In addition, adult long-term HSCs (LT-HSCs) still expressed Hoxa4 at superior levels compared with Hoxb4, although with reduced magnitude [22]. In this respect, it is interesting to note that although adult LT-HSCs are also subjected to self-renewal, the nature of their divisions is asymmetric, giving rise to only 1 HSC postmitosis instead of 2 for fetal HSCs, and serves to maintain the HSC population throughout the lifespan of the organism. The high expression levels of Hoxa4 in HSCs combined with the high homology and functional redundancy found within Hox paralog groups [17,23,24] suggest not only that Hoxa4 might have the ability to expand HSCs, but also that the oncogenic potential of Hoxa4 could be negligible or absent. A publication by Iacovino et al. showing an increase in hematopoietic progenitors from HOXA4-overexpressing ES cells further support the potential capacity of HOXA4 to expand HSCs [25]. Moreover, some studies attributed to HOXA4 a role as potential tumor suppressor rather than as oncogene, which was based on the observation that the HOXA4 promoter was frequently hypermethylated in acute myeloid leukemia samples and that resultant low HOXA4 expression in particular combined with high Meis1 expression was found to be an adverse prognostic marker [26,27]. As the expansion of human severe combined immunodeficiency repopulating cells (SRCs) followed by transplantation induced by HOXB4 was rather moderate [12] to the HSC expansion observed in mice [5], there is a need to find other genes inducing larger regeneration of HSCs. For this reason, we examined Hox candidate HOXA4 for its capacity to expand HSCs in vitro.
Using retroviral overexpression, we showed that HOXA4-overexpressing HSCs expanded 6.6-fold during a week culture. In addition, HOXA4 induced massive proliferation of myeloid progenitors with relative higher impact on the very primitive progenitors. HOXA4 HSCs produced mature myeloid and lymphoid progeny without bias in irradiated recipient mice and thus is not interfering with hematopoietic differentiation. Further, we provide evidence that in vivo HOXA4 is preferentially expanding interleukin (IL)-7–responding B-cell progenitors above myeloid progenitors. These data show that regulatory mechanisms controlling hematopoiesis within its natural context are still intact in the presence of HOXA4, allowing largest expansions in populations that are most permissive to proliferation.
Materials and Methods
Animals
For transplantation assays, congenic mice C57BL/6 (CD45.2) and B6SJL (CD45.1) (The Jackson Laboratories, Bar Harbor, ME) were used. Mice were bred and maintained in a specific pathogen-free animal facility of the HMR Research Center. All mouse experiments protocols were approved by the Animal Care Committee of the HMR Research Center.
Retroviral constructions
Human HOXA4 cDNA (NM_002141.3) was purchased from OriGene Technologies (OriGene Technologies Inc., Rockville, MD). To allow detection of the retrovirally overexpressed protein, a FLAG (DYKDDDDK) sequence was added C-terminal of HOXA4 together with EcoRI (GAATTC) sequences by PCR using KlenTaq LA DNA Polymerase (Sigma-Aldrich Co., St. Louis, MO). The amplicon was digested by EcoRI (Invitrogen Corporation, Carlsbad, CA) and inserted in the MSCV-pgk-GFP retroviral vector.
Retroviral overexpression and in vitro expansion
B6SJL (CD45.1) mice were intravenously injected with 150 mg/kg of 5-fluorouracil (5-FU; Mayne Pharma (Canada) Inc., Montréal, QC, Canada) to recruit HSCs into cell cycle. After 4 days, bone marrow (BM) cells were isolated from these mice and cocultured for 2 days on irradiated confluent layers of packaging cell line (GP+E-86) stably producing MSCV-HOXA4-GFP or MSCV-GFP retrovirus [28] in Dulbecco's modified Eagle's medium (DMEM; Wisent Inc., St-Bruno, QC, Canada) supplemented with 15% heat-inactivated fetal bovine serum (FBS; PAA Laboratories Inc., Etobicoke, ON), 6 ng/mL IL-3, 10 ng/mL IL-6, 100 ng/mL stem cell factor (SCF), 10–5 M 2-mercaptoethanol (Mallinckrodt Baker Inc., Phillipsburg, NJ), 50 μg/mL Gentamycin (Wisent Inc.), 10 μg/mL Ciprofloxacin (Wisent Inc.), and 6 ng/mL polybrene (Tekniscience Inc., Terrebonne, QC). HOXA4-GFP– or MSCV-GFP–transduced BM cells were sorted on a fluorescence activated cell sorting (FACS) Vantage with DiVa software (BD Bioscience, Mississauga, ON) and seeded at different doses in duplicate in BM expansion medium [DMEM, 15% FBS, 6 ng/mL IL-3, 10 ng/mL IL-6, 100 ng/mL SCF, 10–5 M 2-mercaptoethanol, and 10 μg/mL Ciprofloxacin (Wisent Inc.)].
Clonogenic progenitor assays
Clonogenic progenitor assays for myeloid progenitors were performed by plating cells from HOXA4-transduced and control BM cultures or from long-term repopulated chimeras in DMEM containing 1% Methocel MC (Sigma-Aldrich) supplemented with 10% FBS, 5.7% bovine serum albumin (Tekniscience Inc.), 10–5 2-mercaptoethanol, 5 U/mL erythropoietin, 10 ng/mL IL-3, 10 ng/mL IL-6, 50 ng/mL SCF, 2 mM glutamine (Invitrogen Corporation), and 200 mg/mL transferrin (Sigma-Aldrich). Clonogenic progenitor assays for B-cell progenitors were performed by plating cells from BM isolated from HOXA4 or control chimeras in DMEM containing 1% Methocel MC supplemented with 30% FBS selected for B-cells (Stem Cell Technologies, Vancouver, BC), 10–4 2-mercaptoethanol, 2 mM glutamine, and 10 ng/mL IL-7 (Invitrogen Corporation). Colonies were scored as previously described [29]. For serial replating experiments, individual GFP+ granulocytic/erythrocytic/monocytic/megakaryocytic (GEMMs) colonies were harvested after 7 days, dispensed, and further grown in 1 mL of methylcellulose medium supporting myeloid progenitors.
Transplantation assays
Competitive repopulation unit (CRU) assays were performed by transplanting HOXA4-GFP- or GFP-transduced 5-FU BM cells derived from B6SJL (CD45.1) mice intravenously in lethally irradiated (800 cGy) C57Bl/6 (CD45.2) congenic mice at limiting dilution in competition with 2×105 total BM cells derived from a healthy C57Bl/6 (CD45.2) mouse. Peripheral blood (PB) repopulation was monitored every 4 weeks by flow cytometry for the presence of GFP+ cells. Twenty weeks post-transplantation, mice were sacrificed and analyzed by FACS for the contribution of GFP+ cells to the myeloid, lymphoid, and erythroid lineages, using following conjugated antibodies: CD4-PE, CD8a-APC, CD45.1-PE, CD45.2-biotin, Ly-6G(Gr-1)-biotin, CD3e-Biotin, TER119-biotin CD45R/B220-PE, CD11b(MAC-1)-PE, CD45R/B220-APC, and CD45.2-Pacific Blue (BioLegend, San Diego, CA). Biotinylated antibodies were stained with APC, APC/Cy7, or PerCP-Cy5.5–conjugated streptavidin (BioLegend or BD Pharmingen, Mississauga, ON, Canada). FACS data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Mice with ≥1% of GFP+ cells contributing to both myeloid and lymphoid (B- and T-cells) lineages were considered reconstituted. HSC frequencies were calculated using the extreme limiting dilution analysis method for stem cells (
Clonal analysis by Southern blotting
Genomic DNA was isolated from BM, spleen, and thymus cells derived from HOXA4 or GFP chimeras using DNAzol (Invitrogen Corporation), digested with BglII or EcoRI (New England Biolabs, Pickering, ON, Canada), and analyzed by Southern blotting using a probe against the GFP gene.
Western blot analysis
Cells were lysed in cell lysis buffer [2.5 mM Tris, 50 μM EDTA, 0.00001% NP-40, and 0.835 mM KCL (Mallinckrodt Baker Inc.)]. Proteins were subjected to SDS-PAGE and transferred to nitrocellulose membrane. Membranes were incubated with a polyclonal anti-Flag antibody (1:1,000 dilution; Sigma-Aldrich) followed by a secondary polyclonal anti-rabbit antibody coupled with horseradish peroxydase (Jackson ImmunoResearch Laboratories Inc., Westgrove, PA).
Quantitative RT-PCR
Total RNA was isolated from total GFP+ BM of HOXA4 and control chimeras using Trizol reagent (Invitrogen Corporation) and DNase-I treated (Invitrogen Corporation). cDNA was prepared from 5 μg total RNA using MMLV-RT (Invitrogen Corporation) and random primers (Invitrogen Corporation) according to the manufacturer's protocols. Q-PCR was carried out using SYBR Green (Applied Biosystems, Streetville, ON, Canada) and thermal cycler ABI 7500 (Applied Biosystems). Oligonucleotides for all Hox genes were used according to previously validated sequences [32]. Triplicates were accepted in a 0.5 CT range.
Results
HOXA4 BM cultures
To evaluate the effect of HOXA4 on the growth of primitive hematopoietic cells in response to cytokines, a series of BM cultures were performed with BM cells overexpressing HOXA4-GFP or GFP only. Generation of these cells was achieved by stable transfection of BM cells derived from 5-FU–treated wild-type mice (CD45.1) with either MSCV-HOXA4-Flag-pgk-GFP or control MSCV-pgk-GFP bi-cistronic retroviral vectors (Fig. 1A, B). The expression of HOXA4 in those cells was confirmed by western blot analysis (Fig. 1D). GFP+ cells were sorted and seeded in duplicate at 50,000 or 100,000 cells in BM expansion medium and cultured for 3 weeks (Fig. 1B). At the initial phase of the culture period, the numbers of cells in both HOXA4 and control cultures increased at similar rate (Fig. 1C and Supplementary Fig. S1; Supplementary Data are available online at
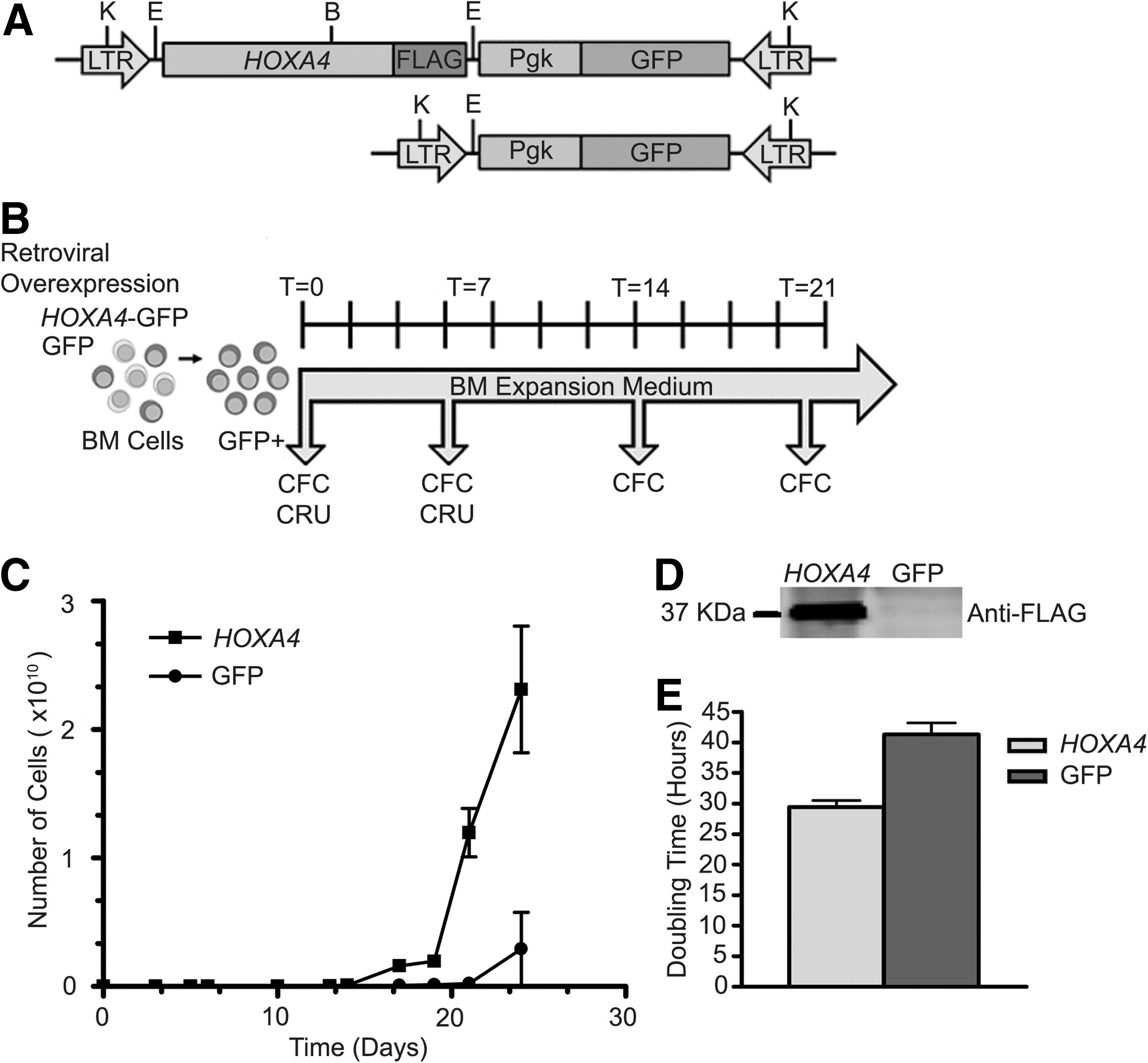
Growth analysis of HOXA4 and control BM cultures.
HOXA4 overexpression in myeloid progenitors
BM cells in culture undergo a massive proliferation and differentiation in response to growth factors, leading to exhaustion of the culture over time. The enhanced growth of HOXA4 BM cultures was apparent only after 7 days, suggesting that HOXA4 might act on primitive progenitors. Weekly performed colony-forming cell (CFC) assays showed an increase in absolute number of myeloid progenitors over the initial values with the progression of both HOXA4 and control cultures (Fig. 2A). The expansion of the HOXA4 progenitor population was significantly larger than control in all experiments (n=4), reaching maximum fold expansions of more than 1 million versus 30,000, respectively, after 3 weeks of culture. Analysis of differential counts showed that, in particular, numbers of multipotent progenitors [granulocytic/monocytic (GM)-CFC and GEMM-CFC] were higher in HOXA4 than in control cultures (Fig. 2B, C). Although 39 times more GM-CFC progenitors were measured in HOXA4 than in control cultures, GEMM-CFC progenitors were not detected after 7 days in control cultures, contrarily to the increase of these progenitors in HOXA4 cultures. As GEMM-CFCs are representative for the presence of multipotent primitive hematopoietic cells, the self-renewal potential of these progenitors was evaluated by serial replating of GEMM colonies. Close to 100% efficiency in colony formation was observed for HOXA4 after each replating, compared with 50% for control GEMMs (Supplementary Table S2). HOXA4 GEMM colonies gave rise to secondary and tertiary GEMMs, but no GEMMs were detected among secondary colonies of controls, preventing further replating (Fig. 2D). In addition, HOXA4 GEMM colonies generated significantly more colonies after the first plating than controls (32.5±17.9 vs. 2.5±3.5; HOXA4: n=10; control: n=2; P<0.0005), but this number decreased with each replating (Fig. 2E). Thus, enhanced growth of HOXA4 BM cultures is supported by larger expansions of the progenitor populations, especially the most primitive. Moreover, regeneration of GEMM colonies after serial replating suggests self-renewal activity of HOXA4-expressing primitive progenitors in vitro.
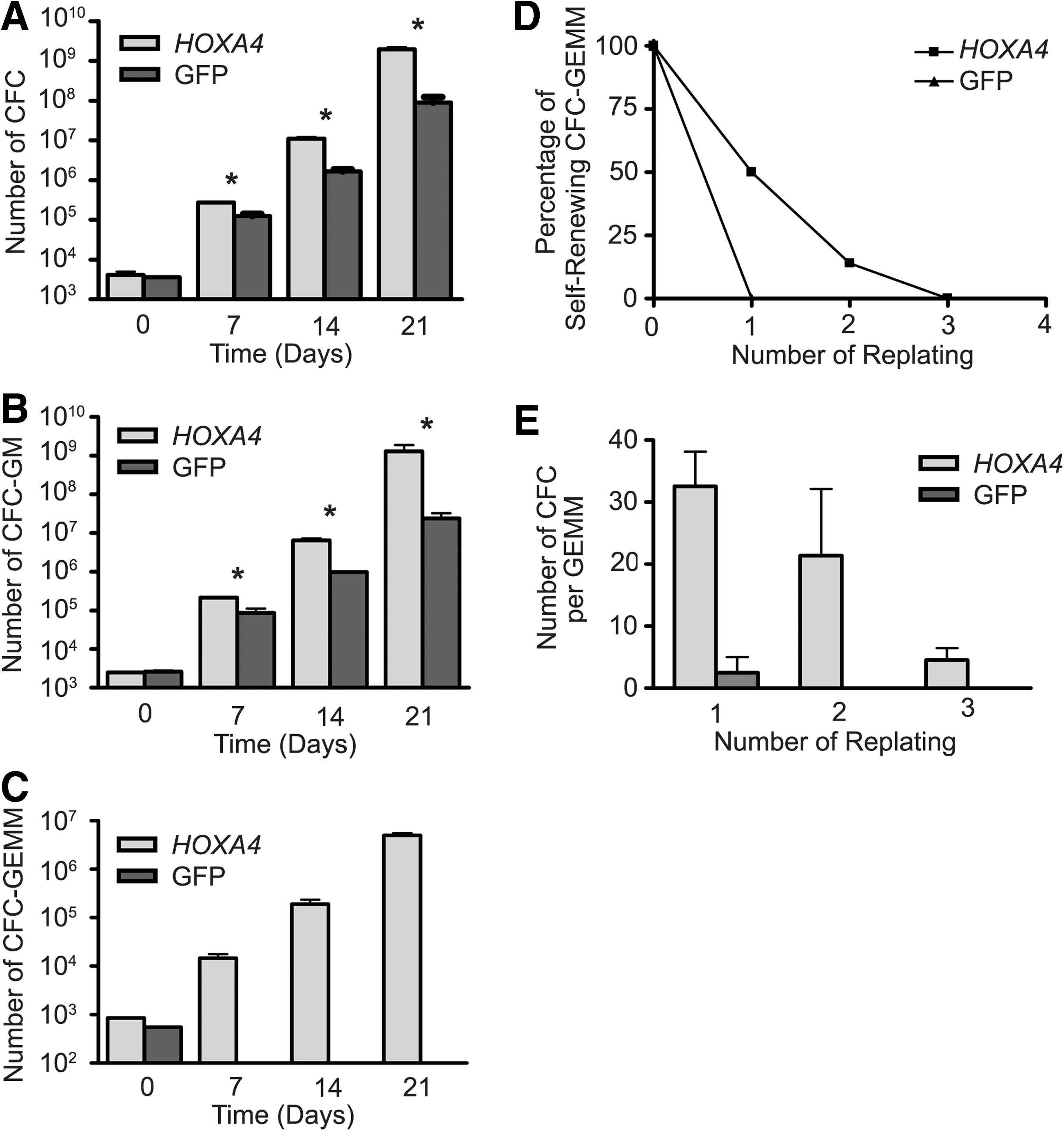
Progenitor analysis of transduced BM cultures. Increase of
Evaluation of HOXA4 HSC expansion in vitro
To examine whether HOXA4 HSCs underwent self-renewal divisions in vitro, CRU assays were performed on HOXA4 and control BM cultures at day 0 (after coculture on retroviral producers) and at day 7 (Fig. 1B). HOXA4 and control cell mixtures used to initiate BM cultures had comparable frequencies of HSCs (1:1013 and 1:1571, respectively; Fig. 3A), meaning that ∼50 and ∼30 HSCs were present in HOXA4 and control cultures. After 7 days of culturing, the frequency of HOXA4 HSCs was significantly higher than control HSCs (1:28.775 and 1:4.822.616, respectively), resulting in a 6.6-fold net increase of HOXA4 HSCs and a 10.0-fold net loss of control HSCs (Fig. 3B). The level of chimerism in the periphery of long-term repopulated recipient mice transplanted with HSC doses superior than 10 from T0 cultures is comparable between HOXA4 and control (Supplementary Fig. S2A). Interestingly, transplantation of <10 HSCs resulted in higher progeny contribution in repopulation derived from HOXA4 HSCs than from control HSCs, indicating that HOXA4 HSCs have an increased proliferation/differentiation potential. Therefore, the mean activity of stem cell (MAS) was calculated as the engraftment per HSC in recipient mice that received <10 HSCs. The MAS of HOXA4 HSCs in T0 cultures was 2.3-fold superior to control HSCs (10.37±4.77 vs. 4.5±3.76; Supplementary Table S3). Expanded HOXA4 HSCs showed reduction at high doses, but similar levels of chimerism as nonexpanded HSCs for the low doses (compare dose 0.5 T0 with equivalent dose 0.3 T7 of Supplementary Fig. S2B). Overall, the calculated MAS value for expanded HOXA4 HSCs at T7 was slightly but not significantly decreased compared with HOXA4 HSCs of T0 (Supplementary Table S3). Levels were reduced in mice that received grafts containing >2 HSCs. The measured expansion of HOXA4 HSCs in culture is only relevant to in vivo HSC populations when multiple HSC clones are involved and not when it is due to proliferation of a single clone. Southern blot analysis on genomic DNA from long-term (>20 weeks) repopulated primary HOXA4 BM chimeras transplanted at day 0 showed the presence of multiple HSC clones at initiation of the BM culture that were transplanted in different mice (Fig. 3C, mice #191, 192, and 200). The contribution of the same clones to the repopulation of BM, spleen, and thymus (eg, Clones a and b in mouse #191; Fig. 3C) enforces their HSC origin, although progeny composition by different clones may vary per organ (eg, Clone f, #200, is relatively more present in spleen and thymus than in the BM). Multiple HSC clones were also detected in mice that received cells from HOXA4 BM cultures at day 7 (mice #238, 245, and 250), indicating a polyclonal expansion of HOXA4 HSCs in vitro. Detection of identical clones in recipients of both T0 and T7 cultures (Clones c and d, mice #192 and 250) indicates that the HSCs underwent a self-renewal division during the coculturing on retroviral producers. These data show that constitutive HOXA4 overexpression results in a polyclonal expansion of HSCs in culture, which retains a strong proliferation/differentiation potential.
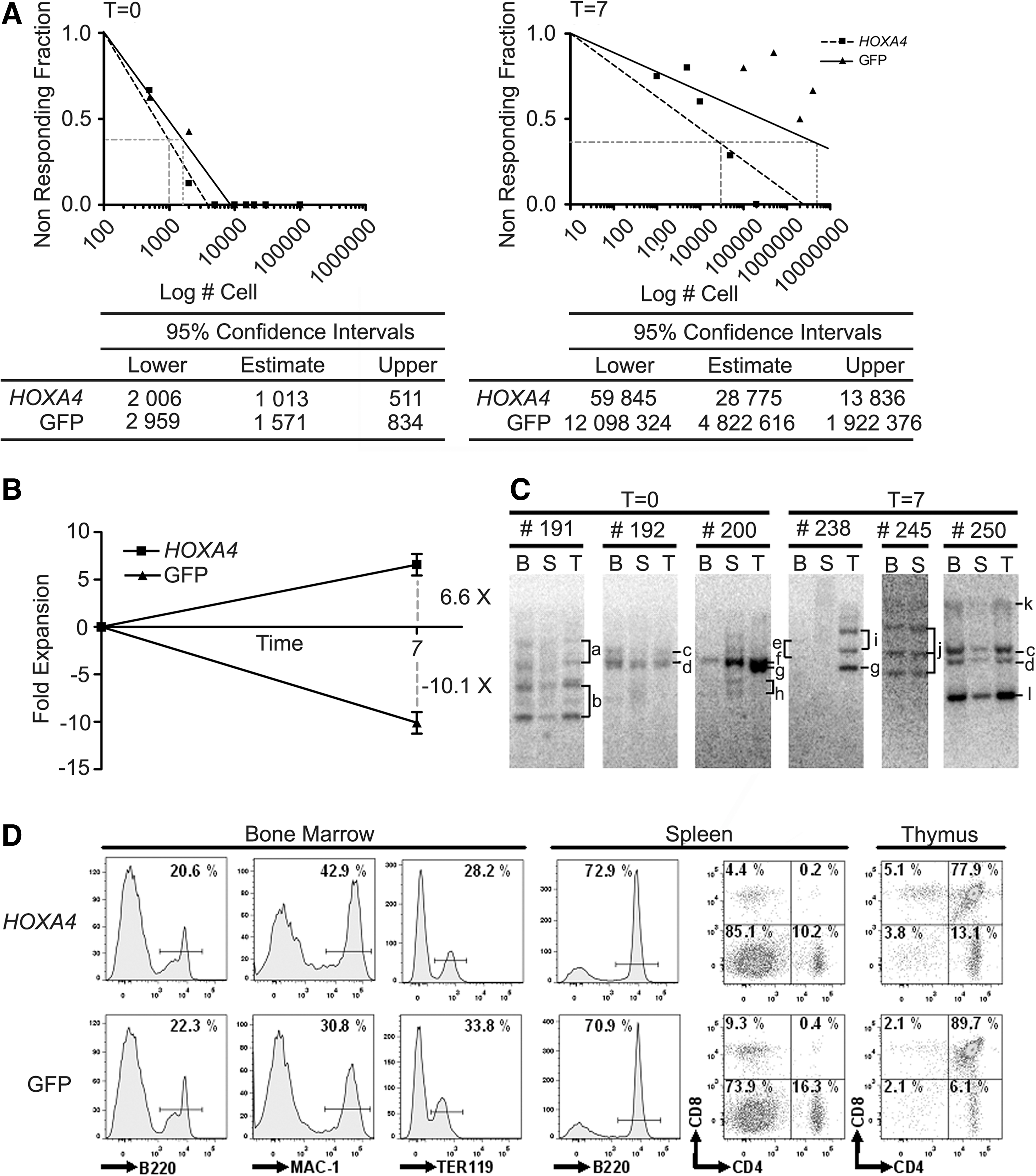
Evaluation of HSC expansion in vitro.
Evaluation of HOXA4 HSC progeny in vivo
FACS analysis using antibodies against surface markers specific for B-cell, T-cells, myeloid cells, and erythroid cells in combination with GFP fluorescence confirmed the contribution of HOXA4 HSCs to all lineages in hematopoietic organs of long-term repopulated recipients (>20 weeks) (Fig. 3D). To test whether the constitutive overexpression of HOXA4 introduced a bias in engagement to lineage differentiation, BM chimeras of well-reconstituted mice were monitored for repopulation of lymphoid and myeloid cells in their PB. FACS analysis showed that population sizes of HOXA4 B-, T-, and myeloid cells were comparable to control at both short term (4 weeks) and long term (20 weeks) post-transplantation (Fig. 4A). In addition, no significant changes in the proportions of myeloid and lymphoid cells were found in hematopoietic organs of long-term (>20 weeks) repopulated HOXA4 and control chimeras, although myeloid populations tend to be higher in the BM (Supplementary Table S4). The increase in myeloid cells led to a significant reduction of erythroid cells, likely through space restraints in the bone. To measure whether HOXA4 overexpression affects the number of myeloid progenitors in vivo as observed in vitro, CFC assays were performed on BM and spleen derived from long-term repopulated (>20 weeks) chimeras. Interestingly, myeloid progenitor population was 2.0-fold enhanced in BM and 17.9-fold in spleen derived from HOXA4 compared with control chimeras (Fig. 4B, C). Differential analysis of colonies showed that except for macrophage colonies (M-CFC) all progenitor populations were significantly enlarged in BM of HOXA4 chimeras (Supplementary Fig. S3A). Similar to results from the BM cultures, the largest increase was observed for the GEMM progenitors (20×, P<0.05). Interestingly, evaluation of the B-cell compartment in these chimeras by CFC assays showed that HOXA4 BM had 10.2-fold more B-cell progenitors than control (Fig. 4D), and that these HOXA4 B-cell colonies were significantly larger (Supplementary Table S5). Determination of more primitive B-cell progenitors in BM fractions of HOXA4 and control chimeras by Whitlock-Witte-initiating cell (WW-IC) cultures showed significantly higher numbers of short- to medium-term B-cell progenitors in the presence of HOXA4, but no difference in long-term WW-IC numbers (Supplementary Fig. S3B). Thus, the presence of more primitive WW-IC B-cell progenitors at normal frequencies together with an augmentation of clonogenic B-cell progenitors suggest that only B-cells in specific differentiation stages, likely those associated with proliferation, are permissive to HOXA4-induced expansion. Importantly, despite the bias in myeloid versus lymphoid progenitors expansion, HOXA4 HSCs repopulate recipients with normal distribution of mature hematopoietic cells, indicating that interactions of HOXA4 BM cells with their environment are intact.

Analysis of long-term reconstituted BM chimeras.
Expression of cluster and paralog family Hox genes in HOXA4 hematopoietic cells
Cross- and autoregulation are very common for Hox genes [33 –35]. To determine whether the observed phenotype in HOXA4 BM chimeras was due to HOXA4 and not mediated through activation of other Hox genes indirectly, we measured the expression of neighboring Hoxa genes and paralog group 4 genes in GFP+ cells derived from total BM of HOXA4 and control chimeras. Q-RT-PCR showed a tendency for Hoxa genes in direct vicinity (Hoxa2, -a3, -a5, -a6) as well as for Hoxb4 to be highly expressed, but did not reach significance (Supplementary Table S6). Thus, the enhancement of B-cell and myeloid progenitors as well as the in vivo expansion of HSCs is likely to be a direct effect of HOXA4 overexpression.
Discussion
Results presented in this article demonstrate for the first time that the overexpression of HOXA4 increases the self-renewal activity of HSCs and myeloid progenitors in vitro. These culture-expanded HOXA4 HSCs remained functionally intact as individual clones and contributed to the generation of terminal differentiated lymphoid and myeloid cells upon adoptive transfer into recipient mice. HOXA4 HSCs generated balanced myeloid and lymphoid populations in the periphery of chimeras, supporting the ability of ex vivo HOXA4-transduced HSCs to normally respond to physiological signals upon return in their natural microenvironment. Our data also show that the HOXA4-induced expansion is highest in primitive multipotent myeloid progenitors and decreases with maturation of progenitors. These findings indicate not only that multipotent myeloid progenitor with highest proliferation/differentiation potential, naturally expressing higher Hox genes levels than unipotent progenitors [3,22], are more permissive to elevated HOXA4 gene levels, but also that HOXA4 does not interfere with normal regulatory mechanisms restricting the response to HOXA4 in mature cells. Interestingly, in contrast to the myeloid lineage, more mature clonogenic B-cell progenitors responded with larger expansion to HOXA4 than their precursors, the WW-ICs.
Although serial replating of individual GEMM colonies clearly showed that the incidence of self-renewing divisions of the colony-initiating cell overexpressing HOXA4 is increased compared with control, the replating potential appeared limited. This might be explained by the ability of HOXA4 to normally engage in hematopoietic differentiation; if the colony initiating cells first undergo a differentiation division, consequently, it will fail to produce a GEMM colony in the next generation and therefore limit the replating. Thus, HOXA4 promotes self-renewal without loss of differentiation, which could lead to malignancy.
Further, chimerism level evaluation in PB showed that proliferation/differentiation potential of single HOXA4 HSCs is superior to control HSCs (Supplementary Fig. S1) and is maintained after ex vivo expansion. The capacity of HOXA4 HSCs to expand in vivo following transplantation in myeloablated mice, contrary to control HSCs (data not shown), is likely to contribute to higher chimerism levels at low doses of transplanted HSCs. Although the level of engraftment of single HOXA4 HSCs to the periphery was intact after in vitro expansion, it is not clear why engraftment levels by higher doses of expanded HSCs appeared to be reduced compared with equivalent doses of HOXA4 HSCs from T0. Others have previously claimed that engraftment by HOXB4 HSCs decreases with prolongation of the culture time, but these studies did not take into account the decrease in frequency of HSCs due to stronger growth of total BM cultures [36].
The presence of several HSC clones contributing to recipients of T7 cultures showed a polyclonal expansion of HSCs without selective advantage that could cause a risk for leukemia development. In that respect, only few HOXA4-positive leukemias of either T-cell or myeloid phenotype have been observed (5 of 69 mice), which developed only after a long latency (5–6 months; data not shown), suggesting that HOXA4 might have some weak oncogenic activity or could function as an oncogenic collaborator. In line with these results, some oncogenic activity has been attributed to HOXB4 in a large animal model [37], and mouse studies reported collaboration of HOXB4 with Meis1 and E2A-PBX1 in myeloid and lymphoid leukemia [38,39], respectively. Thus, it would not be surprising that HOXA4 has similar properties. However, we cannot exclude that leukemias were caused by retroviral integrations near oncogenes and its subsequent activation.
The amplitude of HSC expansion by HOXA4 during 7 days of culturing appeared to be in the same range as found for HOXB4 [11], indicating that these genes are likely to activate a similar pathway leading to self-renewal of HSCs. Minor increase in expression levels of some Hoxa genes and Hoxb4 confirmed that the expansion of HSCs and progenitors overexpressing HOXA4 was not mediated through Hoxb4 or other Hoxa genes.
The specific expansion of IL-7 responsive pro-B-cell clonogenic progenitors by HOXA4 in vivo is unique for Hox genes, so far. HOXB4 chimeras are known to have increased myeloid and B-cell clonogenic progenitors as well [5], but similar magnitude of expansion for both cell types suggests that this expansion is the result of increased numbers of common myeloid/lymphoid precursors and not through its direct effect on B-cell progenitors. Similarly, studies with human HOXB4-transduced CD34+ stem cells and progenitors showed that CD19/CD10 B-cells did not respond to constitutive HOXB4 expression, but enhanced B-cell progeny could be obtained from lympho/myeloid CD34+ cultures [40]. In addition, modulation of other Hox genes in BM cells in knockout and retroviral overexpression mouse is often associated with B-cell deficiencies rather than with an increase in B-cell populations [6,41]. In this respect, Hoxa9 expression has been shown to be inversely related to early B-cell factor, a gene crucial for B-cell development [42]. Thus, our results show for the first time a Hox gene specifically acting on IL-7-responsive B-cell progenitors. As regeneration of the B-cell compartment in patients with lymphomas, who underwent BM transplantation, is very slow [43,44], a B-cell complementation therapy might dramatically reduce infection related to post-transplantation complications. Thus, regeneration of B-cells using HOXA4 could have an important impact on infectious complications in the clinic.
Based on the clear expansion of HSCs in vitro with HOXA4, we predict that human CD34+ hematopoietic cells are likely to expand as well when overexpressing HOXA4, as has been reported for HOXB4 [12,45]. However, regarding the observed differences between human SRC and mouse HSCs in response to HOXB4 overexpression [5,12], it is of interest to further investigate the capacity of HOXA4 to expand human HSCs as well as B-cell progenitors. In conclusion, our results put forward HOXA4 as a new promising candidate for therapeutic expansion of HSCs and B-cell progenitors.
Footnotes
Acknowledgments
The authors thank Martine Dupuis and Nathalie Henley from the HMR flow cytometry platform for cell sorting and assistance in FACS analysis. Felix Jules is thanked for generating the western blot. The authors also thank the staff of the animal care facility for taking care of the animals.
Author Disclosure Statement
The authors declare that no potential conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.