
Abstract
Alzheimer's disease (AD) is a leading cause of age-related dementia that is characterized by an extensive loss of neurons and synaptic transmission. The pathological hallmarks of AD are neurofibrillary tangles and deposition of β-amyloid (Aβ) plaques. Previous research has investigated how Aβ fragments disrupt synaptic mechanisms in the vulnerable regions of the brain. There is a tremendous potential for stem cell technology to extend upon this research, not only in terms of developing therapeutic applications, but also in modeling AD. Indeed, the advent of induced pluripotent stem cell technology has opened up exciting new avenues for generating patient and disease-specific cell lines from somatic cells that may be used to model AD. Amyloid precursor protein (APP) is a key protein in neuronal development and this article reviews the role of APP in AD. Stem cell technology offers the opportunity to make use of APP in the directed differentiation of induced pluripotent stem cells into functional neurons, a process that may help generate a model of AD and thereby facilitate an understanding of the mechanisms underlying this disease.
Introduction
Overview of Alzheimer's disease
Alzheimer's disease (AD) is characterized by a progressive neuronal degeneration that leads to an impairment of memory and cognitive abilities as well as other neuropsychiatric symptoms and behavioral disturbances [1 –3]. The World Health Organization cites AD as the sixth most common cause of death in the world, having estimated that approximately 18 million people worldwide suffered from the disease in 2010 and expecting this number to double by the year 2025 [4,5]. The etiology of disease is not fully understood and a definitive medical diagnosis can only be made on the basis of characteristic neuropathological changes in brain tissue obtained at autopsy [6].
Neuropathological overview of Alzheimer's disease
As originally described by Alzheimer in 1906 [7], AD is characterized by 2 types of neuropathological lesions: dense intraneuronal bundles of filaments known as neurofibrillary tangles (NFTs) and darkly staining amyloid plaques comprised primarily of amyloid β (Aβ), a 40–42 amino acid peptide formed by the cleavage of the type-1 transmembrane protein called amyloid precursor protein (APP). NFT are essentially intraneuronal bundles of paired helical filaments comprised of abnormally phosphorylated microtubule-associated protein tau. Whereas NFTs are not specific to AD, being found in various other neurological disorders like frontotemporal dementia and Parkinson's disease [8], there is considerable support for an amyloid hypothesis for AD.
Following the discovery of Aβ plaques, the prevailing view throughout most of the 20th century was that the Aβ deposition was merely a marker of AD progression and that it played only a minimal role in neurodegenerative processes underlying the disease. This view changed in the 1980s with the advent of the amyloid hypothesis, which postulates that an overproduction of Aβ and its aggregation into plaques is the primary etiology of the neurodegeneration and the cognitive decline associated with AD. The amyloid hypothesis has continued to be the leading hypothesis for understanding of the etiopathogenesis of AD. A multitude of studies have since produced a wealth of information on the role of Aβ in the genesis and biophysiology of AD. Figure 1 illustrates the neuropathological processes that occur in the cells of an AD-affected brain.
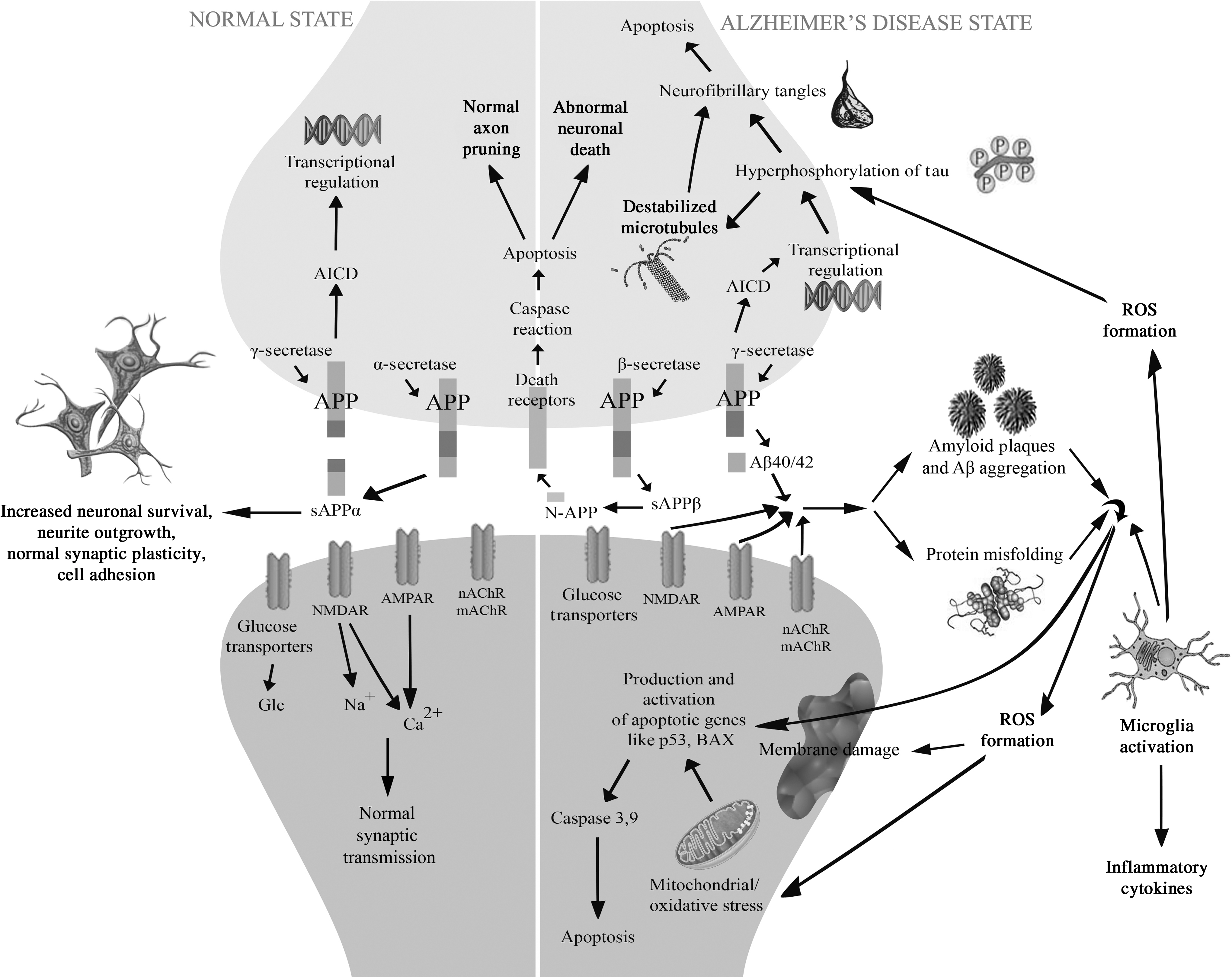
Neuropathological processes in the cells of an Alzheimer's disease (AD)-affected brain. AD is clinically characterized by the presence of β-amyloid plaques and intracellular neurofibrillary tangles that lead to neuronal dysfunction and cell death. The primary neuropathological process associated with AD is the differential processing of the integral membrane protein APP (amyloid precursor protein). In the normal state APP is cleaved via α-secretase to generate large soluble fragment called sAPPα and a C83 carboxy-terminal fragment (not shown in the figure), with sAPPα facilitating normal synaptic signaling, synaptic plasticity and neuronal survival. In an AD, the APP is cleaved sequentially by β- and γ-secretase. This releases the extracellular fragments Aβ40/42, which aggregate into neurotoxic plaques (called Aβ plaques or β-amyloid plaques) that interfere with the normal brain functioning by blocking ion channels such as NMDAR (N-methyl D-aspartate receptors), AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), nAchR & mAchR (nichotinic acetylcholine receptors and muscarinic acetylcholine receptors), disrupting calcium equilibrium, increasing mitochondrial activity and oxidative stress via release of reactive oxygen species (ROS), and diminishing energy metabolism. The plaques are also associated with abnormal glucose regulation, and ultimately induce neuronal cell death by upregulation of apoptotic genes like p53 and BAX (Bcl-2–associated X protein). Additionally AD is also characterized by the presence of neurofibrillary tangles that result from the abnormal phosphorylation/hyperphosphorylation of microtubule-associated protein tau, which is caused due to aberrant transcriptional regulation and activity of certain kinases (not shown in the figure). Hyperphosphorylation results in the dissociation of tau from the microtubules causing them to destabilize. The released tau oligomerizes within the cell leading to the formation of neurofibrillary tangles that disrupt normal processes further promote apoptosis.
Processing of APP and formation of plaques
It is the sequential cleavage of APP by the proteases β- and γ-secretases that generates Aβ [9,10]. This proteolytic cleavage gives rise to two major forms of Aβ that are 40 and 42 residues in length. Mutations in the APP gene on chromosome 21 increase the levels of either total Aβ peptides or Aβ1-42 alone, and are associated with the early onset, though relatively rare, familial form of AD [11].
APP is known to be processed in 2 different pathways: amyloidogenic and non-amyloidogenic (Fig. 2). The amyloidogenic pathway involves the sequential cleavage of APP by β-secretase (BACE) and the γ-secretase complex, generating the Aβ fragments known to be the etiological agents in AD. Along with the Aβ fragments, the amyloidogenic pathway also generates a slightly smaller intracellular soluble fragment called sAPPβ [12], the role and function of which have been largely unexplored. In the non-amyloidogenic pathway, the APP is cleaved by α-secretase within the Aβ domain. While ensuring that no Aβ peptides are generated, this pathway releases sAPPα, the large extracellular domain of APP that forms a soluble fragment shown to possess neuroprotective and memory-enhancing effects [13 –16].
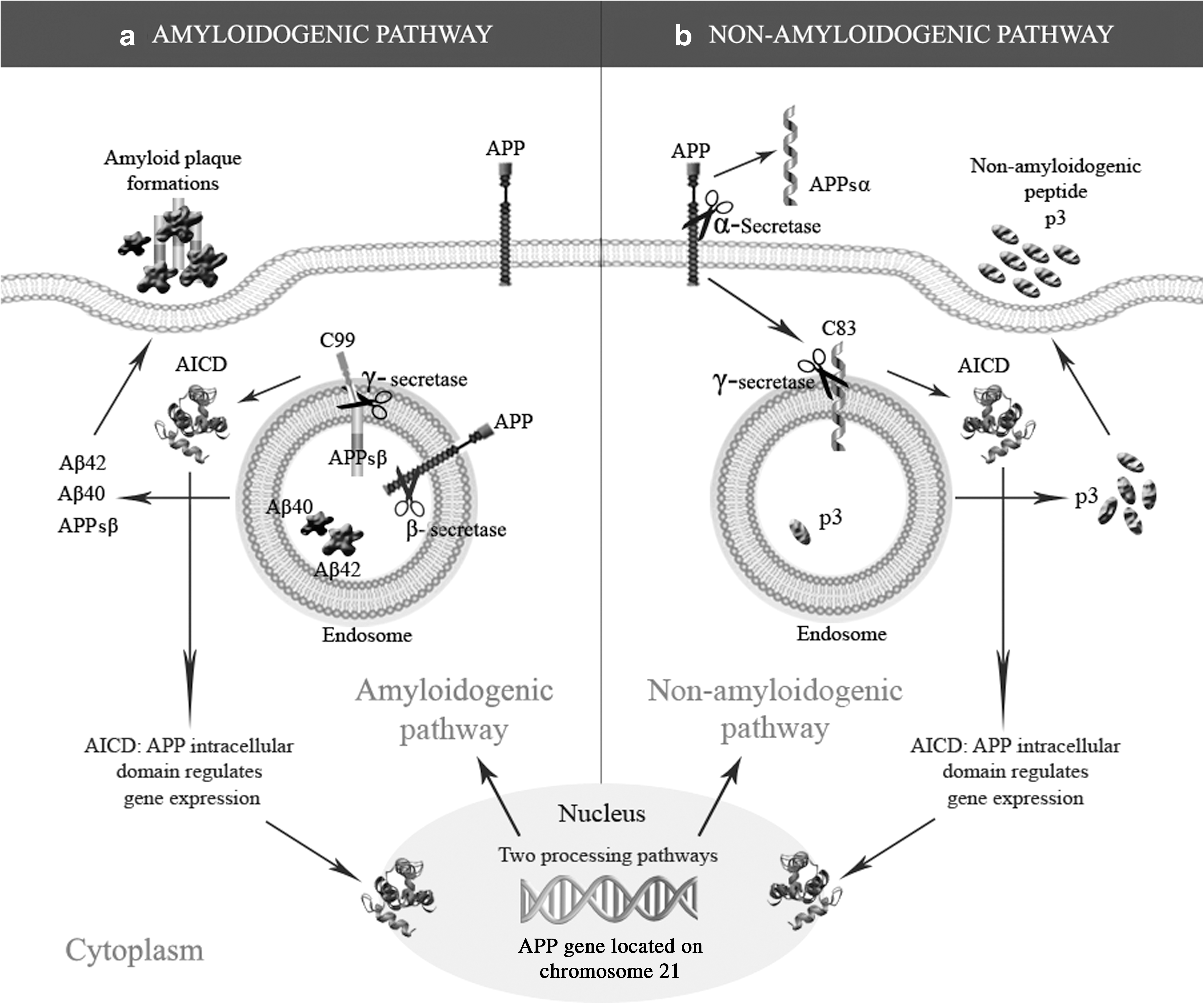
The amyloidogenic
To date no drugs have been successful in treating AD or arresting its progression. Research suggests that there is a potential role for cell replacement strategies, particularly stem cell therapy, in alleviating AD symptoms. The transplantation of stem cells or their differentiated derivatives such as neural precursor cells can improve the functioning of a diseased brain by replacing lost neurons and glia, mediating remyelination, or exerting trophic effects. Moreover, the systematic generation of neurons via the process of differentiation may provide important insights into the pathology and progression of AD. This process of disease modeling may also contribute to the early diagnosis of AD and facilitate the development of drugs or other treatments.
There is increasing evidence suggesting a distinctive role for APP in the development of the central nervous system. These observations have been supported by studies conducted using APP knockout mouse models that show defects in neuronal migration and development [17]. This article describes the trophic-like role of APP in cell development and contrasts this against its deteriorative role in the development and progression of AD. It also discusses the conventional strategies for modeling AD further summarizing the use of human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) and highlights the advantages of stem cells over animals in disease modeling. We address the potential for the trophic properties of APP to facilitate the in vitro differentiation of iPSCs into functional neurons that may be used in modeling and developing therapeutic applications for AD.
Conventional Strategies in Disease Modeling
Non-mammalian models
Drosophila melanogaster
Conventional strategies for disease modeling use animals, particularly when the aim is to understand the underlying genetic mechanisms, epigenetics, and progression of a disease. AD has been modeled using the fruit fly Drosophila melanogaster [18], as this species possessed an APP homologue structurally similar to human APP that is encoded by the β-amyloid precursor protein-like (Appl) gene. The homologue undergoes similar proteolytic processing to human APP, but Aβ is not generated because drosophila lacks endogenous BACE [19]. Several research groups have used the misexpression of Appl gene in drosophila to recapitulate Aβ-plaque formation, and to render a comparatively efficient model for studying mechanisms of Aβ toxicity and identify potential genetic and pharmacological modifiers. Deletion of Appl in drosophila was observed to cause mild defects in locomotor responses that could be reversed by the provision of human APP genes [20]. This suggests that the APP gene is not essential for survival, but may be required for the nervous system to develop and function optimally. Other studies targeting the expression of different forms of Aβ have implicated Aβ1-42 in causing neurodegenerative phenotypes, amyloid deposits, and learning deficits. Conversely, Aβ1-40 was found to be associated only with learning deficits, suggesting an involvement specific to the Aβ1-42 form of Aβ in AD pathology [21].
Caenorhabditis elegans
The nematode worm C. elegans has an APP homolog, APL-1, that is structurally similar to its mammalian counterpart and with which it shares sequence homology in the N-terminal and intracellular C-terminal domains (the latter of which has the highest sequence conservation). The loss of APL-1 in C. elegans is reported to induce a severe molting defect and promote larval mortality, suggesting an importance of APP in general development [22,23].
Zebrafish
Zebrafish possess two homologues of APP with about 70% homology to human APP [24]. Also important is the presence of functional γ-secretase activity and consequent production of Aβ fragments in zebrafish [25]. Additionally, introduction of a mutant form of human tau to the zebrafish is reported to induce cytoskeletal disruptions which closely resemble the tau-based neurofibrillary tangles found in patients with AD [26]. Zebrafish are also valuable tools for studying the effects of oxidative stress, which plays an important role in increasing the risk of AD, as zebrafish, like mammals, have defense mechanisms against toxic chemicals that involve the generation of oxidative stress [27].
Mammalian models
Mice
Another approach to modeling AD has involved the use of transgenic mice expressing mutations of the APP gene. More than 20 autosomal-dominant APP mutations have been linked to AD. In addition, certain mutations in the genes for presenilins and BACE are reportedly sufficient to cause the complete AD phenotype [28]. However, transgenic mouse models have proven to be incomplete models of AD, as not all aspects of AD are replicated when human transgenes with a mutation are expressed in the mouse brains. For example, while APP transgenic mice were seen to develop memory loss and plaques, they did not develop neurofibrillary tangles and underwent very little or no neuronal loss. Various transgenic mice models have been used to study AD (see ref. 29 for a review).
Rats
An advantage of using rats over mice in modeling AD is the greater capacity for rat models to more accurately represent the different progressive stages of AD. Rats have been used to establish toxic effects of Aβ1-42 on the cholinergic system, with direct infusion of Aβ1-42 into the cerebrovascular system, leading to reduced levels of choline acetyltransferase immunoreactive neurons [30 –32] and vesicular acetylcholine transporters [32,33], and behavioral abnormalities in novel environmental conditions [32,34]. Moreover, rats have been routinely used to assess toxicity of novel compounds, thus further aiding in drug development [35].
Primates
Non-human primates undergo reductions in neuronal and cognitive functioning with the age that make them one of the best models for AD and other age-related neurodegenerative disorders [36]. Aged primates with AD-like symptoms exhibit an extensive loss of entorhinal cortex neurons that is not seen in normal aged primates [37,38], consistent with the findings from rodent models of AD [39]. Studies like these enhance our understanding of the neurological differences between normal healthy aging and age-related pathologies like AD.
Stem Cells for Disease Modeling and for Therapeutic Purposes in Neurodegenerative Disorders
Human embryonic stem cells
Human embryonic stem (hESCs) cells are generally obtained from spare IVF embryos[40]. Cells are derived from the inner cell mass (ICM) of the early blastocyst stage of embryonic development, which consists of pluripotent cells that have the capability to self-renew indefinitely and contribute to the formation of the three germ layers [41 –45]. The ability of hESCs to grow indefinitely while maintaining pluripotency promises a role for them in understanding disease mechanisms and curing many presently untreatable diseases [46]. There are several current attempts to develop in vitro protocols for the differentiation of hESCs into progenitor cell populations and to further develop protocols for inducing the directed differentiation into specialized cells [47,48]. This includes attempts to differentiate hESCs into dopaminergic neurons as a potential therapy for Parkinson's disease [49,50]. The aim of other investigations has been to develop motor neurons from hESCs; these have been used to study some of the human motor neuron diseases including spinal muscular atrophy, for which they have proven therapeutically helpful beneficial [51].
While hESCs have significant therapeutic potential, ethical issues associated with the use of human embryos [52] and problems related to tissue rejection hinder the advancement of the hESC-based therapies. Additionally, it is proving difficult to generate the patient or disease specific ESCs required for particular applications [53].
Induced pluripotent stem cells
There is a strong therapeutic potential for autologous cell replacement therapy in treating neurodegenerative diseases for which there are presently no cures [54]. Pioneered by Yamanaka in 2006, the reprogramming of adult somatic cells into iPSC avoids the controversies related to ESCs [46,53]. The very first iPSCs were derived by inducing pluripotency in mouse embryonic fibroblast cells using retroviral transductions of 4 transcription factors: Oct3/4, Sox2, c-Myc, and Klf4 [53]. Since then, many studies have induced pluripotency in human somatic cells, with the objective of using these for autologous transplantations [55 –58]. hESCs and iPSCs are not only being investigated as therapies for neurodegenerative disorders like AD; they are also being used in attempts to generate human neurons for modeling these diorders.
Using Stem Cells Over Animals for Disease Modeling Purposes and the Importance of Using iPSCs
There are various reasons for using stem cells rather than animals or diseased human tissue in disease modeling. One of these is genetic differences between humans and animals that prevent many diseases from being accurately simulated with animal models. Other reasons include the heterogeneity and complexity of animal models; also, the therapeutic effects in the animal models are not always representative of the true human physiological conditions at the molecular or anatomical level [59]. Likewise, the use of diseased human tissue for modeling neurodegenerative diseases is limited by diseased neural tissue being difficult to obtain or representing only the later stages of disease [60].
In contrast, stem cells can be used to model the various stages through which a disease progresses. Fundamental unresolved issues with iPSCs are the extents to which they can be reprogrammed to differentiate into specific target cell lineages, and whether or not those iPSC-derived cells are functional. With hESCs, there is a major concern regarding the formation of teratomas following transplantation in clinical trials, which suggest incomplete differentiation and the presence of pluripotent cells [61 –63]. This concern, the ethical issues, and incomplete understanding of the molecular and cellular processes underlying differentiation, are preventing researchers from performing clinical studies with hESCs. The current research focus on iPSCs is therefore not surprising, and which includes efforts to develop protocols that enable differentiation into specific target neuronal cell lineages and functional neurons that can be used to model neurodegenerative disorders, particularly AD.
With regards to developing efficient differentiation protocols, interestingly, numerous studies have proven that this sAPPα, the large extracellular domain of APP, possesses trophic-like properties [64] and is a critical factor in neuronal maintenance, as it enhances their growth and is involved in neurite outgrowth, neuroprotection, neurotrophism, adult neurogenesis, axonal transport, synaptic function, and transcriptional regulation [65,66]. The studies were conducted in transgenic mouse models and these concepts were then extended to observe the role of APP in differentiation of hESC lines [67], which has been discussed in detail in the section on the role of APP in neuronal development. But prior to that, the following section summarizes the trophic-properties of APP that have been so far extensively observed in the transgenic mouse models.
Transgenic Mouse Models Show Contradictory Evidence for the Role of APP
Transgenic mice expressing the APP gene and other genes associated with familial AD (genes for Presenilin-1 and Presenilin-2) have helped further our understanding of the structural, neurophysiological and behavioral effects of Aβ accumulation in the brain. For example, transgenic Tg2576 mice expressing the APPsw gene mutation (leading to an overproduction of mutant APP) exhibited amyloidosis and behavioral abnormalities, as well as disrupted neurites and decreased dendritic spine density [68]. Furthermore, stereological mapping of neuronal cell density revealed a degree of neuronal loss in the immediate vicinity of Aβ deposits [69].
In another study conducted with Tg-swAPPPrp transgenic mice, aberrant production of Aβ peptides was shown to induce axonal dystrophy and alterations of axonal transport that occur in the early stages of AD and that facilitate the deposition of amyloid peptides [70].
More recently, it was found using APPswe/PS1d9XYFP transgenic mice that neuritic abnormalities later develop in close proximity to Aβ plaques, suggesting a causal relationship between amyloid deposition and neuritic dystrophy [71,72].
While numerous studies have used mouse models to investigate relationship between Aβ deposition and AD progression, very few studies have been conducted to determine whether Aβ accumulation is the cause or the root of developing AD. APP has been detected in the membranes of synaptic preparations, and further localized to postsynaptic densities, axons, and dendrites. However, APP exists in different tissue types (in different isoform ratios) throughout almost the entire body and is therefore likely to play a role in the various body functions [73,74]. Indeed, APP appears to have a critical role in overall development, as suggested by findings that APP knockout mice, if not at first, but at later stages show lower body weight, grip strength, locomotor activity, and synaptic transmission along with hypersensitivity to epileptic seizures and defects in the forebrain commissure, suggesting that APP is critical in overall development [75].
APP acts as a G-protein coupled receptor [76] and is expected to play important roles in cell motility and adhesion [77 –79], though direct experimental support for the later is yet to be found. Studies conducted during the development of the hamster brain suggest that APP can be found in the developing nerve fiber tips [80] and that levels of full-length and secreted APP peak during the end-arbor formation, suggesting that APP is involved in synapse formation and undergoes proteolytic processing at defined stages. Nonetheless, APP is still produced and transported along axons to both central and peripheral synapses even during the stages of adulthood [81,82].
Mice with APP-null mutations show changes in neural structure and function, including but not limited to reactive astrocyte proliferation, lowered neocortical and hippocampal synaptophysin, and reduced length of dendrites in hippocampal neurons; neurons from these mice also exhibit reduced survival in culture neurons and impaired long-term potentiation [83 –86].
Role of APP in Neuronal Development: In Vitro Studies with Stem Cells
A considerable amount of evidence implicates APP in the neurodegeneration underlying AD. However, consistent with its high degree of conservation across phyla, APP plays vital roles in cellular physiological processes during both development and adulthood. APP is structurally similar to growth factors [87], and accordingly exerts several neurotrophic properties, including those associated with neuritogenesis, synaptogenesis, and synaptic plasticity [88].
hESCs are known to express APP, with the type and level of expression influenced by both cell stemness and the pregnancy-associated hormone human chorionic gonadotrophin, suggesting that APP has a functional role in the early stages of human embryogenesis [12]. Some research has investigated the effects exerted by the cleavage products of APP processing in the early stages of embryonic neurogenesis and in the proliferation and differentiation of hESCs. This included detailed studies of α, β, and γ-secretase components to determine the mechanisms by which APP facilitates neuronal development [89].
Experiments with hESC clones have shown that clones over-expressing APP rapidly differentiate towards their neuronal fate without the intervention of exogenous factors. Robust differentiation was observed in these cell lines as early as 4 days, as compared to the 20–30 days associated with conventional differentiation protocols. Interestingly, sAPPα and sAPPβ, the two soluble peptide fragments of APP processing, were revealed to be concentration-dependent drivers of robust and rapid neuronal differentiation. While either peptide fragment is sufficient for neural differentiation, sAPPβ has a stronger ability to cause efficient neural differentiation as compared to sAPPα [67], a significant finding given that the aim of many drug treatments is to inhibit β-secretase activity [90 –92].
Related research has shown that amyloidogenic processing of APP (generating sAPPβ) promotes hESC proliferation, whereas non-amyloidogenic processing (generating sAPPα) leads to the differentiation of hESCs into neural precursor cells [12]. It is now clear that any disturbance in APP processing that leads to altered levels of sAPPβ, sAPPα, or Aβ could result in aberrant neurogenesis. Further studies of the amyloidogenic pathway have shown both of the classic forms of Aβ fragments, Aβ1-40 and Aβ1-42, to be present in hESC and differentiated neural precursor cell cultures. Aβ1-42 was shown to have a predominant role in hESC proliferation, but neither Aβ1-40 nor Aβ1-42 showed a significant role in the differentiation of hESCs. As opposed to Aβ1-42, oligomeric forms of Aβ were shown to drastically decrease hESC proliferation; for reasons likely to involve either reduced cell proliferation or increased toxicity [12]. These results indicate that Aβ fragments are maintaining hESC pluripotency, but that they become toxic once cells have differentiated into a neuronal phenotype.
It has been firmly established that neural stem cells are generated continually from the subependymal zone of the lateral ventricles and from the hippocampal formation, after which they migrate and differentiate into granule cells of the olfactory bulb and dentate gyrus, respectively [93]. The hippocampus of the adult brain shows constant neurogenesis and is the region most affected by AD. Accordingly, the increase in APP processing reported for AD affected brains could be perceived as a mechanism for inducing neural stem cell proliferation and repopulating neurons in and around the hippocampus.
Discussion and Conclusion
It is now well documented that APP plays a critical role in the proliferation and differentiation of neural stem cells, even at the early stages of embryogenesis. Indeed, APP is an important component of current protocols for differentiation of hESCs into neuronal lineages.
There are important aspects surrounding the role of APP in differentiation that we have discussed. One of these involves γ-secretase, which cleaves not only to APP, but to a wide spectrum of type I membrane proteins such as Notch, ErbB4, CD44, E-cadherin, etc, the abnormal proteolysis of which causes familial AD [94]. Notch is a particularly critical protein that has been conserved across evolutionary stages and is involved in neuronal development, neurogenesis, neuritic growth [95], synaptic plasticity, long-term memory [96,97], and neural stem cell maintenance [98]. In addition, Notch plays a critical role in controlling cell fate via local cell-to-cell interaction and suppresses neuronal differentiation during development [99,100]. The Notch signaling cascade was recently reported to contribute to a pathway whereby APP induces differentiation of the neuronal precursor cells into glial, indicating that APP functions in the regulation of cell fate through an alternate pathway involving the Notch signaling cascade [101]. Although detailed studies regarding protein-protein interactions are required, sufficient data suggest that interactions between Notch and APP could be candidates in inducing glial differentiation of neural precursor cells. This suggests that stem cells transplanted into AD patients may have a high chance of differentiating into glial cells rather than neural cells, given the pathological environment of the disease; certainly something to consider prior to commencing clinical trials. A number of studies have attempted to address this issue by inhibiting the Notch pathway [102] via methods that include monoclonal antibodies, RNA interference, antisense Notch, and γ-secretase inhibitors (GSIs) [103]. Of these, the use of GSIs has received the greatest support, as it tends to inhibit the Notch signaling pathway and lower the production of Aβ peptides [104].
There is a current research focus on obtaining iPSCs that can be used to study neurodegenerative disorders. However, it is proving difficult to induce a robust differentiation of iPSCs into defined neuronal lineages. APP has been successfully used in differentiating hESCs, and it would be worthwhile considering a use of APP in also differentiating iPSCs. In doing so, it will be important to remember factors like the Notch signaling pathway that may hinder differentiation protocols. Such protocols can be strengthened by modifying the media components to incorporate all of the conditions required by APP and related proteins to facilitate robust differentiation. Success in these endeavors is likely to help further our understanding of both neurodegeneration and neurogenesis, and could have important implications for disease modeling and the development of therapeutic strategies for AD.
Footnotes
Acknowledgments
I am grateful to Dr. Darrin Lipnicki for his assistance with the article.
Author Disclosure Statement
No competing financial interests exist.