
Abstract
Embryonic stem cell (ESC) differentiation via embryoid body (EB) formation is an established method that generates the 3 germ layers. However, EB differentiation poses several problems including formation of heterogeneous cell populations. Previously, we have enhanced mesoderm derivation from murine ESCs (mESCs) using conditioned medium (CM) from HepG2 cells. We used this technique to direct hematopoiesis by generating “embryoid-like” colonies (ELCs) from mESCs without standard formation of EBs. Two predifferentiation conditions were tested: (1) mESCs cultured 3 days in standard predifferentiation medium (control) and (2) mESCs cultured 3 days in HepG2 CM (CM-mESCs). Both groups were then exposed to primary differentiation for 8 days (ELC-formation period) and 14 days of hematopoietic differentiation. Enhanced mesoderm formation was observed in the CM-mESC group with an almost 5-fold increase in ELC formation (P≤0.05) and higher expression of mesoderm genes—Brachyury-T, Goosecoid, and Flk-1—compared with those of control mESCs. Hematopoietic colony formation by CM-mESCs was also enhanced by 2-fold at days 7 and 14 with earlier colony commitment compared with those of control mESCs (P≤0.05). This early clonogenic capacity was confirmed morphologically by the presence of nucleated erythrocytes and macrophages as early as day 7 in CM-mESC culture using standard 14-day colony-forming assay. Early expression of hematopoietic primitive (ζ-globin) and definitive (β-globin) erythroid genes and proteins was also observed by day 7 in CM-mESC cultures. These data indicate that hematopoietic cells more quickly differentiate from CM-mESCs, compared with those using standard EB approaches, and provide an efficient bioprocess platform for erythroid-specific differentiation of ESCs.
Introduction
E
Blood is derived from mesoderm in direct competition with spontaneous angiogenic and cardiomyogenic differentiation [6 –9]. To recapitulate the process of blood formation from mesoderm in vitro, ESCs are allowed to form EBs for ∼5–8 days followed by terminal hematopoietic differentiation through the use of various supplements and cytokines [3,6,10]. Recent efforts in in vitro red cell production starting from ESCs with a view to clinical applications have resulted in successful serum-free cultures for hematopoietic differentiation and the production of functional nucleated red blood cells [11 –13]. However, these methods have all used EB formation and coculture with feeder cells or conditioned media (CM). Non-EB cultures of hematopoietic cells derived from ESCs can also be achieved with coculture using stromal cells [11,14 –16]. Although coculture provides microenvironmental cues for differentiation of ESCs, the method is labor intense and problematic in the separation of feeders from differentiated cells and the possible formation of heterokaryons within the culture [17]. Recombinant cytokines are used for more defined culture conditions for the controlled differentiation of mesoderm from ESCs. However, the method is costly and can, paradoxically, lead to multilineage development due to the redundant action of cytokines and morphogens, such as BMP-4 and Activin-A [5,18]. Alternatively, CM from the culture of differentiated somatic cells contain soluble factors, although largely undefined, that direct ESC differentiation into layers such as mesoderm in vitro and are less resource intensive, making it a practical alternative for the study of mesoderm derivatives in vitro [5,17,19 –21].
We have previously demonstrated that CM, obtained from cultures of the human hepatocarcinoma cell line HepG2, enhanced mesoderm formation from murine ESCs (mESCs) without EB formation [5]. HepG2 cells have characteristics similar to those of visceral endoderm, an early organizer during embryogenesis that secret signaling molecules specifying cell fate [19,22,23]. The similarities between liver carcinoma cell lines and visceral endoderm have led to the suggestion that HepG2 CM reiterate visceral endoderm-like signaling in this system [5,19]. Differentiation of mESCs cultured in HepG2 CM resulted in a restricted repertoire of cell types comprised of mesoderm and parietal endoderm providing an efficient method for enriched mesoderm formation in vitro [5,24]. Herein, we extend our studies of directed mesoderm differentiation from mESCs using HepG2 CM and present an efficient method for the in vitro development of the hematopoietic lineage, as an alternative process for potential study and application of in vitro erythropoiesis from ESCs.
Materials and Methods
Experimental and control groups were set up as shown in Fig. 1.
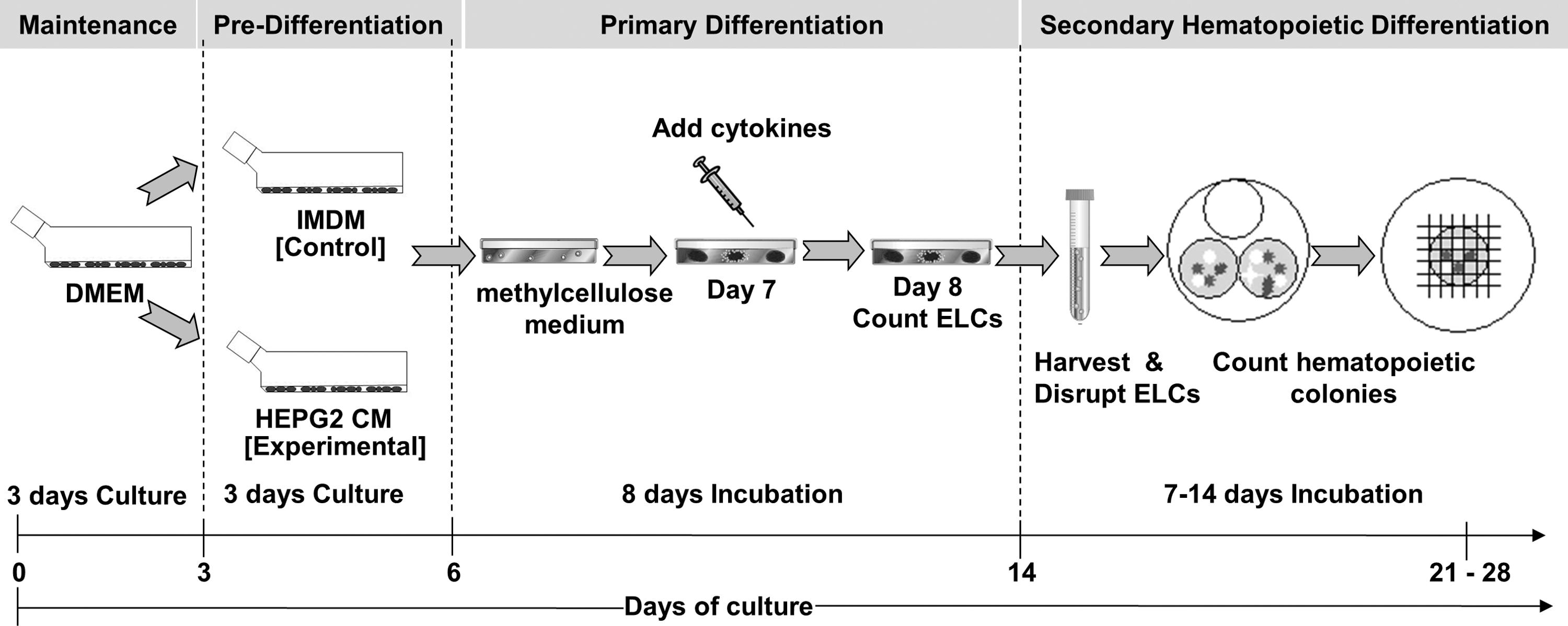
Experimental design: control murine embryonic stem cell (mESC) and conditioned medium (CM)-mESC groups. The control group consisted of normal mESC culture, exposed to standard Iscove's modified Dulbecco's medium (IMDM) prior to differentiation that had undergone an 8-day embryoid-like colony (ELC) formation period using primary differentiation medium followed by a further 14 days in the secondary hematopoietic differentiation medium, as previously described [54]. The experimental CM-mESC group consisted of mESCs previously exposed to HepG2 CM prior to differentiation, followed by the same 8-day ELC formation period using primary differentiation medium and a further 14 days of culture in the secondary hematopoietic differentiation medium, as explained previously. Both culture groups were analyzed simultaneously.
Predifferentiation culture of mESCs (“Control”)
Undifferentiated mESCs (E14/TG2-a cell line; ATCC; P<20) were maintained as previously reported [5]. Predifferentiation medium was prepared by addition of 15% (v/v) fetal bovine serum (FBS; Gibco), 1 mM sodium pyruvate (Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco), 2 mM
HepG2 CM–treated mESCs (“CM-mESCs”)
Culture of HepG2 cells (ATCC; HB-8605) and CM preparation were performed as previously reported [5].
“Embryoid-like” formation in primary differentiation
mESCs (3×103 cells/mL) were plated on low-adherence 35-mm Petri dishes (VWR International Ltd.) with semi-solid media containing 1% basic methylcellulose (Stem Cell Technologies), 15% FBS, 2 mM
Hematopoietic colony forming unit assay
Eight-day-old embryoid-like colonies (ELCs) were disrupted and replated (2×105 cells/mL) on low-adherence 35-mm Petri dishes (VWR) in IMDM (Gibco) supplemented with 15% FBS, 2 mM
MTS assay
Cell proliferation was assessed using the Cell Titre 96 Aqueous One Solution Reagent MTS assay (Promega) as per the manufacturer's instructions. Briefly, 3×104 cells/cm2 were cultured for 1, 2, 3, 4, and 5 days. Absorbance values were read at 490 nm using an enzyme-linked immunosorbent assay reader (ELx808; Bio-Tek).
Live and dead assay
Cells were incubated at room temperature for 30 min with 4 mM ethidium homodimer-1 and 2 mM calcein AM solution (Invitrogen). Cells fixed with 70% alcohol were used as negative control. Images were visualized on an Olympus BX51 microscope (Olympus) and captured using the F-view unit (Soft Imaging System GmbH). Images were not manipulated.
Immunocytochemistry
Cells were fixed in 4% (w/v) paraformaldehyde (VWR), and 0.2% (v/v) Triton X-100 (VWR) followed by blocking with 3% (v/v) goat serum (Santa Cruz Biotechnology) at room temperature. Primary antibodies for Oct-4 (Rabbit polyclonal) and stage-specific embryonic antigen-1 (SSEA-1; murine clone MC-480) (all from Santa Cruz Biotechnology) were incubated at 4°C overnight. Secondary antibodies, goat anti-rabbit fluorescein isothiocyanate and goat anti-mouse Texas Red (all from Santa Cruz Technologies), were applied for 1 h at room temperature. Images were visualized on an Olympus BX51 microscope (Olympus) and captured using the F-view unit (Soft Imaging System GmbH). Images were not manipulated.
Wright-Giemsa staining
Cytospins (1×106 cells/mL) were prepared. Slides were stained for 10 s in Wright-Giemsa stain (Sigma-Aldrich). Images were visualized with an Olympus BX51 microscope and captured using the DP50 camera (Olympus). Images were not manipulated.
Western blot analysis
Total protein was quantified using the BCA™ Assay Kit (Thermo Fisher Scientific). BSA was used as a known protein standard. Protein samples were mixed with 2 × SDS-PAGE sample buffer (Sigma). Protein blotting was performed in the C140 Mini Blot Module (Thermo Electron) at a constant current of 30 mA for 50 min. Membranes were blocked with 5% (w/v) nonfat milk solution (Sigma) for 2 hrs. Primary antibody was incubated overnight followed by application of the appropriate horseradish peroxidase-conjugated (HRP) secondary antibody for 1 h. Proteins were detected using the SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Primary antibodies are as follows: rabbit anti-mGata-1 polyclonal (1:200), rabbit anti-mζ-globin polyclonal (1:200), chicken anti-mβ-major globin polyclonal (1:200), and rabbit anti-mGAPDH polyclonal (1:20,000). Secondary antibodies, anti-rabbit and anti-chicken polyclonal IgG, were conjugated to HRP (1:10,000). All antibodies were from Santa Cruz Biotechnology.
Reverse transcriptase–polymerase chain reaction
Reverse transcriptase–polymerase chain reaction (RT-PCR) analysis was conducted using cDNA with primer sequences as listed in Table 1. PCR conditions were as follows: reverse transcription at 42°C for 45 min, Taq polymerase activation at 95°C for 2 min, thermal cycling at 95°C for 30 s, specific primer annealing temperature at 65°C for 60 s and for 25–35 cycles, elongation at 72°C for 60 s, followed by a final elongation step at 72°C for 5 min. Samples without cDNA were used as negative controls. Images from gel electrophoresis were captured using the Dyversity gel-documentation system using the Geneflash program (SynGene). Images were not manipulated.
Oct-4, octamer-binding transcription factor-4; Rex-1, reduced expression protein-1; FGF-5, fibroblast growth factor-5; Gata-4, glutamyl-tRNA(Gln) amidotransferase subunit A-4; α-feto protein, alpha-1-fetoprotein; Brac-T, brachyury transcription factor T-gene; Flk-1, fetal liver kinase-1; GSC, goosecoid; Sox-1, sex determining region Y-box1; Nestin, neuroepithelial stem cell specific protein; Gata-2, glutamyl-tRNA(Gln) amidotransferase subunit A-2; c-kit, cellular-homolog of protein kinase transmembrane receptor/tyrosine-protein kinase kit; c-myb, cellular-myeloblastosis oncogene/proto-oncogene protein; SCL/Tal-1, stem cell leukemia/T-cell acute lymphocytic protein 1; Gata-1, glutamyl-tRNA(Gln) amidotransferase subunit A-1; βH1-globin, hemoglobin beta-H1 chain; β-major globin, beta-major globin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PCR, polymerase chain reaction.
Statistical analysis
All experiments were done in quadruplicate (n=4) on 5 separate occasions (n=5) and each value represents the mean±standard deviation (SD). Statistical significance of results was evaluated using two-way analysis of variance with a level of significance P≤0.05 or P≤0.01.
Results
CM-mESCs promote early differentiation with higher growth proliferation
mESCs cultivated in standard maintenance Dulbecco's modified Eagle medium (DMEM) formed typical spherical colonies whereas CM-mESC cultures (experimental) using HepG2 CM resulted in formation of clusters with a flattened morphology as described previously (Fig. 2A) [5,24]. Control mESCs in standard predifferentiation IMDM optimized for hematopoietic differentiation [25,26] also resulted in flattened morphology (Fig. 2A). CM-mESC proliferation rate in culture was higher compared with that of control predifferentiation mESCs during the first 3 days of culture (Fig. 2B; P≤0.05), wherein the viability of both mESCs and CM-mESCs was >95% (Fig. 3). Pluripotency expression of Oct-4 and SSEA-1 for all mESC cultures was both uniformly distributed and not restricted to subpopulations of cells within the colonies as previously described [5,19]. Expression of Oct-4 was confirmed with RT-PCR and Rex-1 expression in all culture conditions, indicative of pluripotent capacity. Interestingly, the primitive ectoderm marker FGF-5 was only expressed in CM-mESCs, suggestive of early differentiation.
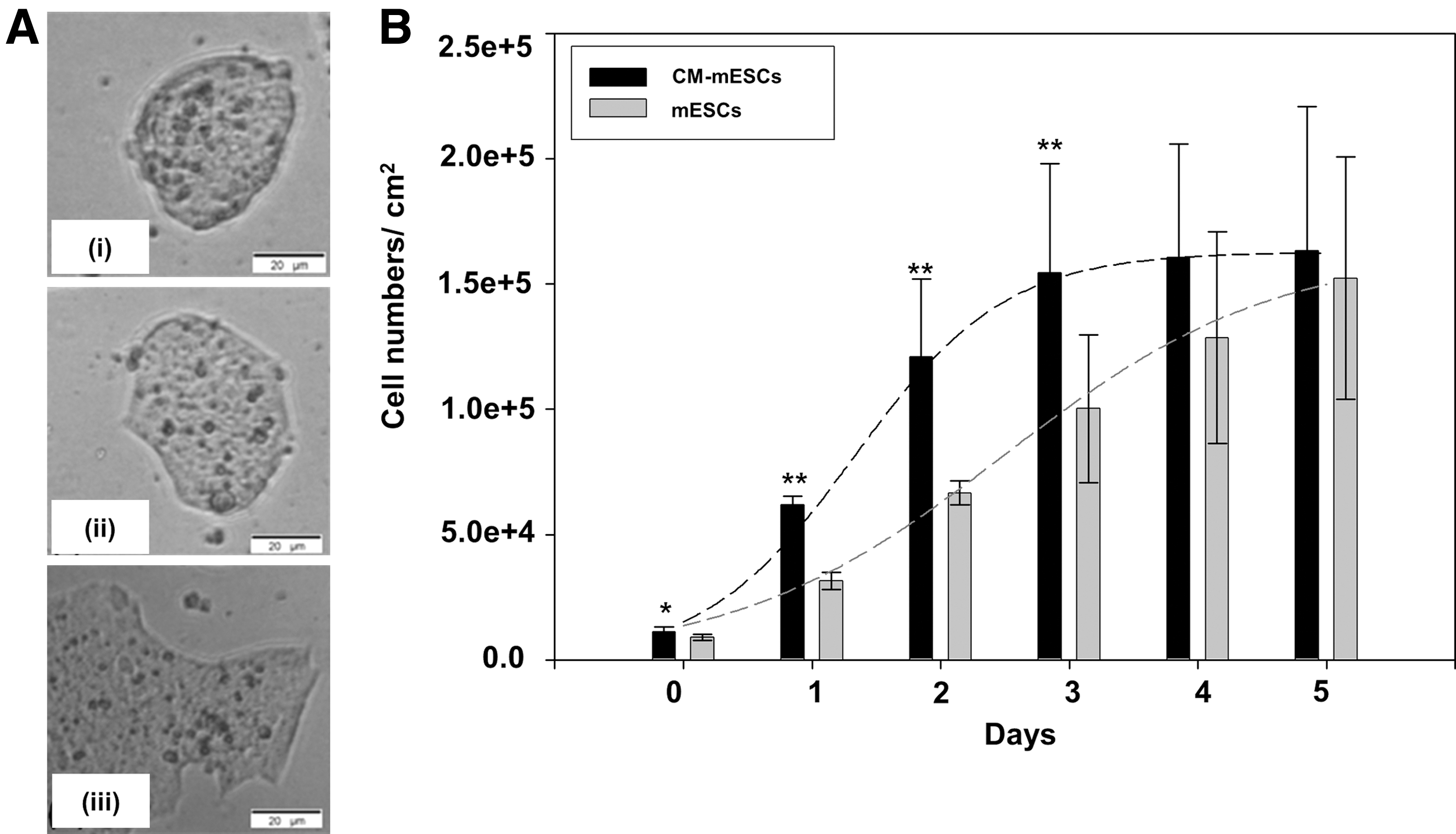
Morphological and growth evaluation of control and CM-mESCs.
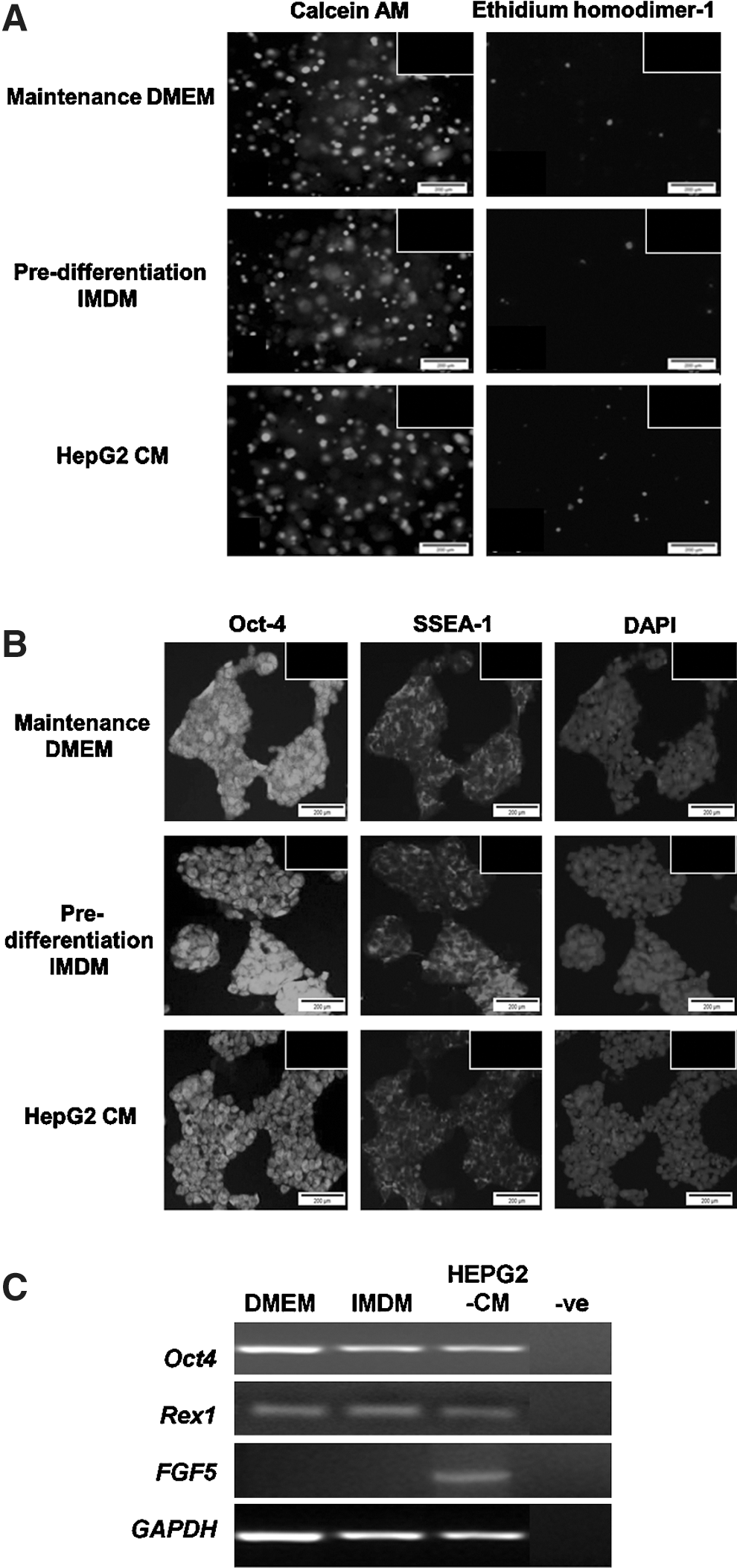
Viability and maintenance of pluripotency of undifferentiated mESCs in different culture conditions at day 3 of culture.
Mesoderm differentiation of ELCs is best with CM-mESCs
ELC formation from the control mESC group by day 8 (Fig. 4A[i]) was larger when compared with that of CM-mESCs (Fig. 4A[ii]), as previously described [5]. Interestingly, culture of CM-mESCs resulted in an almost 5-fold enhancement of ELCs formed (P≤0.01) as compared with that formed by the control group at day 8 (Fig. 4B). Further characterization of the 3 germ layers and early hematopoietic differentiation using RT-PCR indicated that endoderm (Gata-4 and α-fetoprotein), mesoderm (Brachyury-T, Goosecoid, and Flk-1), and ectoderm (Nestin and Sox-2) gene markers were detected in ELCs of the control group (Fig. 4C). In contrast, there was a higher expression of the mesoderm gene Flk-1, but no expression of the endoderm genes Gata-4 and α-fetoprotein in ELCs of CM-mESCs (Fig. 4C). In agreement with this enhanced mesoderm pattern induced by HepG2 CM, ELCs from CM-mESCs displayed a higher expression of Gata-2 and c-Kit, indicative of early hematopoietic differentiation (Fig. 4C).
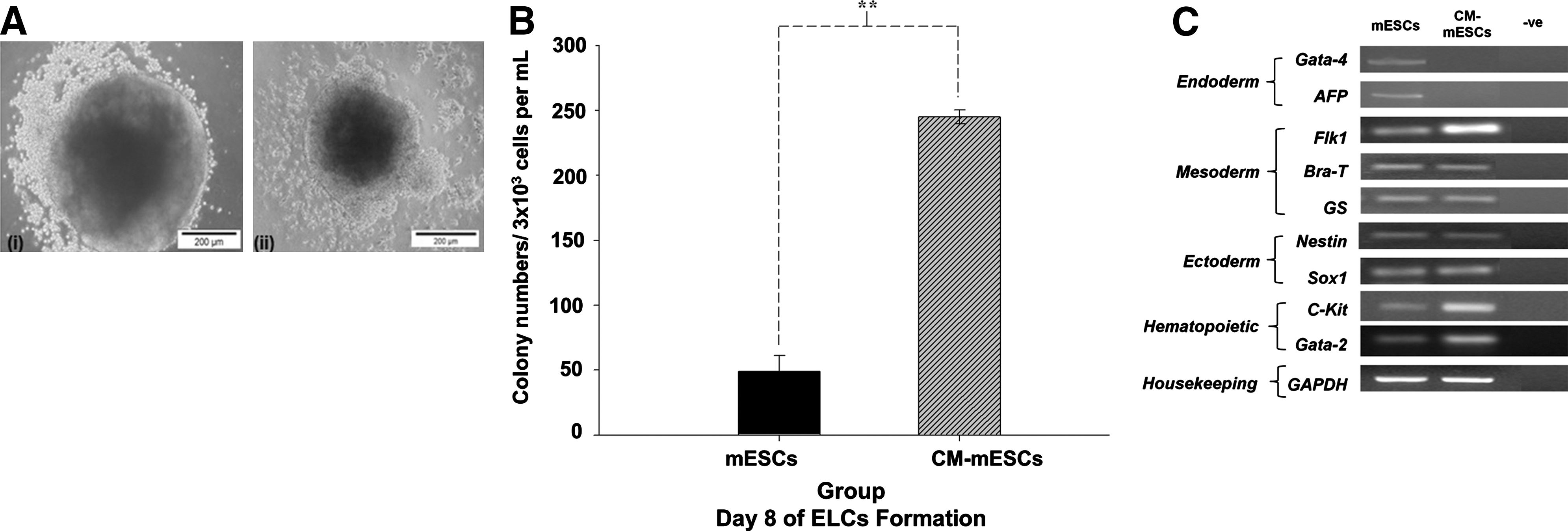
Formation of ELCs in both culture groups at day 8.
CM-mESCs efficiently commit to the hematopoietic lineage in clonogenic assays
Early maturation of hematopoietic progenitor colonies burst-forming unit erythroid (BFU-E), colony forming unit (CFU)-E, CFU-GM, and CFU-GEMM was observed as early as day 7 in the CM-mESC group (Fig. 5A) when compared with that of the control mESC group. Further incubation of the same dishes to the standard 14 days resulted in changes in morphology, with a reduction in the reddish hue of the colonies, associated with a loss of hemoglobin [27,28] in the CM-mESC group, whereas the control mESC group displayed well-matured hematopoietic colonies. A higher number of hematopoietic colonies was also achieved in the CM-mESC group at day 7 as well as at day 14 of incubation (Fig. 5B; P≤0.05) with a 2-fold enhancement of all types of myeloid-erythroid progenitor colonies compared with that of the control mESCs. No morphologic differences were observed between the BFU-E colonies in the different groups. Nucleated erythrocytes and macrophages were identified in the CFUs from CM-mESCs at day 7 (Fig. 5C), as previously noted [29]. Expression of early hematopoietic and myeloid-erythroid genes was not detected in control mESCs yet was distinct in CFUs from CM-mESCs at day 7 of CFU culture (Fig. 5D). Specifically, the definitive erythroid gene β-major globin was detected on day 7, earlier than that observed in control mESCs, with relatively lower expression of the primitive erythroid gene βH1-globin at the same time point. At 14 days of the colony assay, control mESC and experimental CM-mESC groups showed a constant expression of all hematopoietic and myeloid-erythroid genes with lower expression of β-major globin and no expression of βH1-globin (Fig. 5D). This early erythroid maturation of adult type at day 7 of CM-mESC culture was confirmed by detection of Gata1 and β-major globin proteins with a lower expression of the primitive erythroid marker ζ-globin (Fig. 5E). Gata-1 and β-globin markers were maintained in both groups at day 14. Interestingly, the primitive erythroid markers βH1-globin gene and ζ-globin protein were not expressed in the control mESCs at any time point. Overall, the data indicated that CM-mESCs were capable of hematopoietic maturation earlier than control mESCs, indicating a more efficient differentiation system to mesoderm and the hematopoietic lineage.
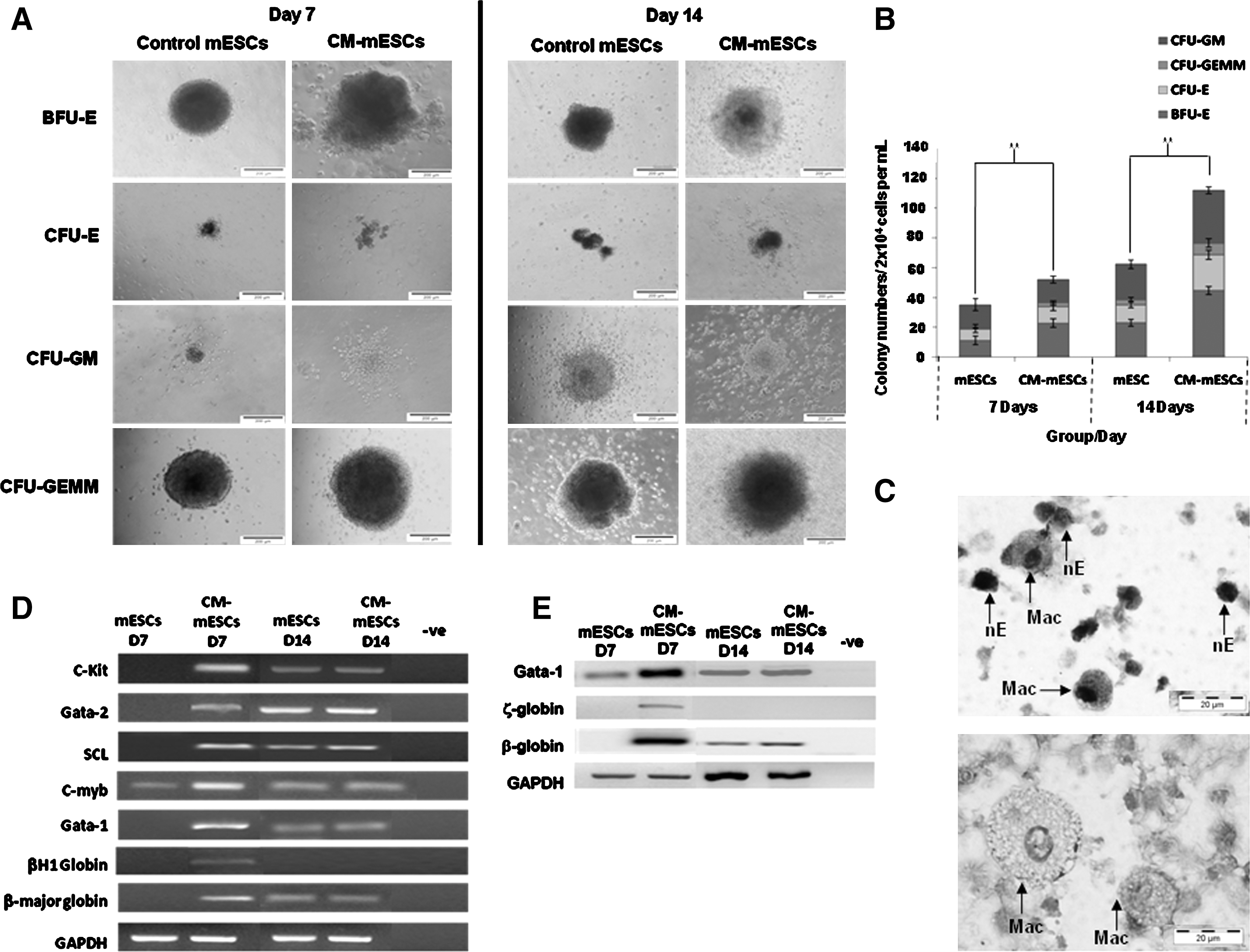
Clonogenic capacity and gene and protein expression of terminally differentiated mESCs at days 7 and 14.
Discussion
We have developed an efficient bioprocess for hematopoietic differentiation toward erythropoiesis from mESCs in the absence of traditional EB formation using HepG2 CM. This method resulted in higher numbers and early hematopoietic maturation, mirroring those found in normal mammalian development. The use of HepG2 CM resulted in a less labor-intensive protocol with reduced use of resources making it a viable alternative for large-scale applications in future.
The enhancement of mesoderm formation using HepG2 CM is consistent with previous findings [5] that were induced by the visceral endoderm-like signaling of the CM. Currently, in vitro differentiation of mESCs toward the hematopoietic lineage is triggered by the addition of hematopoietic growth factors [1 –3,10]. Although cardiogenic differentiation has been considered as a spontaneous effect in mesodermal differentiation [30], our data do not suggest that hematopoietic differentiation occurred spontaneously during mesoderm differentiation using HepG2 CM. Despite the fact that many active factors within the CM are as yet uncharacterized [31,32], initial studies into defining the active properties of the CM have already been undertaken [24]. One of the mesodermal enhancer factors found in HepG2 CM is fibronectin [24] that is expressed in the primitive endoderm and epithelial (epiblast) cells developing into mesoderm during embryogenesis [33]. Serum transferrin—an iron binding glycoprotein that controls the level of iron, the crucial element contained in hemoglobin [34]—is also found in HepG2 CM providing a platform for the enhanced erythropoietic differentiation [24]. Interestingly, erythrocyte membrane proteins, such as α- and β-Spectrin, Ankyrin, and Protein 4.1, are also among the active factors found in HepG2 CM [24,35]. Major protein isoforms such as these, which are produced by developing erythroid cells, are critical in erythroid development, including membrane assembly, expression of erythroid-specific genes, and hematopoiesis generally [35]. The presence of such isoforms in HepG2 CM, in addition to fibronectin, vimentin, and transferrin, may partially explain the enhancement of hematopoietic colonies toward erythropoiesis observed in the CM-mESCs through as yet largely undefined mechanisms, introducing an interesting area of future research using this model. Further efforts into understanding both the protein and peptide active factors found within HepG2 CM and the effects on mESCs during development in vitro could enhance understanding on the mechanisms and biology of both primitive and definitive erythropoiesis. By extension, this model could be used as a tool to offer insight into disease processes and novel targets for treatments in disorders of red cell development and maturation.
CFU numbers presented in this work were highest in cultures using CM-mESCs with 1.5-fold expansion in all types of hematopoietic progenitors on day 7, which increased to 3.2-fold at 14 days of incubation, without requiring EB-stage formation. These CFU data compare favorably with other differentiation protocols directing mESCs to hematopoiesis via EB formation, such as those using cytokine-enhanced media resulting in formation of only erythroid progenitor colonies with inhibition of myeloid and pro-erythroblast colony formation after 16 days of culture [36], and using OP9 stromal cells resulting in a 2.6-fold increase in CFUs after 14 days of incubation [16]. Our current findings indicate that hematopoietic differentiation may be controlled and enhanced toward erythropoiesis specifically in the absence of standard EB formation or coculture methodologies, through streamlining mesoderm differentiation and simplifying the culture process with HepG2 CM. The differentiation time to erythropoiesis starting from mESCs is also truncated using HepG2 CM and can be as little as 12–13 days (3 days in HepG2 CM, followed by 2–3 days in primary differentiation and then 7 days in secondary differentiation medium) [37 –39]. This expedited maturation provides the opportunity for an efficient bioprocess for both the study of erythropoiesis and exploitation of the system in vitro as a potential novel platform for ex vivo blood cell bioprocessing and manufacture once the active factors critical for enhanced erythropoiesis are clarified.
Myeloid-erythroid markers were present as early as day 7 of incubation in CM-mESC cultures [3]. Gata-1, found at an early stage in these cultures, is expressed in primitive and definitive erythroid cells, and is essential for normal erythropoiesis, erythroid cell survival, and proliferation [40,41]. Gene and protein expression of βH1-globin and primitive ζ-globin, respectively, present in primitive erythroid cells (primitive erythroblasts) from E7.5 through E8.5 of mouse gestation [11,28,42,43], and β-major globin, an adult hemoglobin gene correlating with definitive β-globin protein normally expressed in both circulating primitive erythroblasts and differentiating definitive erythroblasts in the fetal liver [11,28,42,43], was also found in the CM-ESCs at day 7 of CFU formation. These findings are consistent with the known downregulation of βH1-globin and upregulation of β-major globin prior to the maturational globin switch from primitive erythroblast to definitive erythroblast between E8.5 and E12.5 [28]. In contrast, low expression of the early hematopoietic progenitor gene c-myb in control mESCs at day 7 suggested that the hematopoietic program had just started and that commitment toward specific blood progenitors was not yet well-developed in culture [44 –47]. By using ESC differentiation as a model, cells with hemangioblast potential have been identified, known as blast colony-forming cells (BL-CFCs; possibly the in vitro equivalent of the hemangioblast), and represent a transient population that appears in cell aggregates before the establishment of any other hematopoietic lineages [12,48 –50]. Our work has also shown an early maturational stage in the CM-mESC group during the 8 days of ELC primary differentiation with high expression of Flk-1 together with Brachyury-T, SCL, Gata-2, and c-kit, suggesting that hemangioblast-type colonies might be identified [9,51 –53], representing an area for future research using this model.
Herein, we have demonstrated that the use of HepG2 CM-mESCs enhanced hematopoietic differentiation and promoted specific erythroid lineage differentiation, making the protocol a useful tool in the study of hematopoiesis. These data support the hypothesis that HepG2 CM enhances mesoderm formation and its derivatives from ESCs and may enable the development of an efficient and defined differentiation method for directed erythrocyte production from ESCs.
Footnotes
Acknowledgments
This work was supported by the Higher Ministry of Education Malaysia, Universiti Malaysia Terengganu, and The Richard Thomas Leukemia Fund.
Author Disclosure Statement
The authors have no conflicts of interest.