
Abstract
Podocalyxin-like protein (PODXL) is a member of CD34 family proteins. It is the protein that carries many post-translational epitopes responsible for various pluripotent surface markers including TRA-1-60, TRA-1-81, GCTM2, GP200, and mAb84. However, PODXL has not attracted the attention of stem cell biologists. Here, we report several features of PODXL mRNA and protein in pluripotent stem cells. Similar to the modification-dependent pluripotent epitopes, PODXL transcripts and carrier protein are also features of pluripotency. PODXL is highly expressed in early human embryos from oocytes up to four-cell stages. During reprogramming of human cells to pluripotency, in contrast to TRA-1-60 and TRA-1-81, PODXL is activated by KLF4 at a very early time of reprogramming. Although TRA-1-60 and TRA-1-81 are completely lost upon differentiation, a residual PODXL+ population exists even after extended differentiation and they were identified by the universal human PODXL epitope 3D3. Unlike TRA-1-60 and TRA-1-81 epitopes that are unique to primate pluripotent stem cells (PSCs), PODXL carrier protein can be used as a murine surface marker. Most importantly, antibody to 3D3 epitope causes massive necrosis and apoptosis of human PSCs (hPSCs). We suggest that 3D3 antibody could be employed to eliminate the tumorigenic pluripotent cells in hPSC-derived cells for cell transplantation.
Introduction
H
PODXL is a member of the CD34 family, which also includes CD34 and endoglycan. PODXL is an integral transmembrane protein heavily modified with O-glycosylation, N-glycosylation, sialylation, and sulfation [17]. It is highly expressed in kidney epithelium, and it is also expressed in several other cell types, including hematopoietic progenitors, endothelium, platelets, and some neural cells. Its well-known role is in the development of kidney epithelia and maintenance of the podocyte filtration slit. PODXL knockout mice die within 24 h after birth due to anuria [18]. It plays various roles in different cells including antiadhesion, adhesion, cell matrix interaction, morphogenesis, and cell signaling. It is also associated with more than 10 human malignancies [15,19 –25]. However, PODXL receives little attention in the field of pluripotent stem cells although numerous pluripotent surface markers are associated with it.
Here, we report several characteristics of PODXL in the context of hPSCs: (a) not only the post-translational modifications are a human pluripotent feature, PODXL is also a general pluripotent marker; (b) PODXL is a mouse ESC marker as well although its modification-dependent epitopes are not shared between human and mouse; (c) KLF4 activated PODXL at an early stage of reprogramming; (d) 3D3, a universal antibody of PODXL, identifies a residual PODXL+ population after differentiation of hPSCs, whereas other modification-dependent PODXL antibodies failed to do so; (e) the antibody 3D3 is cytotoxic to human pluripotent stem cells.
Materials and Methods
Cell culture and reprogramming
Lenti-X 293T (#632180; Clontech) and Hela cells were maintained in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin, and 0.1 mM MEM NEAA. Human fibroblasts (BJ: ATCC CRL-2522™) were cultured in fibroblast medium: DMEM (#12800-058; Gibco) supplemented with 10% heat-inactivated FBS (#10437; Gibco), 0.1 mM 2-mercaptoethanol (#194834; MP), 100 U/mL penicillin and 100 μg/mL streptomycin (#15140-122; Gibco), 0.1 mM MEM NEAA (#11140-050; Gibco), and 4 ng/mL human FGF2 (Nacalai USA, NU0005
For reprogramming, BJ cells were seeded into 24-well plates at 2.0 × 104 cells/well. To determine the multiplicity of infection (MOI) for BJ transduction, cells from a spare well were counted 24 h postplating. The amount of viruses for the reprogramming factors is OCT4, 10 MOI; SOX2, 5 MOI; and KLF4, 5 MOI. Cells were transduced in the presence of 6 μg/mL hexadimethrine bromide (#107689; Sigma). Viruses were removed by medium change after overnight incubation. Cells were reseeded into Matrigel (A1413302; Geltrex)-coated 6-well plates 48 h post virus transduction in 1:10 to 1:12 ratio. Reprogramming was conducted in E7 medium: DMEM/F12 (#12400-024; Gibco) supplemented with 64 mg/L L-ascorbic acid 2-phosphate sesquimagnesium (A8690; Sigma), 13.6 μg/L sodium selenium (S5261; Sigma), 1.7 g/L NaHCO3 (BP328-1; Fisher Scientific), 1 g/L sodium chloride (BP358-10; Fisher Scientific), 10 ng/mL FGF2, 20 μg/mL insulin (I2643; Sigma), and 10 μg/mL holo-transferrin (T0665; Sigma) every day from day 3–13 post virus transduction. E8 medium (E7 + 2 μg/L TGFβ1 [8915LC; Cell Signaling]) was used from day 14–21.
Human ESCs (H1 and H9, WiCell, Wisconsin) and human iPSCs (3RiPSC2, 3RiPSC3, 3RiPSC4, and CD34iPSC6, generated in our lab) were maintained in E8 medium. Mouse ESCs (E14) were cultured in mouse ES cell growth medium: Knockout DMEM (#10829; Invitrogen) supplemented with 15% ES-certified FBS (#10439; Invitrogen), 2 mM L-glutamine (#21051-024; Gibco), 0.1 mM MEM NEAA, 0.5% COS-LIF supernatant medium, and 0.1 mM 2-mercaptoethanol.
Lentivirus packaging, concentration, and titration
Lenti-X 293T cells were seeded in 15-cm dishes at a density of 1.0 × 107 cells/dish 24 h before transfection. When cells reached ∼70–80% confluence, medium was refreshed 1 h before transfection. A total of 60 μg of plasmid DNA (30 μg transfer plasmid, 10.5 μg envelop plasmid pMD2-G, and 19.5 μg packaging plasmid ps-PAX2) was mixed with 150 μL of 0.25 M calcium chloride and 1.35 mL sterilized HPLC water. The DNA-calcium solution was further mixed with equal volume of 2× HBS (pH 7.05). Formation of DNA-calcium phosphate complexes was facilitated by vortexing for 20 s before adding into the dish. The transfection culture medium was changed with fresh fibroblast medium 16 h post-tranfection. Viruses were collected between 48 and 72 h post-transfection. Cell debris was removed from the virus-containing medium at 3,000 g for 10 min at 4°C followed by filtration through a 0.45-μm filter (SCHVU01RE; Millipore). Lentivirus was precipitated with 8.5% PEG-6000 (final concentration, from 50% stock) (5288771KG; Millipore) and 0.4 M sodium chloride in a 50-mL tube for 3–5 h at 4°C. Virus pellet was obtained by centrifugation at 4,000 g for 40 min at 4°C, which was consecutively resuspended in 50 mM Tris-HCl (pH 7.4) with a concentration ratio of around 100–150×. The concentrated virus stock was divided into small aliquot (20 μL), and kept in −80°C. Viral titer was determined with flow cytometry for the expression of GFP in virus-transduced Hela cells.
Plasmid construction, transient transfection, and luciferase assay
PODXL promoter region (−2198 to −163) was amplified from genomic DNA of hiPS cell lines using forward primer (PODXL-PROM3-F): 5′-CCAGCCTCATTCCAGAAAGC and reverse primer (PODXL-PROM3-R): 5′-GCCGTGAACAATGAGCAGAG. The PCR fragment was inserted into Sma I site of pGL3-Basic vector (E1751; Promega) to generate P2K plasmid. A shorter P0.5K plasmid was constructed by amplifying PODXL promoter region (−500 to −163) from P2K using forward primer (pPODXL-0.5k.F): 5 ′-TCAACGCGTGGGAGATGAGCTTCTCCG and reverse primer PODXL-PROM3-R. PODXL proximal promoter region (PODXL-TSS: −455 to −1/+1) was synthesized and cloned onto Sma I and Bgl II sites of pUC57 by Genewiz. Such fragment was double digested with Sma I and Bgl II and inserted into the previous intervening plasmids to generate pGL3-PODXL.500 and pGL3-PODXL.2198 plasmids; Plasmids overexpressing OCT4, SOX2, KLF4, and c-MYC under CMV promoter were constructed using PCR-amplified human cDNAs.
Hela cells were seeded in 24-well plates at a density of 5.0 × 104 cells/well 24 h before plasmid transfection. Cells were co-transfected with O/S/K/M (1 μg/well), PODXL promoter plasmids (pGL3-PODXL.500 or pGL3-PODXL.2198: 0.5 μg/well), and internal control plasmid pRL-TK (25 ng/well) using Lipofectamine 3000 (L3000; Invitrogen) per manufacturer's instructions. Luciferase assay was performed 48 h post-transfection with Dual-Luciferase Kit (E1910; Promega).
Western blotting
Whole cell lysates were prepared by rotating cells in RIPA buffer: 100 mM Tris-HCl (pH 7.4), 150 mM sodium chloride, 1 mM EDTA, 1% Triton-X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1× Pierce Halt Protease Inhibitor cocktail (#78429; Pierce) for 30 min at 4°C. Proteins were fractionated on 7.5% SDS-PAGE, and transferred onto polyvinylidene difluoride membranes (#1602177; Bio-Rad). Membrane was blocked with 5% nonfat milk in TBS-T (0.2‰ Tween-20 in 1× Tris-buffered saline) for 45 min, and then was blotted for 2 h with primary antibodies: human PODXL (sc-23904, 3D3; Santa Cruz Biotech); mouse PODXL (AF1556; R&D); GAPDH (sc-25778; Santa Cruz Biotech); P21 (#2947, Cell Signaling), or β-actin (#4970; Cell Signaling). Membrane was further incubated with secondary antibodies: anti-mouse IgG-HRP (#7076; Cell Signaling), or anti-rabbit IgG-HRP (#7074; Cell Signaling). Protein blots were analyzed with Image Lab software on Bio-Rad ChemiDoc MP Imaging System.
Reverse transcription-quantitative polymerase chain reaction
Cells were lysed in TRIzol reagent (#15596-018; Invitrogen). Total RNAs were extracted using the Direct-zol™ RNA Miniprep Kit (R2052; Zymo Research). cDNAs were synthesized with M-MLV reverse transcriptase (#28025-013; Invitrogen). Quantitative PCR was performed in triplicates on ViiA 7 Real-time PCR system (Applied Biosystem) using SYBR-Green Master PCR mix (#639676; Clontech). Primers used for qPCR were listed in Supplementary Table S1 (Supplementary Data are available online at
Antibody treatment
Human pluripotent stem cells were seeded in 12-well plates at a density of 5.0 × 104 cells/well 24 h before antibody treatments. Cells were incubated with vehicle (0.1% sodium azide stock), control mouse IgG (10400C; Life Technologies), or different antibodies: SSEA-4 (MAB4304; Millipore), TRA-1-60 (MAB4360; Millipore), and PODXL (sc-23904; Santa Cruz Biotech or MABD89; Millipore) at a concentration of 1 μg per 1.0 × 105 cells, or 1–3 μg/mL. Cell numbers were counted every day using replicated wells of the same treatments as references to calculate the amount of antibodies to be added for each treatment on that day. Cell growth curve was generated for treatments for four continuous days, and fresh antibodies were added every 24 h together with daily medium change.
Embryoid body formation and in vitro differentiation
PSCs were grown to 70% confluence in six-well plates. Cells were collected and aggregated in AggreWell™ 400 plate (#27845; STEMCELL) per manufacturer's instructions. Cell aggregates were transferred to Ultra-Low Attachment Surface six-well plates (#3471; Corning) on day 4, and were further cultured in suspension for another 10 days before being seeded on 0.1% gelatin-coated six-well plates. Embryoid body (EB) differentiation was induced by FBS for more than 30 days. Culture medium was refreshed every other day, with the concentration of FBS increasing gradually from 2% to 10%.
Teratoma formation for in vivo differentiation
PSCs were grown to 70% confluence in six-well plates. Cells were harvested and divided into small aliquots (1.0 × 106 cells in 100 μL cold E8 medium supplemented with 30% Matrigel). Bilateral subcutaneous injection was performed on 6–8 week immunocompromised NSG mice. Teratoma was harvested 8 weeks after initial injection. UAB Institutional Animal Care and Use Committee approved the mouse procedure used in this study.
Flow cytometry
Harvested cells were resuspended in 1 mL fluorescence-activated cell sorting (FACS) buffer (2% BSA, 2 mM EDTA, and 0.05% sodium azide in PBS), and cell clumps were removed by a cell strainer. The individualized cells were collected by centrifugation. Cell pellet were resuspended in FACS buffer. About 2.0 × 105 cells in 100 μL FACS buffer were incubated with primary antibody at 4°C for 30 min. After washing cells with 3 mL FACS buffer, cells were further incubated with Alexa Fluor 488-labeled secondary antibody at 4°C for 30 min. The cells were then washed with 3 mL FACS buffer. Cell pellet was resuspended in 600 μL FACS buffer. Then, 10 μL 7-AAD (20 μg/mL stock) was added before assay with flow cytometer (LSRFortessa™; BD Bioscience). Antibodies used are hPODXL (sc-23904, 3D3; Santa Cruz Biotech) or mPODXL antibodies (AF1556; R&D), goat anti-mouse IgG (A10667; Invitrogen), Alexa Fluor 488-donkey anti-goat IgG (A11055; Invitrogen), PE-SSEA4 (#560128; BD Pharmingen), PE-TRA-1-60 (#560193; BD Pharmingen), or PE-SSEA1 (#560886; BD Pharmingen).
Apoptosis assay
Cellular apoptosis and necrosis were analyzed on flow cytometer using Annexin V PE Apoptosis Detection Kit (#88-8102; eBioscience) per manufacturer's instructions.
Chromatin Immunoprecipitation
Cells were collected and divided into aliquots (3–5 million) in 500 μL PBS. Then, 13.9 μL 37% formaldehyde (final concentration 1%) was added into the tube to cross-link DNA-protein for 8 min at RT. Formaldehyde was then quenched with 27 μL of 2.5 M glycine (final concentration 125 mM) for 5 min. Cells were collected by centrifugation at 500 g for 10 min. Cell pellet was then washed with 1 mL ice-cold hypotonic buffer (20 mM HEPES-KOH pH 7.5, 20 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, 0.1 mM PMSF, and 1× Halt Protease Inhibitor cocktail). Washed cells were collected by centrifugation at 500 × g for 10 min. The cells undergo three rounds of freeze-thaw cycle between liquid nitrogen and RT. Cells were resuspended in 1 mL ice-cold lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton-X-100, 1 mM DTT, 0.1 mM PMSF, and 1× Halt Protease Inhibitor cocktail). Cell suspension was rocked for 10 min at 4°C. Nuclei were collected by centrifugation at 750 g for 10 min at 4°C and washed with 1 mL ice-cold wash buffer (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 mM DTT, 0.1 mM PMSF, and 1× Halt Protease Inhibitor cocktail). After wash, the nuclei were lysed for 30 min on ice in 300 μL sonication-lysis buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-lauroylsarcosine, 1 mM DTT, 0.1 mM PMSF, and 1× Halt Protease Inhibitor cocktail). Chromatin sonication was then performed using diagenode bioruptor (3 × 10 cycles: 30 s ON/OFF). Sheared chromatin was collected by centrifugation at 14,000 g for 15 min at 4°C, and it was quantified using Nanodrop Spectrophotometer (Thermo Scientific).
For each chromatin immunoprecipitation (ChIP), 50 μL protein G beads slurry (#29398; Pierce) were washed thrice with 1 mL blocking buffer (0.5% BSA in 0.2‰ PBS-Tween). The beads were saturated with 10 μg HA antibody (Abcam, ab9110) or corresponding rabbit IgG control (ab37415; Abcam) in 1 mL blocking buffer by rotating at 4°C for 6 h. ChIP was performed by mixing 80 μg of sheared chromatin with antibody-beads and rotating for another 16 h at 4°C. The beads were transferred to fresh prechilled tubes and washed five times with 1 mL ice-cold Li-RIPA buffer (50 mM HEPES-KOH pH 7.5, 500 mM LiCl, 1 mM EDTA, 1% NP-40, and 0.7% sodium deoxycholate). The ChIP'd chromatin was eluted from beads with 200 μL elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, and 1% SDS) by gentle agitation for 1 h at 65°C. Cross-linking of protein to chromatin was reversed by incubating the samples at 65°C for 18 h. Equal volume (200 μL) of TE buffer (10 mM Tris-HCl pH 8.0 and 1 mM EDTA) was added to the tube to dilute SDS. Proteinase K (0.2 mg/mL) treatment was carried out for 2 h at 55°C to digest any residual protein in the sample. DNA was purified using PureLink PCR Purification kit (K3100; Invitrogen) and resuspended in 50 μL TE buffer. DNA concentration was determined using Qubit fluorometer (Invitrogen).
RNA-Seq
mRNA-sequencing was performed on the Illumina HiSeq2500 using the sequencing reagents and flow cells providing up to 300 Gb of sequence information per flow cell. Briefly, the quality of the total RNA was assessed using the Agilent 2100 Bioanalyzer followed by two rounds of polyA+ selection and conversion to cDNA. We used the stranded mRNA library generation kits per manufacturer's instructions (Agilent). Library construction consists of random fragmentation of the polyA mRNA, followed by cDNA production using random primers with inclusion of actinomycin D in the first strand reaction. The ends of the cDNA are repaired, A-tailed and adaptors ligated for indexing (four different barcodes per lane) during the sequencing runs. The cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for library quantitation (Kapa Biosystems) before cluster generation. Clusters were generated to yield ∼725K–825K clusters/mm2. Cluster density and quality were determined during the run after the first base addition parameters were assessed. We ran paired end 2 × 50 bp sequencing runs to align the cDNA sequences to the reference genome.
Bioinformatics
All the reads from our RNA-seq (GSE66798) [26] and the reads from the experiments GSE71645 [27], GSE59528 [28] and GSE36552 [47] were mapped to the human reference genome (GRCh37/hg19) using the STAR aligner [29] guided by a gene transfer file (Ensembl GTF version GRCh37.70).
Read count tables were generated using HTSeq (v0.6.0) [30] and were normalized based on their geometric library size factors. Deferential Expression (DE) analysis was performed using DESeq (v3.0) [31] due to its better performance than the widely used RPKM normalization [32].The Read Per Million (RPM) normalized BigWig files were generated using BEDTools (v2.17.0) [33] and bedGraphToBigWig tool (v4) [34]. For the DE analysis of the microarray data all the raw CEL files from GSE34200 were analyzed using the Limma R package (v3.0) [35]. All the downstream statistical analyses and data visualizations were performed in R (v3.1.1;
Immunohistochemistry
Teratomas were fixed in formalin and were sectioned at UAB histology laboratory. Thin tissue sections (5 μm) are rehydrated and treated for heat-induced antigen retrieval in citrate buffer (pH 6.0). Tissue sections were treated with 3% hydrogen peroxide. They were probed with anti-PODXL (sc-23904, 3D3; Santa Cruz Biotech) and anti OCT4a (mouse, IgG2b, sc5279) overnight at 4°C per manufacturer protocol, followed by treatment with biotinylated anti mouse (A10519; IgG1, Invitrogen) and detected with streptavidine-HRP cy3-conjugated tyramide (SAT04A001EA; Perkin-Elmer). The same slides were again treated with 3% hydrogen peroxide and probed with hydroxyl peroxidase-conjugated anti-mouse IgG2b antibody followed by fluorescein-conjugated tyramide (Perkin Elmer NEL701001KT) to detect OCT4(a/b). Tissue sections were stained with DAPI (H1200; Vector). Images were captured using Olymous BX3 microscope with Olympus camera and software.
Results
High PODXL transcription is a signature of human pluripotent stem cells
PODXL encodes a protein that carries epitopes for several well-established hPSC markers including TRA-1-60, TRA-1-81, GP200, GCTM-2, and mAb84 [8,15,36]. These marker antibodies recognize epitopes of distinct post-translational modifications. It is possible that only the differential post-translational modifications are features of pluripotency considering the extensive and cell type-specific protein modifications as shown by huge difference in the apparent molecular weights in western blots. Specifically, the calculated MW is 58 kD. In hPSCs and somatic cells the protein has mobility around 200 kD, or 100–170 kD, respectively [16,37,38]. We asked whether the transcription of PODXL is characteristic of pluripotency. PODXL is among the lists of genes whose expression is elevated in human embryonic stem cells (hESC) based on several previous transcriptome profiling of hESCs using limited samples [39 –43]. To address this question firmly, we analyzed a recent large pluripotent dataset GSE34200, which is a microarray transcription profiling of 21 hESC lines registered at NIH, 8 human iPSC lines and 20 human tissues [44]. Impressively, the expression of PODXL in hPSCs is higher than that of the housekeeping gene GUSB, and comparable to that of beta actin and GAPDH (Fig. 1A). Its expressions in hPSCs are even higher than that of the master pluripotent genes OCT4 (POU5F1), SOX2, and NANOG. In somatic tissues, PODXL expression levels are significantly lower, but higher than that of the master pluripotent gene NANOG in hPSCs. This significant expression in somatic tissues can be explained by the high expression of PODXL in vascular endothelial cells, kidney podocytes, hematopoietic progenitors, some neural cells, platelets, and bone marrow stromal cells [45,46].
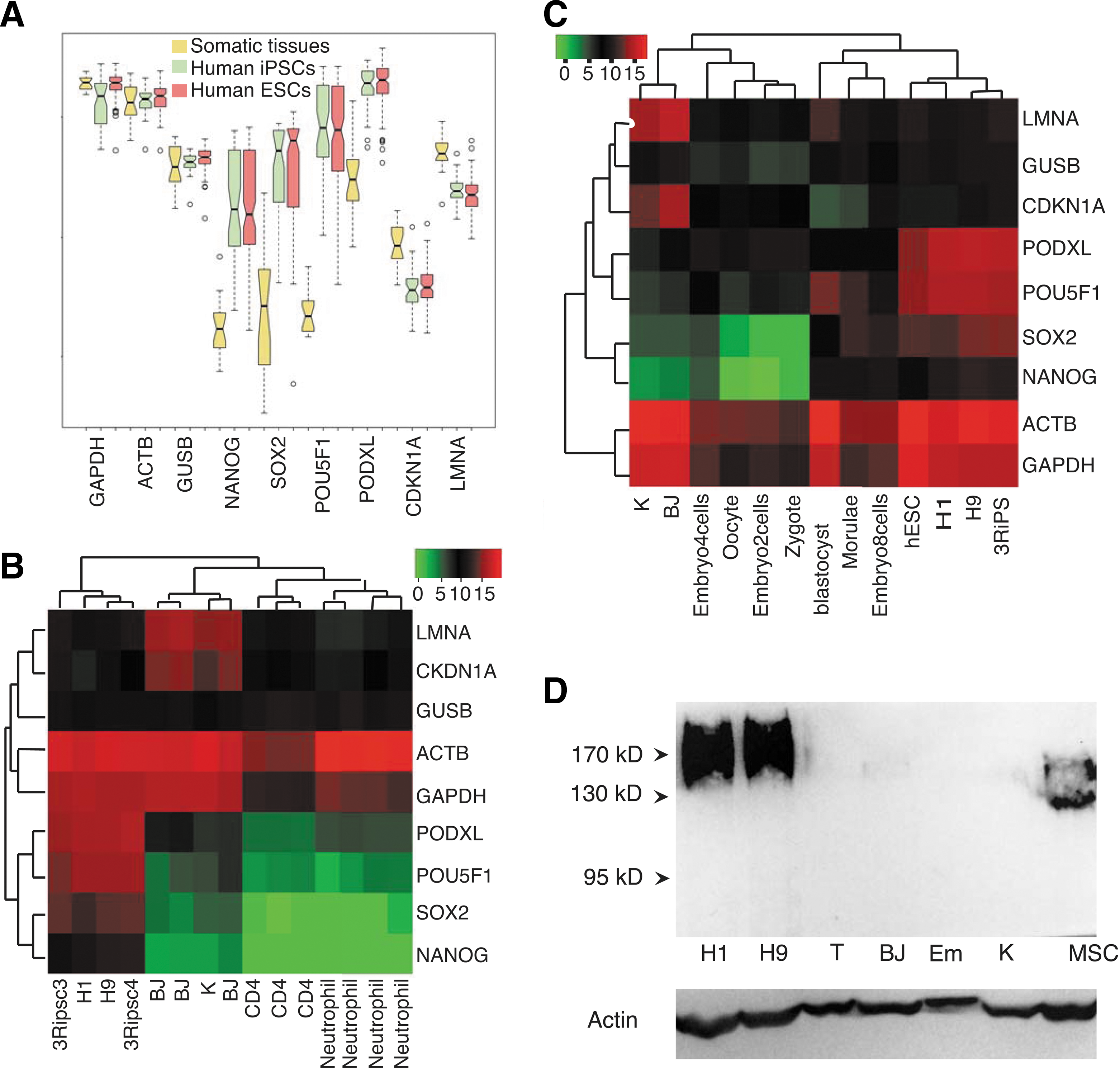
PODXL is a pluripotent feature at transcription levels.
To avoid noise from the PODXL-expressing cells in tissue samples, we performed RNA-seq of several somatic cell lines and compared to the expressions in PSCs. Our RNA-seq dataset GSE66798 confirmed the array data in that PODXL has a higher expression in hPSCs than pluripotent genes POU5F1, SOX2, and NANOG (Fig. 1B). In human fibroblasts and keratinocytes, expression of PODXL is insignificant, and human CD4 and neutrophils do not express PODXL (Fig. 1B). One of the PODXL epitopes 3D3 represents the primary unmodified sequence. Therefore, 3D3 antibody can detect all glycoforms of PODXL. We then used 3D3 to compare the total protein expression levels between hPSCs and somatic cells. Our results show that PODXL is expressed in hPSCs but not in human fibroblasts, T cells, and keratinocytes, consistent with the mRNA expression pattern (Fig. 1D). In agreement with previous observations with TRA-1–81 antibodies (Fig. 2C), PODXL western bands are diffusive (Lanes H1 and H9 in Fig. 1D), indicating a spectrum of various post-translational modifications. 3D3 antibody does not recognize murine PODXL (lane Em in Fig. 1D; see below), which can serve as a negative control as well. As expected, human bone marrow stromal stem cells express PODXL but with lower molecular weights (Lane MSC in Fig. 1D) [46].
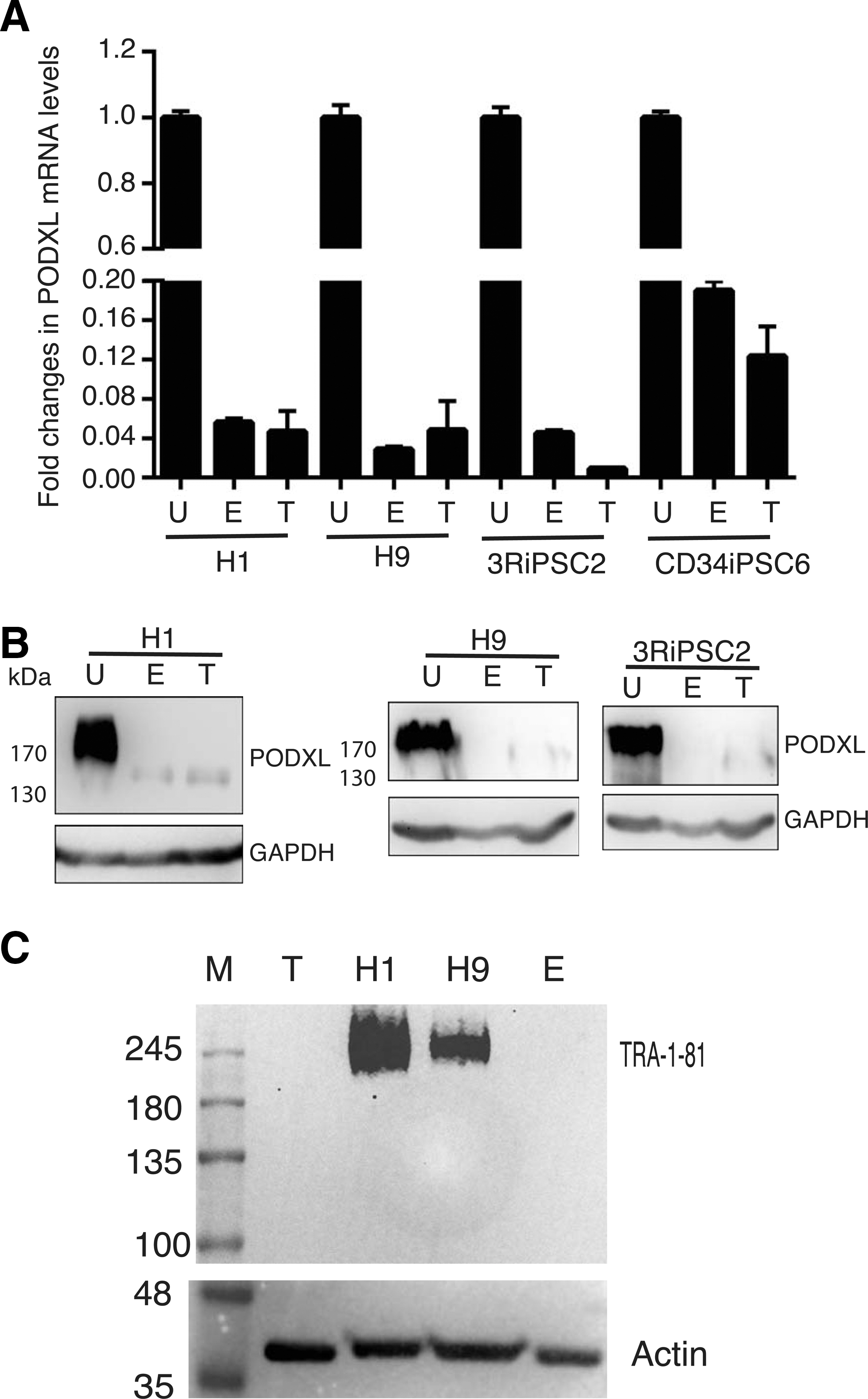
PODXL is a differentiation marker.
With the above observations, we wanted to know the expression status of PODXL in early human embryos. We reanalyzed the single-cell RNA-seq dataset GSE36552 of human embryos [47]. Interestingly, the expression of OCT4 (POU5F) correlates better with PODXL than with NANOG and SOX2 (Fig. 1C). But, PODXL has the highest expression in cells from oocyte up to 4-cell stages, while POU5F1 peaks at morulae and blastocysts (Fig. 1C), and SOX2 and NANOG are not expressed from zygote to four-cell stage. At later stages, PODXL expression levels vary among cells (Supplementary Fig. S1). In summary, PODXL has a similar expression pattern in established pluripotent cells as those of the established pluripotent transcription factors OCT4, SOX2, and NANOG, and even has broader expression in early embryoes. PODXL transcription in hPSCs is comparable to the housekeeping gene beta actin, and significantly higher than its expression in somatic tissues. Its expression is high in early human embryos, and absent from the cultured fibroblasts and keratinocytes.
PODXL is downregulated upon differentiation of hPSCs
We next asked whether PODXL could be used as a pluripotent marker during the differentiation process, as its post-translational modification epitopes do. Two approaches were used. First, we performed in vitro EB-initiated differentiation and examined the expression levels by RT-qPCR and by western blots. PODXL RNAs were significantly reduced upon differentiation for two hESC lines (H1 and H9) (column E, left, Fig. 2A). Similar results were observed for two human iPSC lines established and characterized previously in our laboratory (column E, right, Fig. 2A). Western blot analyses with the universal PODXL antibody 3D3 confirmed the significant decrease of PODXL for both hESCs and HiPSCs upon differentiation (lane E, Fig. 2B). The molecular weight of PODXL in the differentiated cells is lower than those in undifferentiated cells, indicating the loss of post-translational modifications upon differentiations. The species of PODXL with lower molecular weight was previously observed in some somatic cells [16,37,38]. To further confirm the above observations, we used a second differentiation method, in vivo teratoma differentiation in immune compromised mice. Similar downregulation of PODXL were observed at both RNA and protein levels (T in Fig. 2A, B). As a control, we performed western analysis with the antibody TRA-1-81, as expected, we did not observe any band in teratoma and EB samples (Fig. 2C) while a diffused band was observed with both H1 and H9 undiffrentiated cells, indicating a loss of the TRA-1-81 epitope.
Residual PODXL+ population remains after extended differentiation of hPSCs
Although PODXL is significantly downregulated both at mRNA and protein levels upon differentiation using both in vivo and in vitro protocols, there was residual PODXL expression based on western blot analysis. These residual expressions have two possibilities: (a) PODXL is downregulated in all cells; (b) Only some of cells are positive for PODXL upon differentiation. To distinguish these possibilities, we performed flow cytometry to examine PODXL expression in single cells. Our two distinct differentiation protocols stated above are efficient because there were no residual expressions for the two established pluripotent surface markers SSEA4 and TRA-1-60 in multiple hPSC lines analyzed (Fig. 3A and Supplementary Fig. S2). However, there are significant numbers of 3D3+TRA-1-60-SSEA4- cells after both EB and teratoma differentiations (Fig. 3A and Supplementary Fig. S2). This population was observed for both hESCs and HiPSCs. 3D3+ cells ranged from 3.69% to 17.5% (Fig. 3A and Supplementary Fig. S2). We further conducted immunohistochemistry with 3D3 and OCT4 antibodies with teratoma sections. In agreement with flow cytometry data, we observed a 3D3+ population, but did not detect any OCT4+ cells (Fig. 3B). Based on these observations, it can be concluded that after prolonged differentiation the majority of cells are not expressing PODXL, but significant populations are still expressing PODXL at different levels.
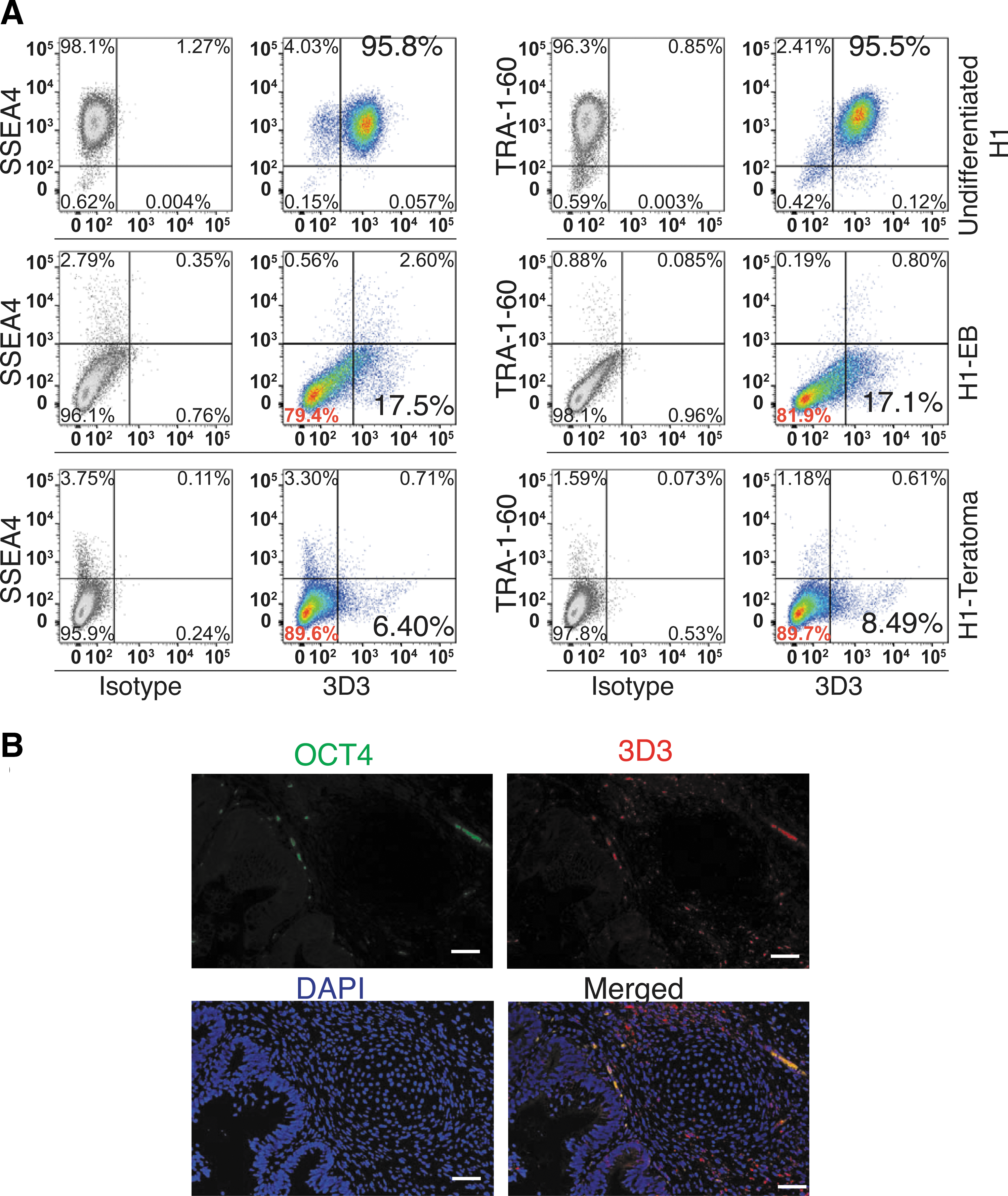
The universal human PODXL antibody 3D3 identifies a residual population of 3D3+TRA-1-60-SSEA4- cells.
KLF4 activates PODXL during early reprogramming
PODXL carrier epitopes are responsible for two well-known pluripotent surface markers TRA-1-60 and TRA-1-81, both of which are regarded as the most reliable markers for the generation of hiPSCs [48]. We were interested in how and when the PODXL gene is activated during human somatic cell reprogramming. We transduced human fibroblasts BJ cells with lentiviral Yamanaka factors (OCT4, SOX2, KLF4, and MYC), and performed RT-qPCR to test the expression of PODXL at 48 and 72 h post-transduction. Impressively, unlike the late emergence of TRA-1-60 and TRA-1-81 during reprogramming [48 –50], PODXL expression levels were increased by >20-fold at both time points (Fig. 4A). Time point analyses showed that PODXL mRNA levels kept increasing up to day 21 (Fig. 4B). Western analyses with 3D3 antibody confirmed a robust and early activation of PODXL during reprogramming (Fig. 4C).
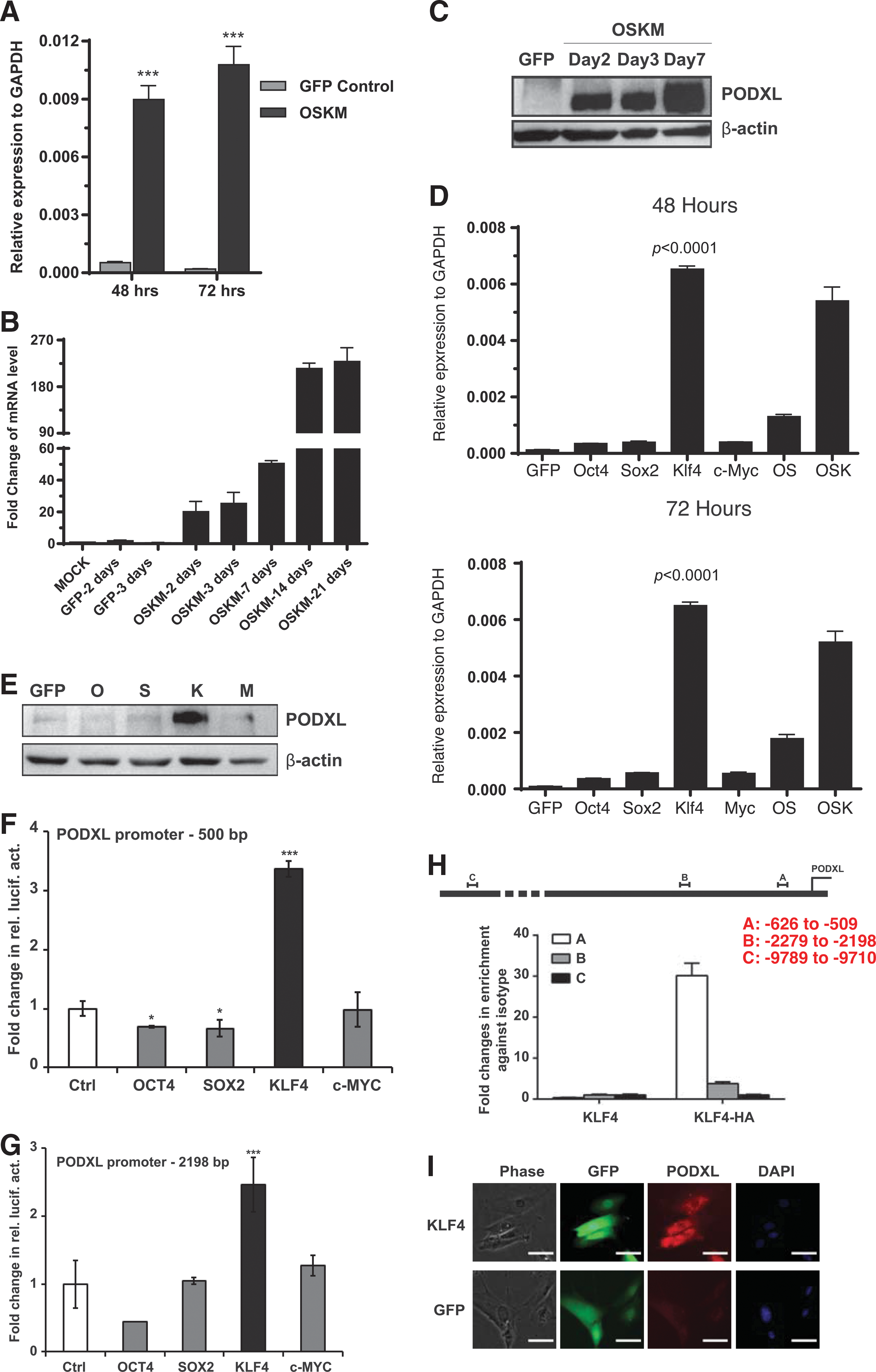
KLF4 activates PODXL at an early time during reprogramming.
Next, we asked whether all four reprogramming factors are required for the early and robust activation of PODXL during reprogramming. As shown in Figure 4D, the combination of OSK demonstrated strong activation of PODXL at 48 and 72 h. Interestingly, removal of KLF4 dramatically affected the activation activity. Moreover, KLF4 alone activated PODXL more efficiently than OSK at both time points. Individually, the other three reprogramming factors alone showed little activation activities of PODXL. KLF4 activation of PODXL was confirmed by western blots (Fig. 4E) and immunocytochemistry (Fig. 4I) with 3D3 antibody. Regulation of PODXL by KLF4, but not the other reprogramming factors, was further substantiated with two luciferase reporter assays with upstream regulatory sequences of PODXL (Fig. 4F, G). Moreover, We also demonstrated that KLF4 directly bound to PODXL promoter using ChIP-qPCR (Fig. 4H). The transcription of PODXL has previously been reported to be positively regulated by WT1 and SP1 [51,52], but negatively by p53 [53]. In agreement with KLF4 and WT1 regulation of PODXL, WT1 and KLF4 are also critical for podocyte functions [54,55].
Mouse PODXL is a pluripotent marker
It is well known that TRA-1-60 and TRA-1-81 are unique surface markers for human PSCs that are not shared with mouse. However, TRA-1-60 and TRA-1-81 represent post-translational modification epitopes of PODXL. It is possible that only TRA-1-60 and TRA-1-81 epitopes are unique to primate, but the carrier protein is also expressed in mouse PSCs. To test this hypothesis, we analyzed mouse ESC line E14 with a mouse PODXL antibody by flow cytometry. As previously known, E14 is positive for the mouse pluripotent surface marker SSEA1 (Fig. 5A), but negative for TRA-1-60 (Fig. 5A). But, PODXL is expressed in almost all mouse E14 cells (Fig. 5A). RT-qPCR further demonstrated that PODXL is highly expressed in E14 compared to mouse embryonic fibroblasts (MEF; compare the first and the last bar in Fig. 5C). We next examine whether KLF4 can also activate PODXL in mouse cells. We overexpressed human KLF4 in MEF. As in human cells, KLF4 but not OCT4 and SOX2 activated PODXL (Fig. 5C).
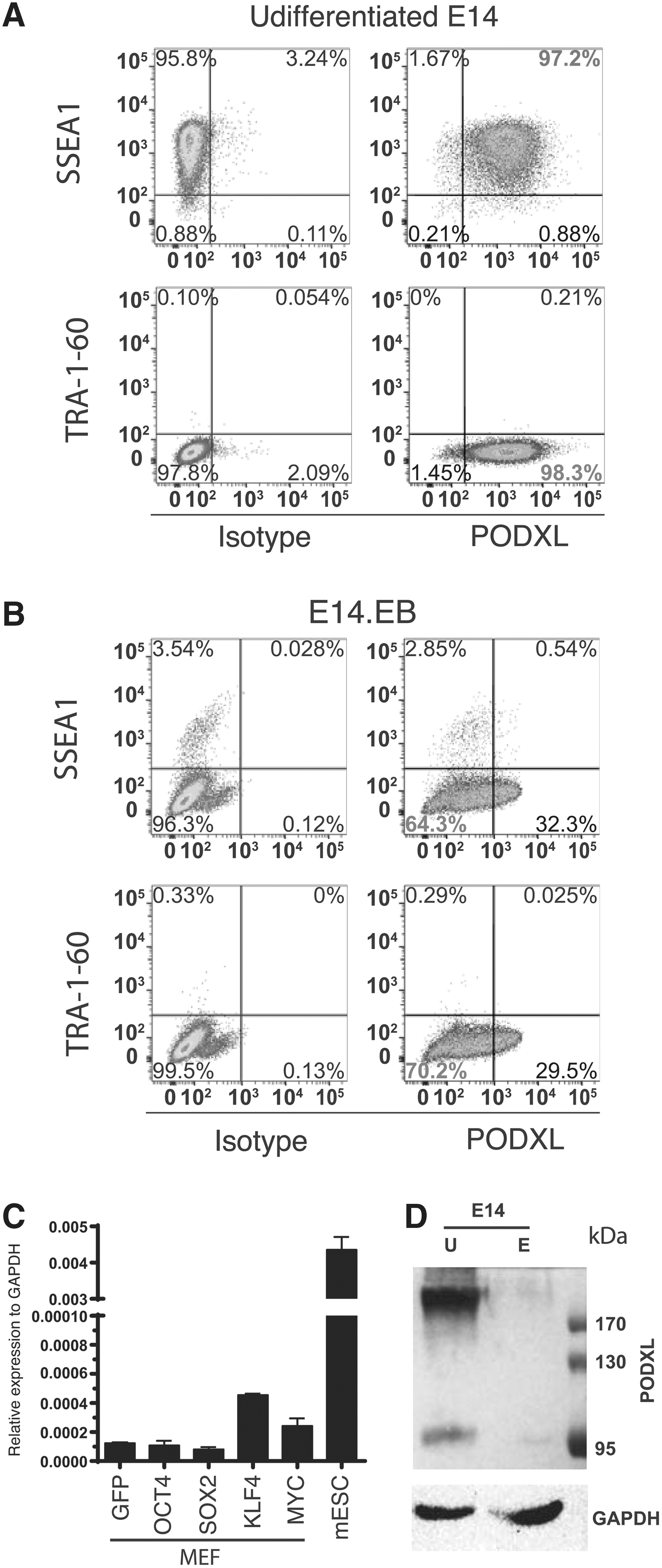
PODXL is a mouse pluripotent marker.
We next asked whether PODXL is also a characteristic of mouse pluripotent cells as it is for human PSCs. To this end, we differentiated E14 using EB-FBS methods. Our protocol efficiently downregulated SSEA1, a mouse pluripotent surface marker (5B). Upon differentiation, most cells became PODXL−. But higher percentage of PODXL+ cells was observed relative to that observed upon differentiation of human PSCs. Nevertheless, the PODXL+ cells expressed a lower level of PODXL based on the intensity of the fluorescence (Fig. 5B). We further conducted western analyses with murine PODXL antibody (Fig. 5D). Undifferentiated mouse embryonic stem cells express high levels of PODXL. Interestingly, a smaller band at around 98 kD was observed. Upon differentiation, intensity of both bands is significantly decreased to the levels slightly above background intensity.
3D3 is cytotoxic to human hPSCs
It is reported that one modification-dependent antibody mAb84 is cytotoxic to hPSCs [14]. This antibody is suggested to be useful in elimination of tumorigenic pluripotent cells in the future hPSC-derived transplants for cell therapy. Using annexin V-7AAD assay, we confirmed the cytotoxic nature of mAb84 when incubated with the detached hPSCs in test tubes for 45 min (right, Fig. 6A). However, when applied to cultured cells, mAb84 is not cytotoxic to hPSCs (right, Fig. 6B, Supplementary Figs. S3 and S4). We performed similar tests for 3D3; in contrast to mAb84, 3D3 is not cytotoxic when incubated for 45 min with detached hPSCs in test tubes (Fig. 6A). However, 3D3 is cytotoxic when added into hPSC culture for two days (Fig. 6B). We performed a growth curve evaluation for several PODXL antibodies including TRA-1-60, 3D3, and AF1658 (R&D) along with mouse and goat IgG controls, and another pluripotent marker SSEA4. Only 3D3 demonstrated cytotoxicity to hPSC. After exposure of human embryonic stem cell line H9 to 3D3 for 4 days, all hPSCs were dead but not other antibodies (Fig. 6D and Supplementary Fig. S3). We observed the same results with a human iPSC line (Fig. 6C). 3D3 cytotoxicity was observed with an additional embryonic stem cell line H1 (Supplementary Figs. S4 and S5). This cytotoxic effect of 3D3 was not seen with Hela cells, which express PODXL (Fig. 6E). Thus, the cytotoxic effect is specific for hPSCs and HiPSCs. mAb84 damages the cell membrane. It is possible that the broken cell membrane is responsible for the positivity for annexin V observed in 3D3-treated cells. To further confirm that 3D3 caused apoptosis, we performed western analysis with p21 antibody. We observed significant increase in p21 level in 3D3-treated H1 cells (Fig. 6F).
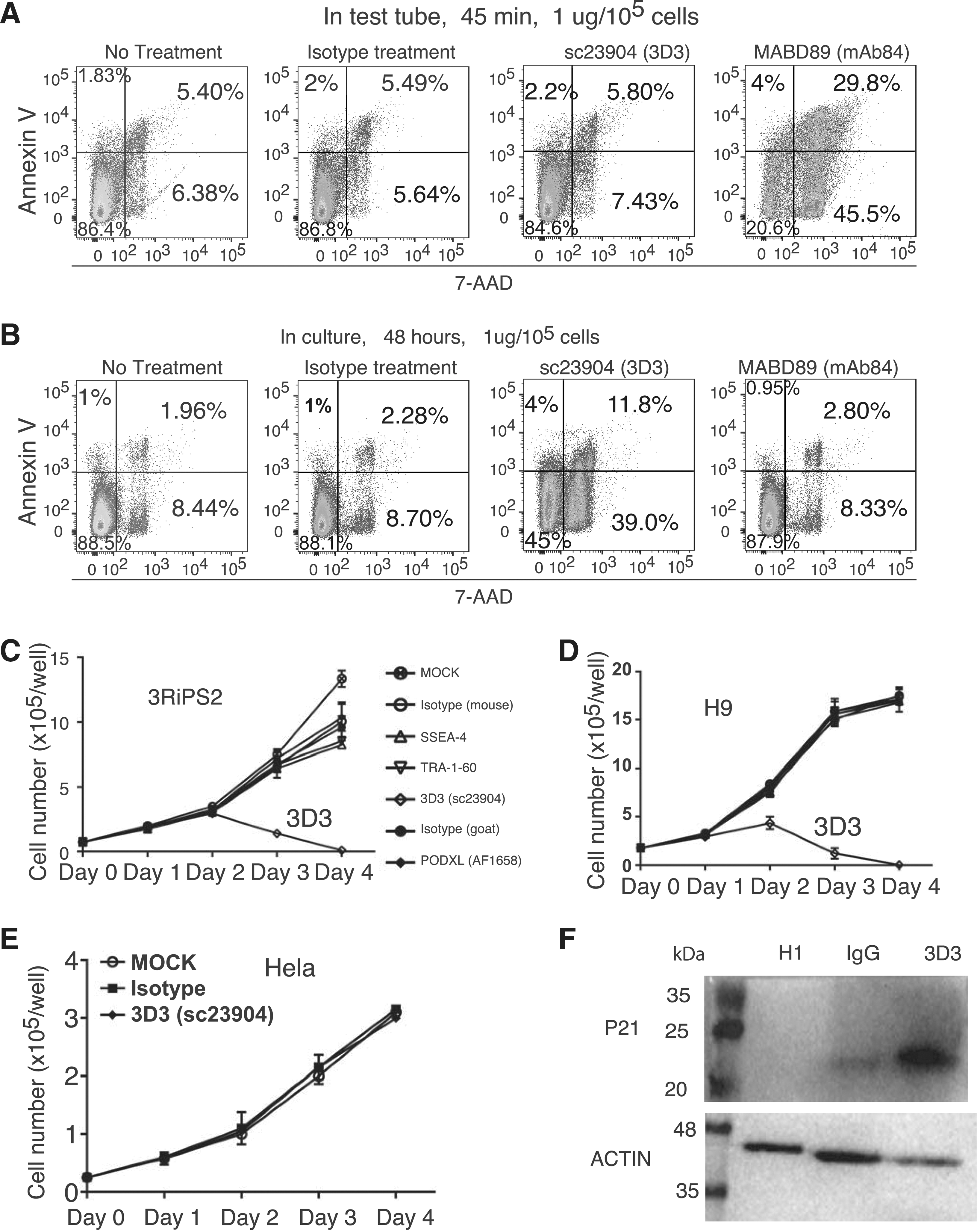
3D3 is cytotoxic to hPSCs.
Discussion
Human embryonic stem cells are defined by a set of surface markers. One interesting fact is that many pluripotent marker antibodies were reported to recognize various epitopes of the same carrier protein PODXL [8,10,15], including TRA-1-60, TRA-1-81, GP200, GCTM2, and mAb84. These markers are expressed in hPSCs and embryonic carcinoma. Upon differentiation of hPSCs, these markers become lost. There has not been much emphasis paid to the carrier protein itself and the PODXL gene. In this report, we demonstrated that a high level of mRNA of PODXL is also a feature of pluripotent stem cells. PODXL expression is extremely high in hPSCs, and comparable to that of housekeeping genes. It was higher than those of the master pluripotent transcription factors OCT4, SOX2, and NANOG. Upon differentiation, PODXL mRNA was downregulated to the basal levels. The modification-independent antibody 3D3 identifies a somatic PODXL species with lower apparent molecular weight and low intensity. Our results show that during differentiation, pluripotent cells lose modifications before downregulation of the gene.
It is well known that TRA-1-60 and TRA-1-81 are pluripotent surface markers specific to hPSCs not shared by mouse PSCs. In this study, we found that Podxl is expressed in mouse PSCs. This is confirmed by RT-qPCR analysis. This observation indicates that the carrier protein is conserved between rodent and human. Furthermore, in both species, KLF4 alone upregulated the transcription of Podxl gene.
One intrinsic feature of hPSCs is their ability to generate teratoma when injected into immunecompromised mice. This tumorigenic nature of PSCs has been a safety concern. hPSC-based cell therapy has entered clinical trials for the treatment of blindness, heart infarction, and spinal cord injury [56 –60]. However, there is no safety regimen to eliminate the tumorigenic cells in hPSC-based transplants. Several suggestions have been made, including FACS or MACS sorting to separate therapeutic cells from the undifferentiated PSCs, the use of a suicide gene preintegrated into hPSCs, the use of cytotoxic antibodies, the use of cytotoxic small molecules, extended differentiation process, and individualization of cells [61 –66]. However, even trace amount of incompletely differentiated cells may undergo a dedifferentiation process and give rise to tumors. This was reported for mouse SSEA1+OCT4− cells [67] and for primate SSEA4+OCT4− cells [63] in PSC-derived transplants.
In this study, we report a pluripotent cytotoxic activity of PODXL antibody 3D3. 3D3 antibody could be an invaluable means to eliminate the residual undifferentiated tumorigenic cells in hPSC-derived cells. 3D3 was raised against a PODXL fragment expressed in bacteria [16]. Therefore, this peptide is devoid of human post-translational modification. With this universal PODXL antibody, we identified a significant population of 3D3+TRA-1-60-SSEA4- cells after extended differentiation. This population has never been revealed by the various modification-dependent pluripotent surface marker antibodies. It is reported that PODXL is involved in more than 10 different human malignancies [15,19 –25], and it is also reported to have roles in metastasis and tumor invasion. With these in mind, there is an urgent need to investigate whether the 3D3+TRA-1-60-SSEA4- cells are tumorigenic, and whether 3D3 antibody can be used to cleanse the tumorigenic cells in hPSC-derived transplants.
PODXL plays various roles including antiadhesion, adhesion, cell–matrix interaction, morphogenesis, cell migration, intracellular communication, and others [17,45,68]. The transcription regulation of PODXL is less clear. It is positively regulated by WT1 and SP1 [51,52], but negatively by p53 [53]. Here, we demonstrated that KLF4 is a new regulator of PODXL. PODXL plays a critical role in the development and functions of the kidney epithelium podocytes.
Footnotes
Acknowledgments
We thank Dr. Kevin Pawlik for his critical reading of our article. Data were deposited into GEO repository with an accession number GSE66798. This research is supported by a UAB faculty development grant and a stem cell seed grant from the Alabama Institute of Medicine (AIM) (AIMG1003).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.