
Abstract
Recently, we reported the isolation and characterization of mesenchymal stem cells from the decidua basalis of human placenta (DBMSCs). These cells express a unique combination of molecules involved in many important cellular functions, which make them good candidates for cell-based therapies. The endothelium is a highly specialized, metabolically active interface between blood and the underlying tissues. Inflammatory factors stimulate the endothelium to undergo a change to a proinflammatory and procoagulant state (ie, endothelial cell activation). An initial response to endothelial cell activation is monocyte adhesion. Activation typically involves increased proliferation and enhanced expression of adhesion and inflammatory markers by endothelial cells. Sustained endothelial cell activation leads to a type of damage to the body associated with inflammatory diseases, such as atherosclerosis. In this study, we examined the ability of DBMSCs to protect endothelial cells from activation through monocyte adhesion, by modulating endothelial proliferation, migration, adhesion, and inflammatory marker expression. Endothelial cells were cocultured with DBMSCs, monocytes, monocyte-pretreated with DBMSCs and DBMSC-pretreated with monocytes were also evaluated. Monocyte adhesion to endothelial cells was examined following treatment with DBMSCs. Expression of endothelial cell adhesion and inflammatory markers was also analyzed. The interaction between DBMSCs and monocytes reduced endothelial cell proliferation and monocyte adhesion to endothelial cells. In contrast, endothelial cell migration increased in response to DBMSCs and monocytes. Endothelial cell expression of adhesion and inflammatory molecules was reduced by DBMSCs and DBMSC-pretreated with monocytes. The mechanism of reduced endothelial proliferation involved enhanced phosphorylation of the tumor suppressor protein p53. Our study shows for the first time that DBMSCs protect endothelial cells from activation by inflammation triggered by monocyte adhesion and increased endothelial cell proliferation. These events are manifest in inflammatory diseases, such as atherosclerosis. Therefore, our results suggest that DBMSCs could be usefully employed as a therapeutic strategy for atherosclerosis.
Introduction
M
Endothelium comprises of a squamous epithelium that lines the lumen of all blood vessels that is highly active metabolically. It plays a major role in vascular homeostasis and acts as an endocrine organ by producing a variety of molecules including hormones, growth factors, coagulation factors, and adhesion molecules [11]. Moreover, endothelium is an active biological interface between blood and tissues, which regulates the delicate balance between vasoconstriction and vasodilatation, coagulation and fibrinolysis, proliferation and apoptosis. Finally, endothelium acts as an interface for the transient adhesion and subsequent diapedesis of blood-borne leukocytes [12]. Inflammatory diseases, such as atherosclerosis, are characterized by endothelial cell activation as a result of the accumulation of high amounts of low-density lipoprotein (LDL) and immune cells [13]. When LDL is modified by oxidation, cells in the vessel wall sense this modification and respond to this threat by recruiting immune cells from the blood circulation [13]. These events upregulate the expression of adhesion molecules in the endothelial cells, particularly intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) [14]. Subsequently, the recruitment of leukocytes, such as lymphocytes and monocytes is initiated [15]. Moreover, the transmigration of monocytes, upregulation of adhesion molecules on a variety of cells, and production and release of chemotactic factors are also enhanced [15]. These changes in the endothelium and surrounding environment are necessary for the transfer of monocytes to the intima and the simultaneous differentiation of monocytes into macrophages [15]. The accumulation of LDL is a requirement for further transformation of macrophages into foam cells (lipid-loaded macrophages), which are major cells formed beneath the endothelium of the vessel wall [15]. LDL modification is also associated with inflammatory reactions that intensify the proinflammatory events initiated previously because of the adherence and transmigration of monocytes and lymphocytes into the intima [15]. If endothelial cell activation is intense and sustained, eventually the endothelium will become damaged [11,12]. This damage could be manifested through major phenotypic changes, such as increased expression of inflammatory markers in the endothelium, and dysfunction as a result of increased endothelial proliferation that is characteristic of atherosclerosis [15].
An essential prerequisite before the use of DBMSCs in cell-based therapy is to determine their effects on the phenotypic and functional characteristics of endothelial cells. There is certainly potential for DBMSCs to interact with endothelial cells and monocytes, either directly or indirectly. We recently showed that DBMSCs (also called DMSCs) reside in a vascular niche in the decidua [16] where they are closely associated with endothelial cells and the various cell types, including monocytes, in the maternal blood circulation. A vascular niche for MSCs is found in many organs and tissues [17], where MSCs and endothelial cells reside on either side of a common basement membrane and can potentially be in direct contact with each other through gap junctions or through discontinuities in the basement membrane [18]. Studies provide evidence for interactions between MSCs and endothelial cells either by direct contact [17] or by paracrine signaling [19].
In this study, we examined the consequences of the direct contact between DBMSCs and endothelial cells and the effects of DBMSCs on endothelial cells through monocytes. We characterized the expression of adhesion and their inflammatory molecules in endothelial cells following direct interaction with DBMSCs, or after exposure to monocytes that were pretreated with DBMSCs. In addition, we carried out a functional analysis of endothelial cells where we examined their proliferation and migration responses to DBMSCs, monocytes, and monocytes that were pretreated with DBMSCs. Moreover, the effect of DBMSCs on the adhesion of monocytes to endothelial cells was also examined. Finally, we investigated the mechanism underlying endothelial cell proliferation in response to monocytes, and monocytes pretreated with DBMSCs. Our study demonstrated that the interaction between DBMSCs and monocytes had an antiproliferative effect on endothelial cells. In addition, DBMSCs, monocytes, monocyte-pretreated with DBMSCs and DBMSC-pretreated with monocytes increased endothelial cell migration. Moreover, DBMSCs reduced monocyte adhesion to endothelial cells. Furthermore, DBMSCs reduced endothelial cell expression of adhesion and inflammatory proteins either in a direct effect on the endothelial cells or through modulating the effects of monocytes on the endothelial cells. Finally, we report that DBMSC reduced endothelial cell proliferation through monocytes by inducing arrest in the cell cycle through the tumor suppressor protein p53. These data suggest that MSCs from decidua basalis have a protective effect on endothelial cells and suggest that DBMSCs may be a good candidate for stem cell-based therapy to treat inflammatory diseases, such as atherosclerosis by protecting endothelial cells from injury initiated by inflammatory cells. However, more studies are necessary to further elucidate this both in vitro and in vivo.
Materials and Methods
Ethics of experimentation
This study was approved by the Institutional Research Board (ref. no. IRBC/246/13) at King Abdulla International Medical Research Centre/King Abdulaziz Medical City, Riyadh, Saudi Arabia. All normal term human placentae including umbilical cords were obtained with informed consent.
Placentae
Human term placentae were obtained from uncomplicated pregnancies following normal vaginal delivery (38–40 weeks of gestation). The gestational age and fetal viability of all pregnancies were confirmed by early ultrasound examination before 20 weeks of gestation. The placentae were used within 2 h of delivery.
Isolation and culture of mesenchymal stem/multipotent stromal cells from decidua basalis of human placenta (DBMSCs)
DBMSCs were isolated from the decidua basalis of human placenta as previously described [10]. Briefly, 10 grams of the decidua tissue was dissected from the maternal surface of the placenta and washed thoroughly with sterile phosphate-buffered saline (PBS, pH 7.4) to remove excess blood. The tissue was then finely minced and washed with PBS until the fluid was free of blood. After centrifugation at 300g for 5 min, the tissue pellet was digested using a digestion solution containing 0.3% collagenase type I (Life Technologies, Grand Island, USA) diluted in PBS, 100 μg/mL streptomycin, 100 U/mL penicillin, and 271 U/mL DNase I (Life Technologies) at 37°C in a water bath for 1 h. The mixture was then filtered through a 100-μm nylon filter (Becton Dickinson, New Jersey, USA), centrifuged, and incubated with red blood cell lysis buffer (cat. no. sc-3621, FCM Lysing solution; Santa Cruz Biotechnology, Inc., CA, USA) for 45 min at room temperature (RT). After centrifugation, the cells were washed and 1 × 105 cells were cultured in T25 flasks (Becton Dickinson) in complete DBMSC culture medium containing Dulbecco's modified Eagle medium nutrient mixture F-12 (DMEM-F12), 10% MSC certified fetal bovine serum (MSCFBS) (Life Technologies), 100 μg/mL
Isolation and culture of human umbilical vein endothelial cells
Primary human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cord veins as previously described with some modifications [20]. Cannulated umbilical veins were gently massaged and rinsed with sterile PBS, pH 7.4 several times to remove clots and blood. The veins were then filled with a disgestive solution containing 6 mg/mL collagenase type II (cat. no. 17101015; Life Technologies) diluted in PBS, and incubated at 37°C in a cell culture incubator for 25 min. After incubation, the collagenase solution containing the HUVEC was collected by perfusion of the cord with Vascular Cell Basal Medium (PCS-100-030™; ATCC, USA) and cells were then collected by centrifugation at 300g for 10 min. Cells were then resuspended in PBS and incubated with red blood cell lysing buffer for 45 min at RT to lyse red blood cells. After centrifugation at 300g for 10 min, the cell pellet was resuspended in complete HUVEC growth medium [no. ATCC® PCS-100-041™, Endothelial Cell Growth Kit-VEGF; ATCC] and cultured in T25 flasks at 37°C in a cell culture incubator. When cells reached 75% confluency, they were harvested with TrypLE Express detachment solution and characterized by flow cytometry using a CD31 endothelial cell marker (R&D Systems). Samples with a purity >95% were used in experiments. Cells from passages 3 to 5 were used in subsequent experiments. Thirty umbilical cords were used in this study.
Culture of monocytes with DBMSCs (physical contact and conditioned medium)
Human monocytes (THP-1) from ATCC (no. TIB-202™) were cultured with DBMSCs in physical contact and conditioned medium (CM) experiments. For physical contact experiments, DBMSCs were seeded on a plastic surface of a six-well culture plate until they were fully adhered and monocytes were then added to DBMSC culture at ratio 1DBMSC:5 monocytes (Fig. 1). For CM experiments (CMDBMSC), supernatant from unstimulated DBMSCs was prepared as previously described [21] and then added to monocyte culture (Fig. 1). To produce CMDBMSC, 1 × 105 DBMSCs were cultured in T75 flasks containing DMEM-F12 medium with 10% MSCFBS, 100 μg/mL of
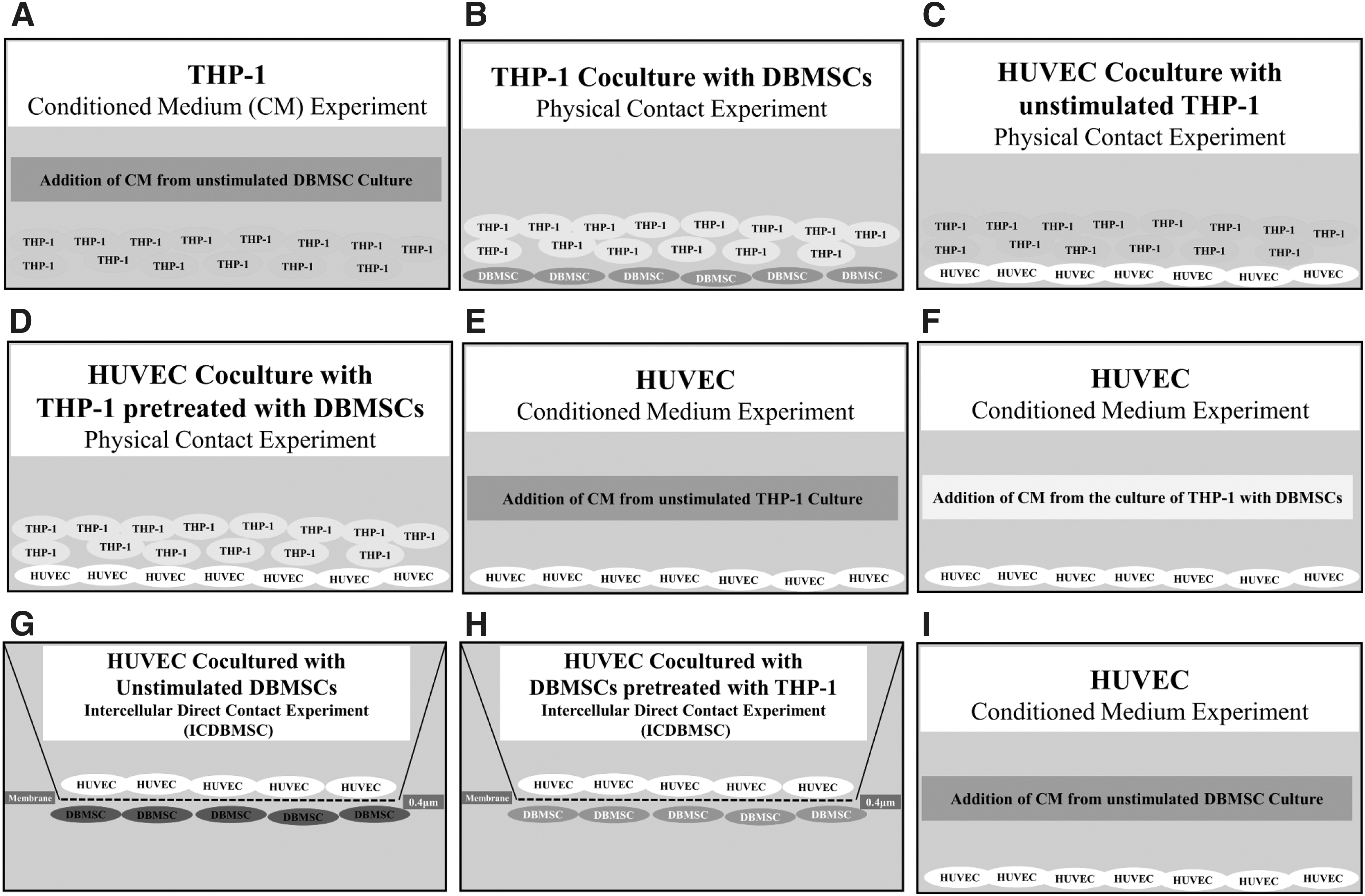
Culture systems used in this study. Monocytes (THP-1) cultured with CM obtained from unstimulated DBMSC culture
Culture of endothelial cells with different treatments of DBMSCs and monocytes
Endothelial cells were cocultured with different treatments of DBMSCs (DBMSCs alone, DBMSC/THP-1, and CMDBMSCs) and THP-1 (THP-1 alone, THP-1/DBMSC, CMTHP-1, and CMTHP-1/DBMSC) prepared as previously described. For HUVEC and DBMSC experiments, 1:1 ratio was used for both HUVEC:DBMSC and HUVEC:DBMSC/THP-1. For HUVEC:THP-1 experiments, 1:10 ratio was used for both HUVEC:THP-1 and HUVEC:THP-1/DBMSC. For CM experiments, 25% DBMSCs was used for all experiments of HUVEC with CM from DBMSC culture, THP-1, and THP-1/DBMSC. Intercellular direct contact (IC) experiments (ICDBMSC) were used as previously described for the coculture of endothelial cells with DBMSCs and DBMSC/THP-1 [21] (Fig. 1). Briefly, the 0.4 μm pore size transwell chamber membrane culture system (Greiner Bio-One, Germany) was used to seed DBMSCs on the reverse side of the membrane of the chamber until they were fully adhered and HUVEC were then seeded on the upper side of the membrane. This culture system prevents the contamination of HUVEC with DBMSCs and facilitates harvesting of HUVEC without DBMSC contamination. For the coculture of endothelial cells with THP-1 and THP-1/DBMSC, HUVEC were seeded in a six-well culture plate until they were fully adhered. Then, THP-1 and THP-1/DBMSC were added (Fig. 1). For the CM experiments, CM collected from the culture of DBMSCs, THP-1, and THP-1/DBMSC was added to fully adhered HUVEC in a six-well culture plate (Fig. 1). Cells were then incubated at 37°C in a cell culture incubator for 24 h. Then, HUVEC were harvested as described above and characterized by flow cytometry. Each experiment was performed in duplicate and repeated with five independent preparations of HUVEC and DBMSCs. HUVEC cultured in HUVEC culture medium without DBMSCs, DBMSC/THP-1, THP-1, THP-1/DBMSC, and CM from different treatments of DBMSCs or THP-1 were included as a negative control for all experiments.
Cell proliferation assay
To examine the proliferation of HUVEC in response to different treatments of cells and CM prepared as described above (DBMSCs, DBMSC/THP-1, THP-1, THP-1/DBMSC, CMDBMSC, CMDBMSC/THP-1, CMTHP-1, and CMTHP-1/DBMSC), HUVEC were cultured with different ratios of cells and CM. For HUVEC and DBMSC experiments, different HUVEC:DBMSC ratios were used ranging from 1:1, 2:1, 5:1, and 10:1 HUVEC:DBMSC ratios. For HUVEC and DBMSC/THP-1 experiments, different HUVEC:DBMSC/THP-1 ratios were used ranging from 1:1 and 2:1 HUVEC:DBMSC/THP-1. For HUVEC and THP-1 experiments, different HUVEC:THP-1 ratios were used ranging from 1:1, 1:2.5, 1:5, and 1:10 HUVEC:THP-1. For HUVEC and THP-1/DBMSC experiments, HUVEC and THP-1/DBMSC were used at a ratio of 1HUVEC:10THP-1/DBMSC. For CM experiments, 1%, 5%, and 25% were used for DBMSC, DBMSC/THP-1, and THP-1 while 5% and 25% were used for CMTHP-1/DBMSC. Briefly, HUVEC were seeded at a density of 5 × 103 per well in 96-well tissue culture plates containing complete HUVEC culture medium for 24 h at 37°C in a cell culture incubator. Following removal of the culture medium and washing cells with sterile PBS to remove unattached cells, DBMSC, DBMSC/THP-1, THP-1, THP-1/DBMSC, and CM were added to HUVEC culture at the indicated ratios and then cultured for 24 h at 37°C in a cell culture incubator. To examine the proliferation of THP-1 in response to different treatments of DBMSCs, different THP-1:DBMSC ratios were used ranging from 2.5:1, 5:1, 10:1, and 20:1 THP-1:DBMSC, and 1%, 5%, and 25% CMDBMSC were also used. Briefly, THP-1 were cultured at a density of 50 × 103 per well in 96-well tissue culture plates with DBMSCs and CMDBMSC at the indicated ratios of cells and percentage of CM. Cells were then cultured in complete RPMI-1640 culture medium for 24 h at 37°C in a cell culture incubator. To examine the proliferation of DBMSCs in response to different treatments of THP-1, different DBMSC:THP-1 ratios were used ranging from 1:1, 1:5, 1:10, and 1:20 DBMSC:THP-1, and 1%, 5%, and 25% CMTHP-1 were also used. Briefly, DBMSCs were seeded at a density of 5 × 103 per well in 96-well tissue culture plates containing complete DBMSC culture medium for 24 h at 37°C in a cell culture incubator. Following removal of the culture medium and washing cells with sterile PBS to remove unattached cells, THP-1 and CMTHP-1 were added to DBMSC culture at the indicated ratios of cells and percentage of CM then cultured for 24 h at 37°C in a cell culture incubator. Cell proliferation was then assessed using a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] kit (no. G5421, CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay; Promega, Germany) according to the manufacturer's instructions. Briefly, MTS solution was added into each well of the 96-well assay plate containing cells in complete culture medium in presence or absence of different treatments, and incubated for 4 h. The absorbance at 490 nm was recorded using an ELISA plate reader (Spectra MR; Dynex Technologies, Denkendorf, Germany). Results are presented as means (±standard deviation) obtained from triplicate cultures. MTS solution in medium not exposed to cells was used as blank. Before the addition of DBMSCs and THP-1 to the coculture experiments, DBMSCs and THP-1 were treated with 25 μg/mL Mitomycin C for 1 h at 37°C to inhibit their proliferation and then followed by five extensive washes with culture medium containing FBS as previously described [21]. Different incubation times (24–72 h) for the culture of HUVEC with different treatments of cells and CM were evaluated. Each experiment was performed in triplicate using HUVEC and DBMSCs from passages 3 to 5, from five individual placentae.
Western blot analysis
Treated and untreated HUVEC harvested from the proliferation experiments described above were washed twice with cold sterile PBS, pH 7.4 to remove debris. Following the addition of 100 μL of cell lysis buffer (Cell Signaling Technologies, Beverly, MA) containing protease and phosphatase inhibitors to each well, cells were scrapped using a cell scraper and lysate was collected, centrifuged at 15,000 rpm for 5 min at 4°C to remove the debris, and the supernatant was then collected. Total protein was estimated by the Bradford method. Briefly, after mixing 30 μg of extracted proteins with an equal amount of 2x Laemmli Sample Buffer (Bio-Rad, Hercules, CA, USA), boiled for 7 min and then loaded onto a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad). The separated proteins were then transferred onto a nitrocellulose membrane using Mini Trans-Blot system (Bio-Rad) at 120 V for 100 min. After blocking the membranes for 30 min at RT in Tris-buffered saline with 0.1% (v/v) Tween 20 (TBS-T) containing 5% nonfat dry milk (Bio-Rad), the membranes were incubated with phospho-retinoblastoma (Rb) (Ser795); phospho-p53 (Ser15); phospho-Chk2 (Thr68); phospho-cdc2 (Tyr15); or Phospho-Chk1 (Ser345) rabbit/mouse antibodies (Cell Signaling Technologies) at 1:1,000 dilutions overnight at 4°C. Next day, the membranes were washed thrice with TBS-T and then incubated with horseradish peroxidase-conjugated secondary antibodies (R&D Systems, Minneapolis, MN) at 1:3,000 dilution for 2 h at RT. After washing thrice with TBS-T, specific proteins were visualized using SuperSignal™ West Pico or West Femto Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA) in a ChemiDoc visualization system (Bio-Rad). Densitometry of the bands was performed by the image analyzing software Image Lab (Bio-Rad). Briefly, the size of bands was measured by drawing a rectangle around the bands and getting an arbitrary value pertaining to the overall density of that particular area of the blot where the band was located. A background blank value was obtained from an equal area from a different location on the blot with a clear background. Then, the net density of the band was identified by subtracting the value obtained for the blank area from the value of the area obtained for the actual band. Afterward, the net density was normalized by subtracting the values obtained for internal controls such as, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or β-actin, from the actual values obtained for the test bands. Finally, the resulting values obtained for the different test samples were converted into percentages using the formula: (value obtained for treated band/value obtained for untreated band) × 100. The resulting values were plotted in a graph. Each experiment was performed in triplicate using HUVEC and DBMSCs from passages 3 to 5, from five individual placentae.
Adhesion assay of monocyte to HUVEC
The effect of DBMSCs on the adhesion of THP-1 to HUVEC was examined by growing HUVEC to confluency to form a monolayer in 96-well tissue culture plates. Following coculturing THP-1 with DBMSCs at 5:1 THP-1:DBMSC ratio as previously described, THP-1 were labeled with 5 μM green fluorescent cell tracker stain [5-chloromethylfluorescein diacetate (CMFDA); Molecular Probes, Life Technologies] in RPMI-1640 medium for 4 h, washed thrice with fresh RPMI-1640 medium, and then added to HUVEC culture at a ratio of 5THP-1:1HUVEC. Cells were then incubated at 37°C in a cell culture incubator for 30 min. Following removal of nonadherent THP-1 cells by gentle washing with sterile PBS, pH 7.4, the fluorescence intensity of the THP-1 cells that had adhered to the monolayer of HUVEC was measured at excitation 485 nm and emission 528 nm using a fluorescence microplate reader (Glomax Multi Detection System; Promega). Results were expressed as relative fluorescence intensity. Different ratios of HUVEC to THP-1 were evaluated. Experiments were performed in triplicate using HUVEC prepared from independent umbilical cord tissue and repeated thrice.
HUVEC migration using xCELLigence Real-Time Cell Analyzer
HUVEC migration in response to CMDBMSCs, DBMSC/THP-1, THP-1, and THP-1/DBMSC as described above was examined using the xCELLigence Real-Time Cell Analyzer (RTCA-DP version; Roche Diagnostics, Mannheim, Germany), which continuously monitors the cellular events recording label-free changes in electrical impedance (reported as cell index) [22]. We used the 16-well plates (no. 05665825001, CIM-16; Roche Diagnostics GmbH, Mannheim, Germany) as previously described with minor modifications [23]. The CIM plates have 16-well migration chambers comprising upper and lower chambers separated by a porous (pore size 8 μm) polyethylene terephthalate membrane in conjunction with microelectrodes. Treatments (group 1, 2, and 3 as described in the migration experiment) were made to the desired concentrations (final volume of 160 μL) and loaded in the lower wells of the plate. Following the addition of 50 μL prewarmed media to the wells of the upper chamber, the plates were locked in the RTCA-DP device at 37°C in a cell culture incubator for 1 h to obtain equilibrium as per the manufacturer's instructions and a measurement step was then performed as a background signal, generated by cell-free media. To initiate the experiment, HUVEC were seeded at a density of 20 × 103 in the upper chamber in 100 μL and the plates were then incubated for 30 min at RT to allow the cells to settle onto the membrane as per the manufacturer's instructions. In the migration experiment, HUVEC were seeded into the upper chamber in HUVEC serum-free medium while HUVEC medium supplemented with 20% CMDBMSC, CMTHP-1, and CMTHP-1/DBMSC or with HUVEC medium supplemented with 20% FBS (Untreated HUVEC) was added to the lower chamber. Each test was performed in quadruplicate and after equilibration, the analyzer was programmed to scan the membrane every 15 min for 24 h. The impedance value of each well was automatically monitored by the xCELLigence system for a duration of 24 h and expressed as a confidence interval value. Five experiments were performed in triplicate using HUVEC and DBMSCs from passages 3 to 5, from five independent placentae. Migration observed in the presence of 30% FBS, and with medium alone, served as positive and negative controls, respectively.
Flow cytometry
Cells (DBMSCs, HUVEC, and THP-1) were phenotypically characterized by flow cytometry as previously published [10]. Briefly, DBMSCs, HUVEC, and THP-1 were harvested as previously described and 1 × 105 of cells were then stained with monoclonal antibodies [ICAM-1, VCAM-1, CD11b, CD44, ecto-5′-nucleotidase (CD73), eNOS, IL18Rα, IL18Rβ, TNF-α, IL-6, and IL-8] for 30 min. Cells were washed twice by adding cold PBS, pH 7.4 and then centrifuged at 150g for 5 min at 8°C. To analyze the intracellular expression of proteins, cells were fixed with 4% paraformaldehyde in sterile PBS, pH 7.4 for 10 min at RT and then permeabilized using sterile PBS, pH 7.4 containing 0.1% saponin for 5 min at RT. Unstained and isotype controls were used. Immunoreactivity to cell surface antibody markers or intracellular proteins was assayed by a BD FACS CANTO II (Becton Dickinson) flow cytometer.
Statistical analysis
Data were analyzed using the t-test (unpaired t-test, two tailed). These analyses were performed using GraphPad Prism 5. Results were considered to be statistically significant if P < 0.05.
Results
Isolation and characterization of DBMSCs from the decidua basalis of human placenta
All DBMSC preparations were derived from the decidua basalis attached to the maternal side of healthy human term placenta. At passage 3, DBMSCs were more than 95% positive for MSC markers (CD44, CD90, CD146, CD166, and CD105) and negative for hematopoietic markers (CD19, CD45, HLA-DR, CD80, CD86, and CD40), this was consistent with our previously published study (data not shown) [10]. DBMSCs derived from the placenta in this study were successfully differentiated into bone, fat, and cartilage in vitro using appropriate growth factors and methods as we described recently (data not shown) [10]. Based on the above criteria, we used DBMSCs at passage 3 in all subsequent experiments.
DBMSCs reduced the ability of monocyte to induce HUVEC proliferation
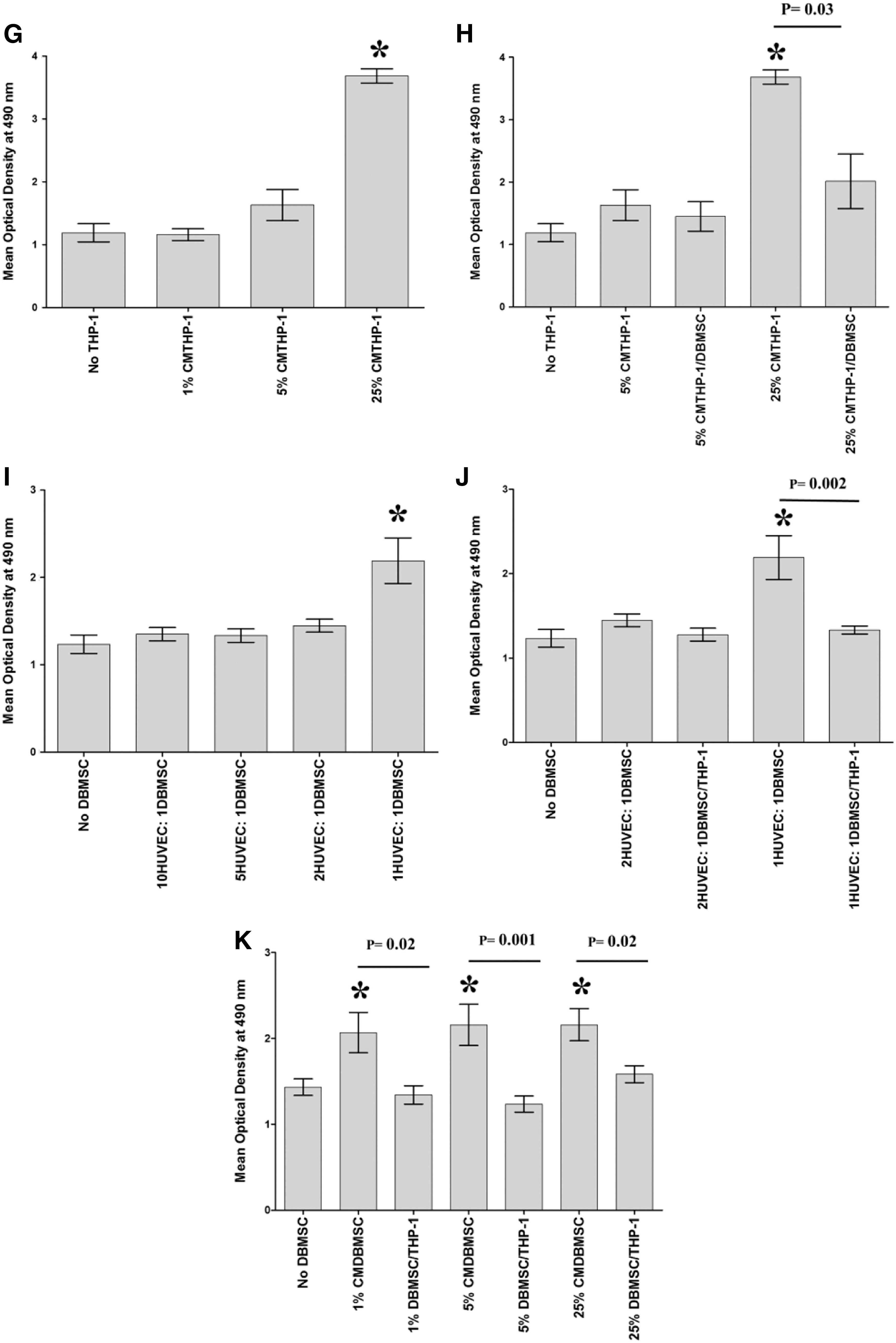
To determine the effect of DBMSCs on the interaction between endothelial cells and monocytes, the proliferation of monocytes in response to DBMSCs and the proliferative response of DBMSCs to monocytes were first examined using the MTS assay. Then, the proliferation of endothelial cells in response to monocytes and monocytes pretreated with DBMSCs was assessed. After 24-h culture with DBMSCs, monocyte proliferation significantly increased at all examined concentrations of DBMSCs (Fig. 2A) or CMDBMSCs, P < 0.05 (Fig. 2B). Similarly, after 24-h culture with monocytes, DBMSC proliferation significantly increased but only with increasing concentration of THP-1 (Fig. 2C) or CMTHP-1 (Fig. 2D), P < 0.05. The proliferative effect of monocytes on DBMSCs or DBMSCs on monocytes is time dependent and reversible (data not shown). After 24-h culture with monocytes, or monocytes pretreated with DBMSCs (THP-1/DBMSC), endothelial cell proliferation in response to THP-1 was significantly increased at a high ratio of THP-1 (Fig. 2E) and this monocyte proliferative effect on endothelial cell was significantly reduced by THP-1/DBMSC, P < 0.05 (Fig. 2F). In addition, endothelial cell proliferation in response to CMTHP-1 was significantly increased at a high percentage of CMTHP-1 (Fig. 2G) and this proliferative effect of CMTHP-1 on endothelial cells was significantly reduced by CMTHP-1/DBMSC, P < 0.05 (Fig. 2H). Similarly, after 24-h culture with DBMSCs, or DBMSCs pretreated with monocytes (DBMSC/THP-1), endothelial cell proliferation was significantly increased at the high ratio of DBMSCs (Fig. 1I) and this DBMSC proliferative effect on endothelial cell was significantly reduced by DBMSC/THP-1, P < 0.05 (Fig. 2I). In addition, endothelial cell proliferation was significantly increased in response to all examined concentrations (1%, 5%, and 25%) of CMDBMSC (Fig. 2J) and this CMDBMSC proliferative effect on endothelial cell was significantly reduced by CMDBMSC/THP-1, P < 0.05 (Fig. 2K). The proliferative effect of DBMSCs or monocytes on endothelial cells was time dependent and reversible (data not shown).
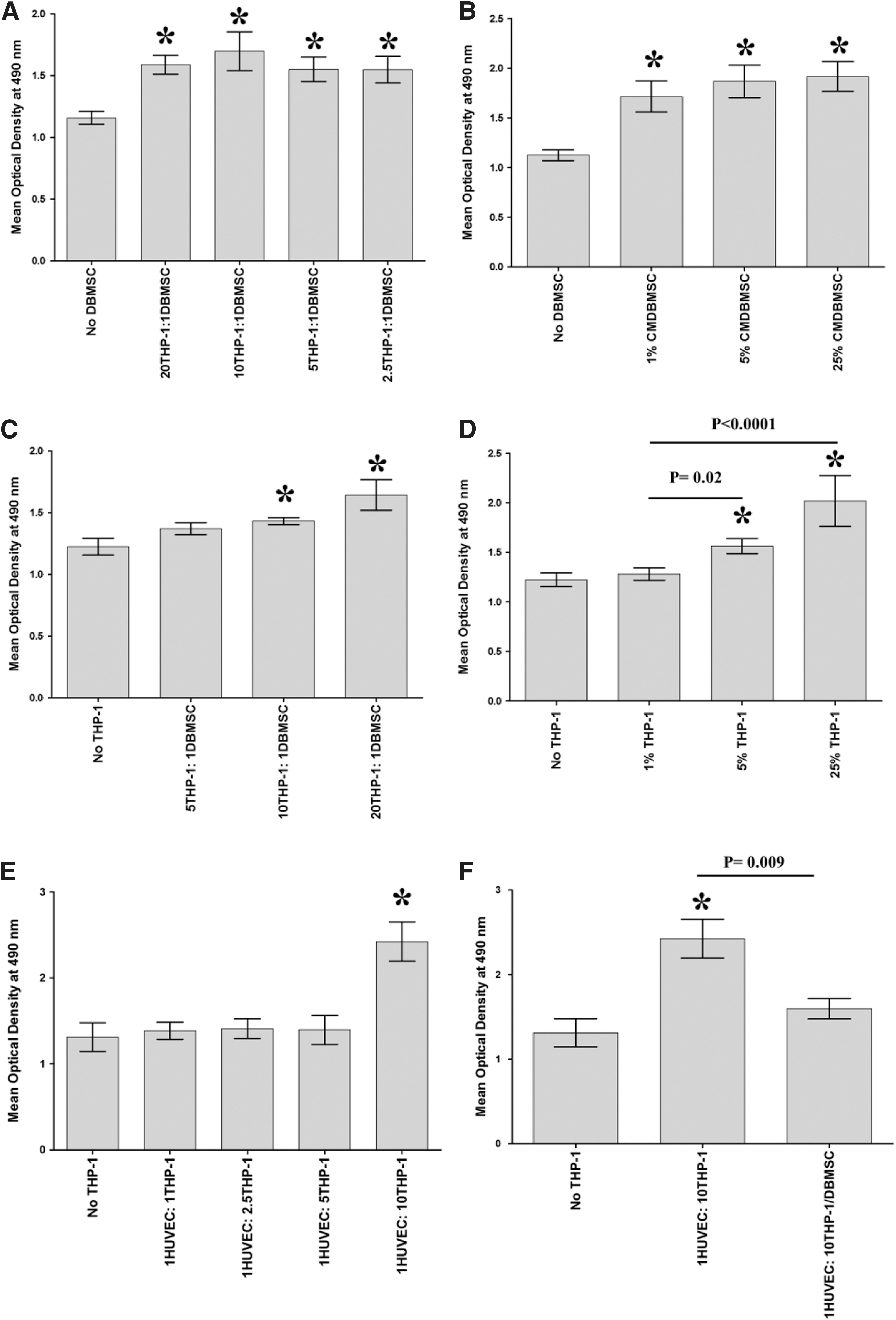
Proliferation of cells measured by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]. The proliferation of monocytes (THP-1) significantly increased in response to different ratios of DBMSC to THP-1
Pretreatment of monocytes with DBMSCs induces cell cycle arrest in HUVEC through the activation of cell cycle checkpoint proteins
To determine the mechanism of reduced endothelial cell proliferation in response to monocytes pretreated with DBMSCs (THP-1/DBMSC), the phosphorylation of cell cycle checkpoint proteins, such as Chk1, Chk2, cdc2, p53, and Rb, which act as key regulators in maintaining the cell cycle and mitosis, were evaluated in endothelial cells by immunoblotting. After 24-h culture with TTHP-1 (monocytes pretreated with DBMSCs), the phosphorylation of Chk1, Chk2, cdc2, p53, and Rb increased 1.8-, 1.29-, 2.62-, 2.67-, and 3.44-fold, respectively in endothelial cells as compared to endothelial cells cultured with THP-1 (untreated THP-1) (Fig. 3). In addition, after 24-h culture with TCM-THP-1 (CM obtained from monocytes pretreated with DBMSCs), the phosphorylation of Chk1, Chk2, cdc2, p53, and Rb increased 1.01-, 1.15-, 1.40-, 1.30-, and 1.42-fold, respectively as compared to endothelial cells cultured with CMTHP-1 (untreated THP-1) (Fig. 3).

Western blot analysis of endothelial cells (HUVEC) cultured with monocytes (THP-1) and monocyte conditioned medium. The proliferation of HUVEC incubated with monocytes pretreated with DBMSCs was reduced due to cell cycle arrest through the activation of cell cycle checkpoint proteins. The mechanism of reduced HUVEC proliferation in response to THP-1-pretreated with DBMSCs (THP-1/DBMSC) was evaluated using the immunoblotting technique to check the phosphorylation of cell cycle checkpoint proteins including Chk1, Chk2, cdc2, p53, and Rb. After 24-h culture with TTHP-1 (THP-1-pretreated with DBMSCs), the phosphorylation of Chk1
DBMSCs reduced monocyte adhesion to HUVEC
To assess the effect of DBMSCs on the adhesion of monocytes to endothelial cells, monocytes were cultured with DBMSCs for 96 h and then labeled with the green fluorescent dye CMFDA and added to endothelial cell in an adhesion assay. Results showed that the adhesion of monocytes to endothelial cells was significantly reduced after culturing monocytes with DBMSCs, P < 0.05 (Fig. 4A). Next, we investigated whether the inhibitory effects of DBMSCs on monocyte adhesion was mediated through the modulation of the expression of adhesion molecules in monocytes or endothelial cells. A variety of adhesion molecules were studied by flow cytometry and expression recorded as median fluorescence intensity. After 24-h culture with DBMSCs, the expression of ICAM-1, VCAM-1, and CD44 was modulated in monocytes but not significantly, P > 0.05 (Fig. 4B–D). Similarly, monocyte expression of ICAM-1, VCAM-1, and CD44 was not significantly modulated by CMDBMSC, P > 0.05 (data not shown). In addition, after 24-h culture with THP-1 or THP-1/DBMSC, HUVEC expression of ICAM-1 and VCAM-1 was significantly increased (P < 0.01) compared to untreated HUVEC (Fig. 4E, G) and HUVEC expression of ICAM-1 was significantly reduced by THP-1/DBMSCs as compared to THP-1 alone (Fig. 4E). Moreover, HUVEC expression of VCAM-1 was reduced by THP-1/DBMSC but not significantly (P > 0.05) (Fig. 4G). In contrast, HUVEC expression of CD44 was not significantly modulated by THP-1 or THP-1/DBMSC, P > 0.05 (Fig. 4I). Similarly, HUVEC expression of ICAM-1, VCAM-1, and CD44 was not significantly modulated by CMTHP-1 or CMTHP-1/DBMSC, P > 0.05 (Fig. 4F, H, J).
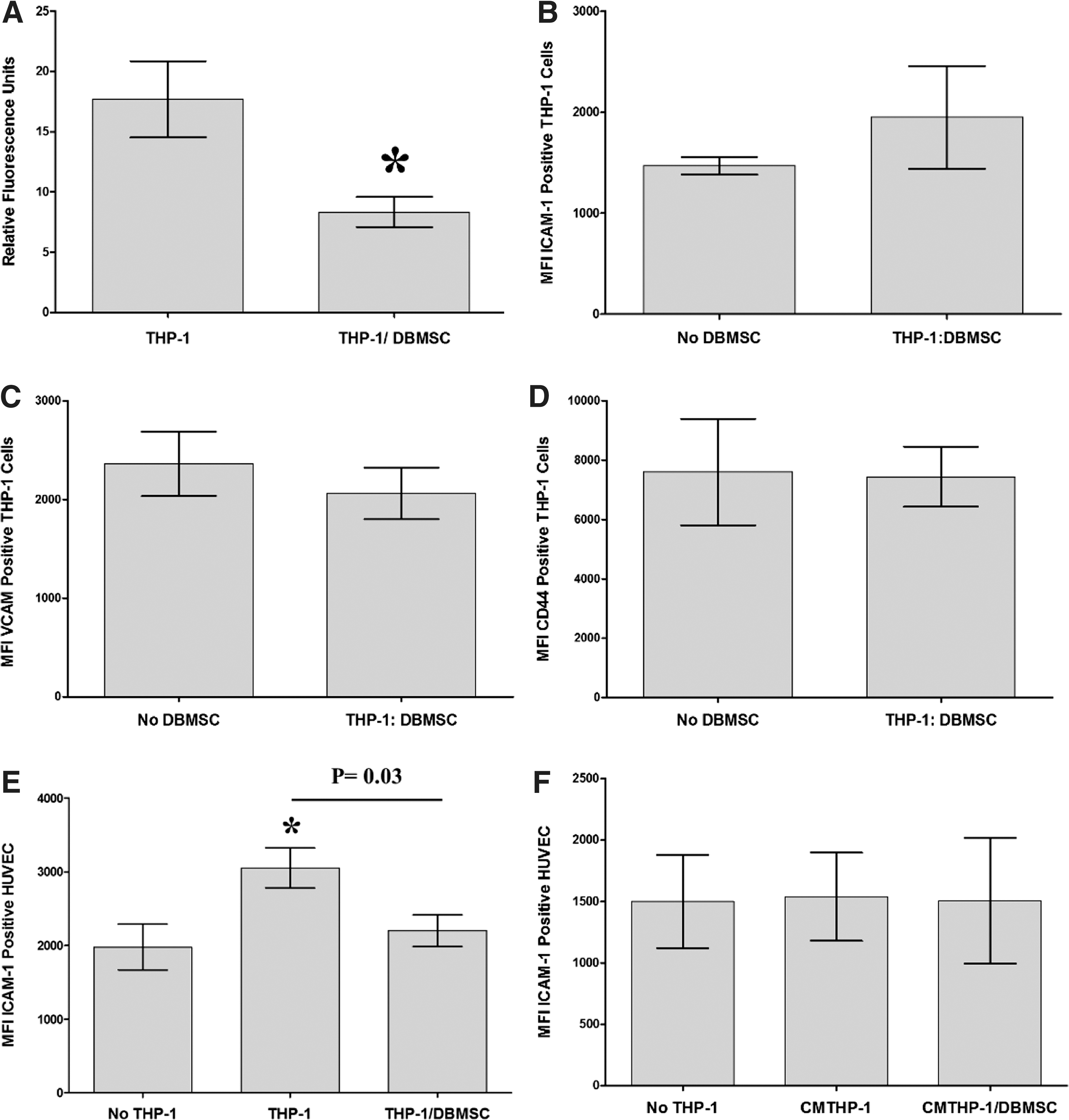
The adhesion of monocytes to endothelial cells (HUVEC) was significantly reduced when monocytes were initially cultured with DBMSCs for 96 h and then added to the monolayer of HUVEC, as compared to untreated monocytes
DBMSCs modulate endothelial cell expression of adhesion and inflammatory markers
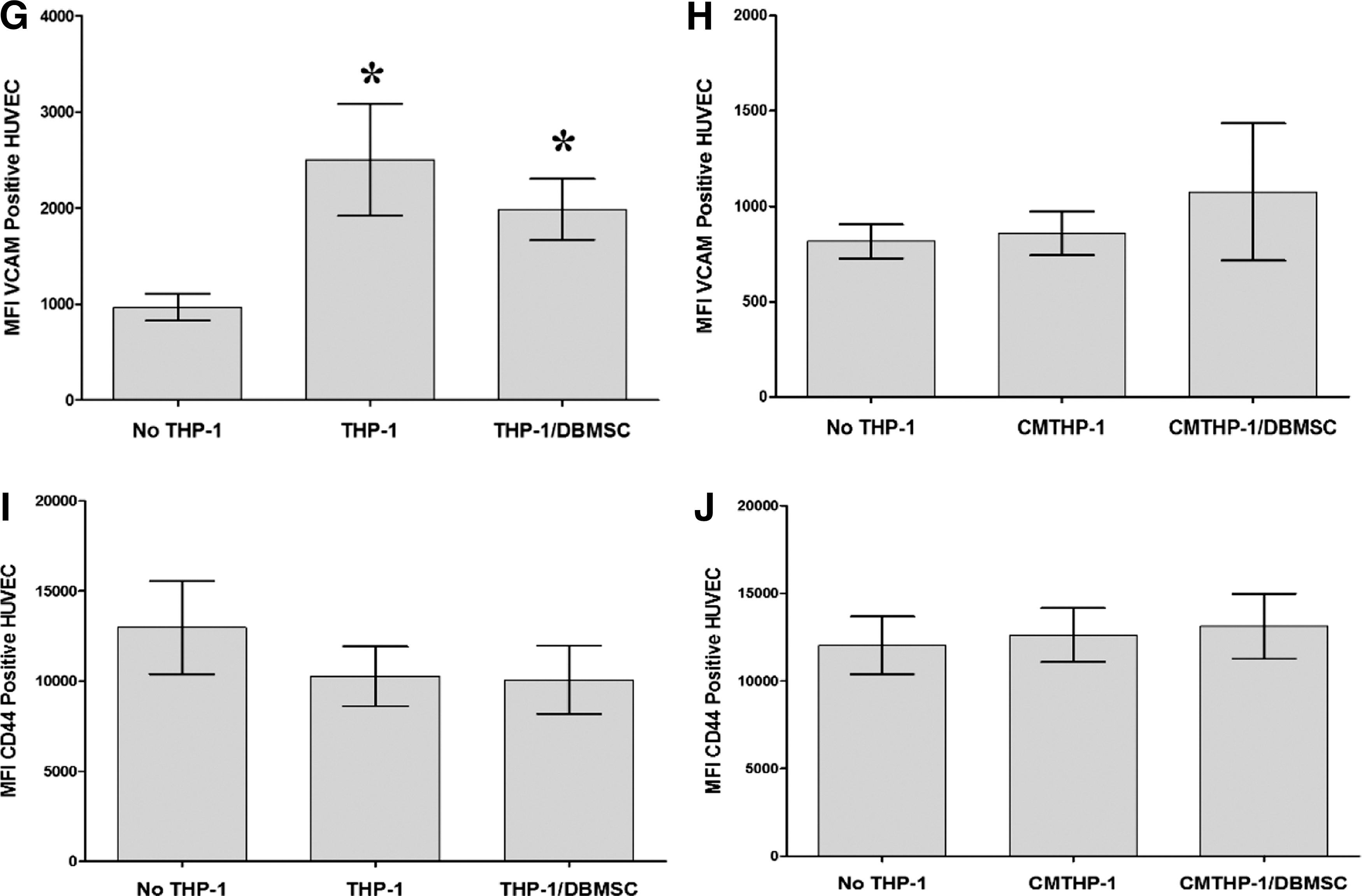
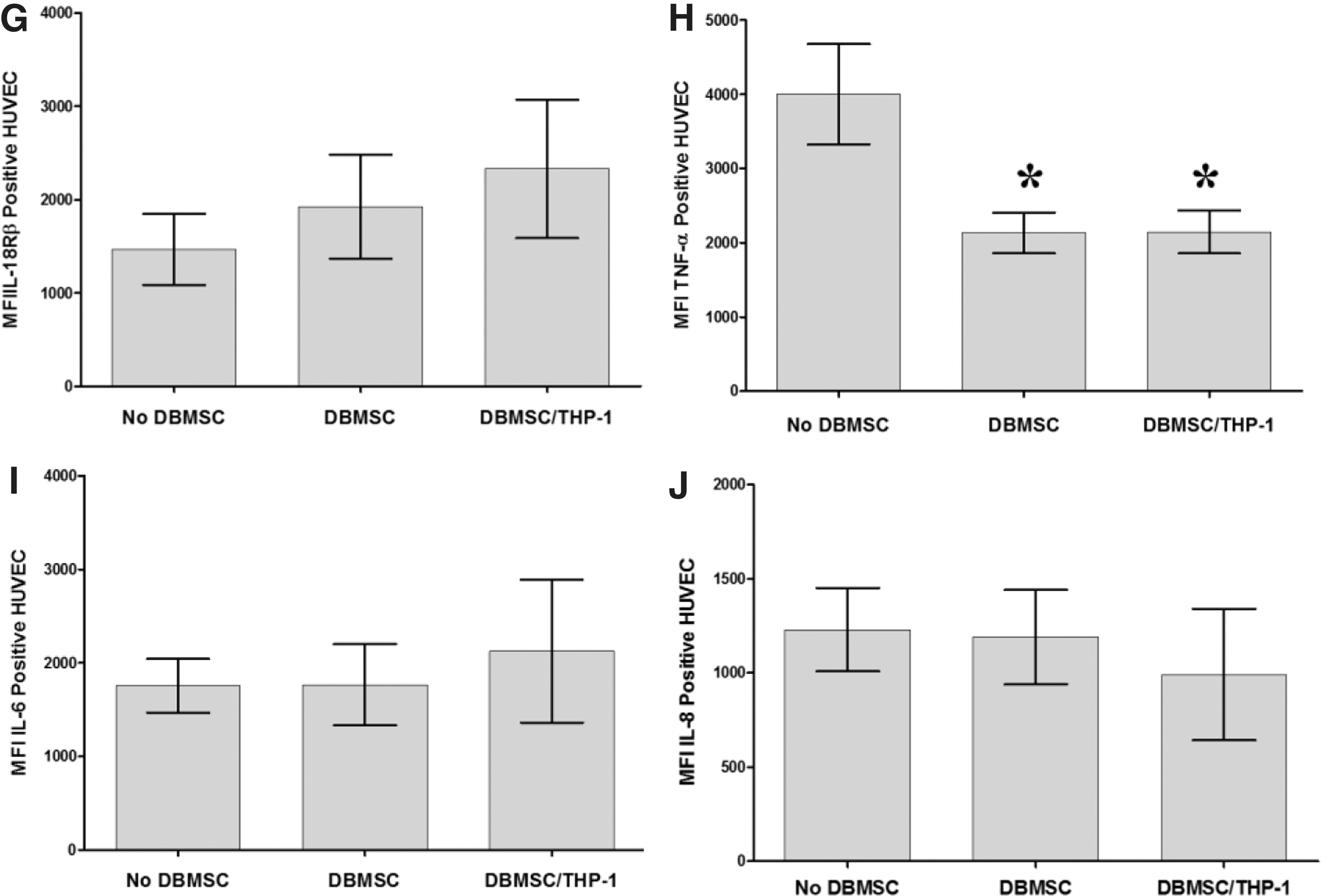
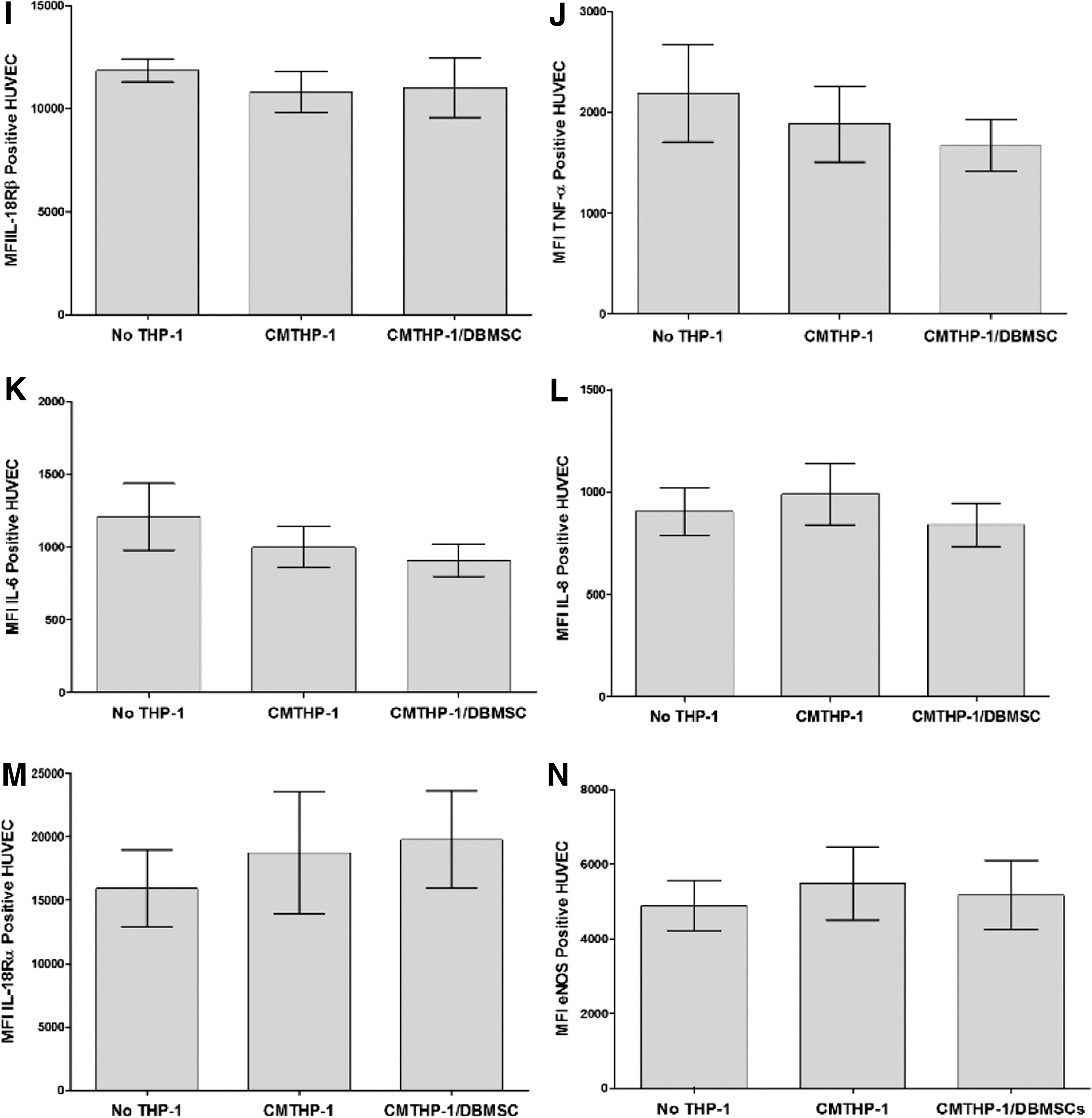
To evaluate the modulatory effects of DBMSCs on endothelial cell functions, a variety of proteins important in endothelial functions were studied by flow cytometry and expression was recorded as median fluorescence intensity, or as a percentage of cells. After 24-h culture with DBMSCs or DBMSC/THP-1, endothelial cell expression of ICAM-1 and CD44 was significantly reduced compared with untreated endothelial cells, P < 0.05 (Fig. 5A, B) and this increased endothelial cell expression of ICAM-1 and CD44 was not changed after exposing DBMSCs to monocytes (DBMSC/THP-1), P > 0.05 (Fig. 5A, B). In contrast, endothelial cell expression of VCAM-1 was not significantly changed after culturing endothelial cells with DBMSCs or DBMSC/THP-1, P > 0.05 (Fig. 5C). In addition, after 24-h culture with DBMSCs or DBMSC/THP-1 endothelial cell expression of ecto-5′-nucleotidase and TNF-α were significantly reduced as compared to untreated endothelial cells, P < 0.05 (Fig. 5E, F, J) and both treatments were not different in terms of their modulatory effects on endothelial cell expression of ecto-5′-nucleotidase and TNF-α, P > 0.05 (Fig. 5D, H). Moreover, endothelial cell expression of IL18Rα was significantly increased in response to DBMSCs or DBMSC/THP-1 as compared to untreated endothelial cells, P < 0.05 (Fig. 5F). Furthermore, endothelial cell expression of IL18Rα was significantly increased in response to DBMSC/THP-1 as compared to DBMSC alone, P > 0.05 (Fig. 5F). In contrast, endothelial cell expression of eNOS, IL18Rβ, IL-6, and IL-8 was not significantly modulated by DBMSCs or DBMSC/THP-1 as compared to untreated endothelial cells, P > 0.05 (Fig. 5E, G, I, J). Similarly, HUVEC expression of ecto-5′-nucleotidase, eNOS, IL18Rα, IL18Rβ, TNF-α, IL-6, and IL-8 was not significantly modulated by CMDBMSC or CMDBMSC/THP-1, P > 0.05 (data not shown). We also evaluated the modulatory effects of DBMSCs on monocyte effects on endothelial cell expression of ecto-5′-nucleotidase, eNOS, IL18Rα, IL18Rβ, TNF-α, IL-6, and IL-8. After 24-h culture with THP-1 or THP-1 initially cultured with DBMSC (THP-1/DBMSC), endothelial cell expression of ecto-5′-nucleotidase, IL18Rβ, TNF-α, IL-6, and IL-8 was significantly increased in response to THP-1 as compared to untreated endothelial cells, P < 0.05 (Fig. 6A–E) and this increased endothelial cell expression of ecto-5′-nucleotidase, IL18Rβ, TNF-α, IL-6, and IL-8 was significantly reduced by THP-1/DBMSC as compared to THP-1 alone (Fig. 6A–E). In contrast, endothelial cell expression of IL18Rα and eNOS was not significantly modulated by THP-1 or THP-1/DBMSC, P > 0.05 (Fig. 6F, G). Similarly, endothelial cell expression of ecto-5′-nucleotidase, eNOS, IL18Rα, IL18Rβ, TNF-α, IL-6, and IL-8 was not significantly modulated by CMTHP-1 or CMTHP-1/DBMSC, P > 0.05 (Fig. 6H–N).
Flow cytometric analysis of endothelial cell (HUVEC) expression of adhesion and inflammatory markers. DBMSC and DBMSCs pretreated with monocytes (DBMSC/THP-1) significantly decreased HUVEC expression of ICAM-1
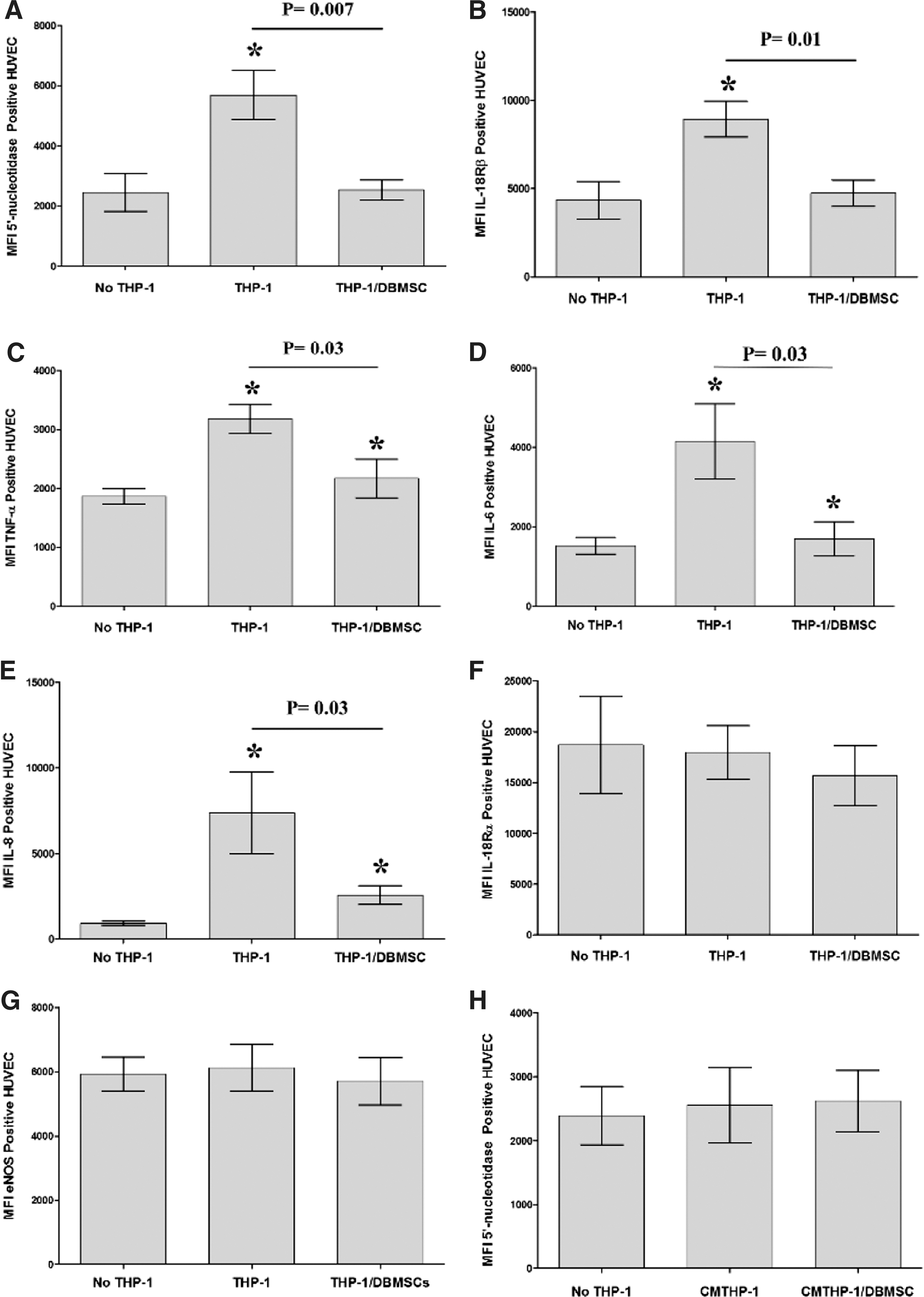
Flow cytometric analysis of endothelial cell (HUVEC) expression of inflammatory markers. Monocytes significantly increased HUVEC expression of ecto-5′-nucleotidase
HUVEC migration in response to DBMSCs, monocytes, and monocytes pretreated with DBMSCs
To further evaluate the effect of DBMSCs on the migration of endothelial cells, endothelial cell migration in response to CMDBMSC, CMTHP-1, and CMTHP-1/DBMSC in the lower chamber of the migration plate was measured using the xCELLigence RTCA. Endothelial cells significantly migrated in response to CMDBMSC, CMTHP-1, and CMTHP-1/DBMSC after 24 h as compared to untreated endothelial cells, P < 0.05 (Fig. 7).
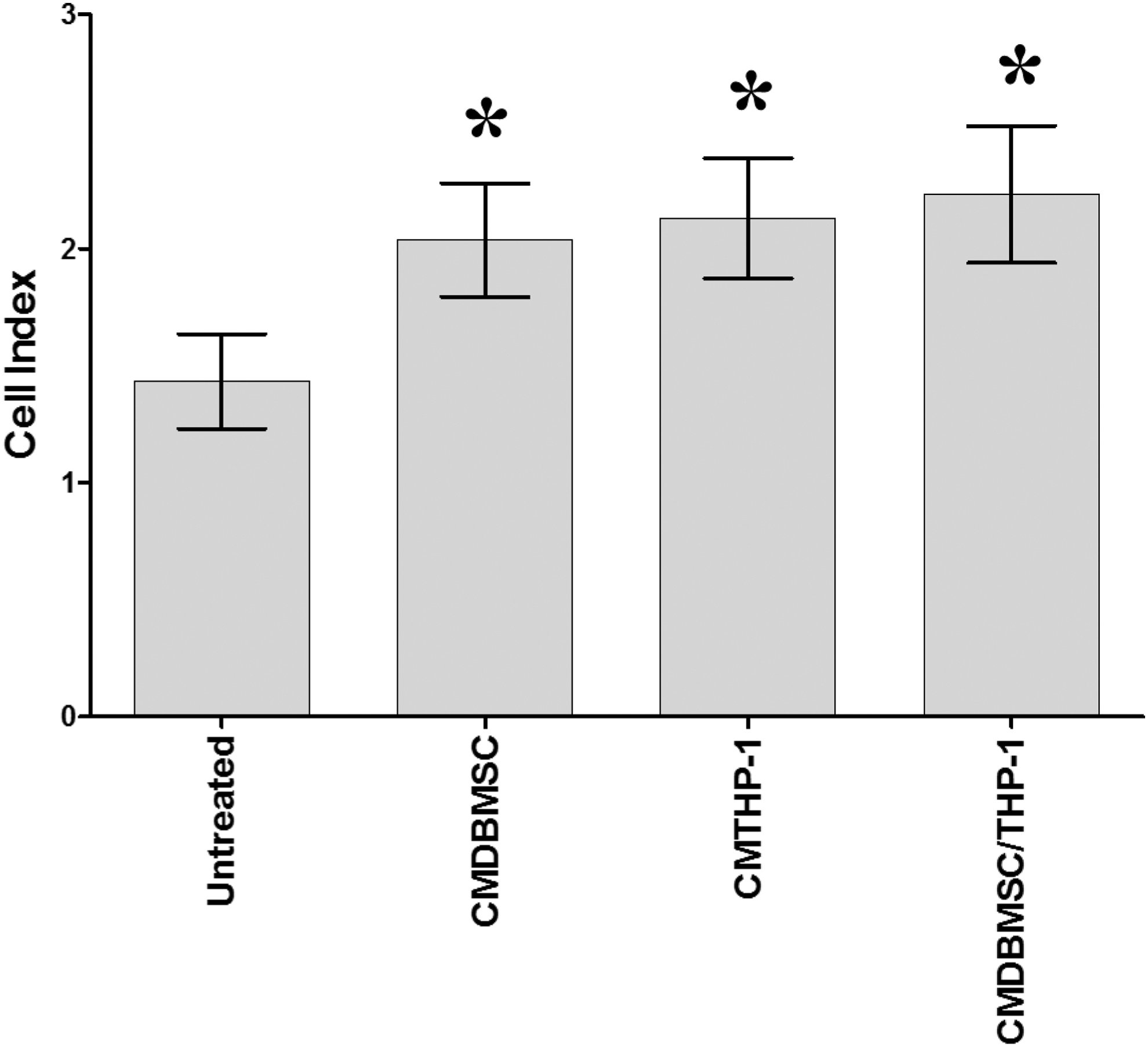
The migration of endothelial cells (HUVEC) in response to CMDBMSC, CMTHP-1, and CMTHP-1/DBMSC in the lower chamber of the migration plate was measured using the xCELLigence Real-Time Cell Analyzer. Endothelial cells significantly migrated in response to CMDBMSC, CMTHP-1, and CMTHP-1/DBMSC after 24 h as compared to untreated HUVEC, *P < 0.05. Experiments were conducted in triplicate using five independent placentae and umbilical cord tissues to prepare DBMSCs and HUVEC, respectively. Bars represent standard errors.
Discussion
We previously reported that DBMSCs have unique phenotypic and functional properties, which could be advantageous for cell-based therapies where modulation of the inflammatory response is important [10]. However, the modulatory property of DBMSCs specifically on endothelial cell functions has not yet been examined. In this study, we present for the first time the ability of DBMSCs to modify the responses of human endothelial cells through direct effects or through modulating the effects of monocytes on endothelial cells. Initially, the consequences of interactions between DBMSCs and monocytes were evaluated. We found that monocytes increased DBMSC proliferation at a high ratio of monocytes to DBMSCs and a higher percentage of monocyte CM to fresh medium (Fig. 2). Similarly, DBMSCs increased monocyte proliferation, but at all examined ratios of DBMSCs to monocytes and percentages of DBMSC CM to fresh medium (Fig. 2). Next, we evaluated the proliferative responses of endothelial cells to DBMSCs, monocytes, DBMSCs pretreated with monocytes (DBMSC/THP-1) or monocytes pretreated with DBMSCs (THP-1/DBMSC). In a cell–cell contact assay, DBMSCs increased endothelial proliferation at a higher ratio of DBMSCs to endothelial cells, while all percentages of DBMSC CM increased endothelial cell proliferation (Fig. 2). Similarly, in the cell–cell contact assay, at high ratios of monocytes to endothelial cells and higher percentages of monocyte CM, endothelial proliferation was increased (Fig. 2). These results suggest that DBMSCs and monocytes express and secrete factors with proliferative activity. Recently, we reported that DBMSCs express and secrete a unique combination of molecules involved in many important cellular functions, including proliferation [10]. For example, DBMSCs express and secrete IL1β, IL-6, granulocyte-macrophage colony-stimulating factor, and hepatocyte growth factor [24 –27]. Monocytes also express and secrete most of these proliferative factors [15,27]. This may explain the enhanced proliferation of endothelial cells by DBMSCs and monocytes. Interestingly, when DBMSCs and monocytes are allowed to interact, this reversed their proliferative effect on endothelial cells, as demonstrated by the reduction in endothelial cell proliferation after their culture with DBMSC/THP-1 or with THP-1/DBMSC (Fig. 2). Mechanistically, we found that the reduction of endothelial cell proliferation is due to cell cycle arrest through the activation of cell cycle checkpoint proteins. Monocytes pretreated with DBMSCs upregulated the expression of phosphorylated forms of cell cycle checkpoint proteins including Chk1, Chk2, cdc2, tumor suppressor protein p53, and Rb protein (Fig. 3). These proteins are key regulators of the cell cycle and mitosis [28 –32]. The enhanced expression of these proteins indicates that the cell cycle is arrested at the G1/S regulation point, as shown by upregulation of the phosphorylated form of p53 in endothelial cells. The phosphorylation of p53 results in transient cell cycle arrest that holds the cell cycle at the G1/S regulation point to allow a correction to occur [28]. This may allow the repair of the DNA to restart the cell cycle, or for more damaged cells it may allow apoptosis and elimination of the dead cells [28]. In our study, cells were viable as shown by the Trypan blue staining where viability of endothelial cells was more than 90%. In addition, the expression of the phosphorylated form of Rb was also upregulated (Fig. 3). This indicates that the progression of cell cycle from the G1 to S phase is not irreversibly inhibited and can continue [29]. Moreover, the phosphorylation of cdc2, Chk1, and Chk2 indicates that mitosis is prevented from progression at a later stage of the cell cycle. These proteins are involved later in the cell cycle at the G2 and M phases and their phosphorylation leads to cell cycle arrest and decreased proliferation [30 –32]
A characteristic feature of diseases, such as atherosclerosis is that monocytes activate endothelial cells as a result of inflammation and this leads to the development and progression of disease [33 –38]. In atherosclerosis, two of the early modifications that occur at the sites of the atherosclerotic lesions are the adhesion of monocytes to the endothelium and the proliferation of endothelial cells [35]. Inhibition of monocyte adhesion and endothelial proliferation reduces the progression of atherosclerotic lesions in animals [35]. Adhesion of monocytes to the vessel wall induces the proliferation of endothelial cells and angiogenesis [36]. The critical role of monocytes in the activation of endothelial cells was confirmed by the infusion of chemoattractant protein-1 (MCP-1) produced by monocytes, which significantly increased the growth of endothelial cells and angiogenesis [37]. Similarly, the recruitment of leukocytes and production of MCP-1 and IL-8 were also associated with endothelial cell activation in experimental animals that underwent bilateral iliac artery balloon denudation [33]. In addition, the adhesion of monocytes to the vessel wall and subsequent production of basic fibroblast growth factor and TNF-α, increased the growth of endothelial cells and angiogenesis in animals following femoral artery occlusion [38]. Therefore, our data indicate that DBMSCs can protect endothelial cells by reducing their proliferation through a mechanism that may involve direct contact between DBMSCs, and endothelial cells or through monocytes.
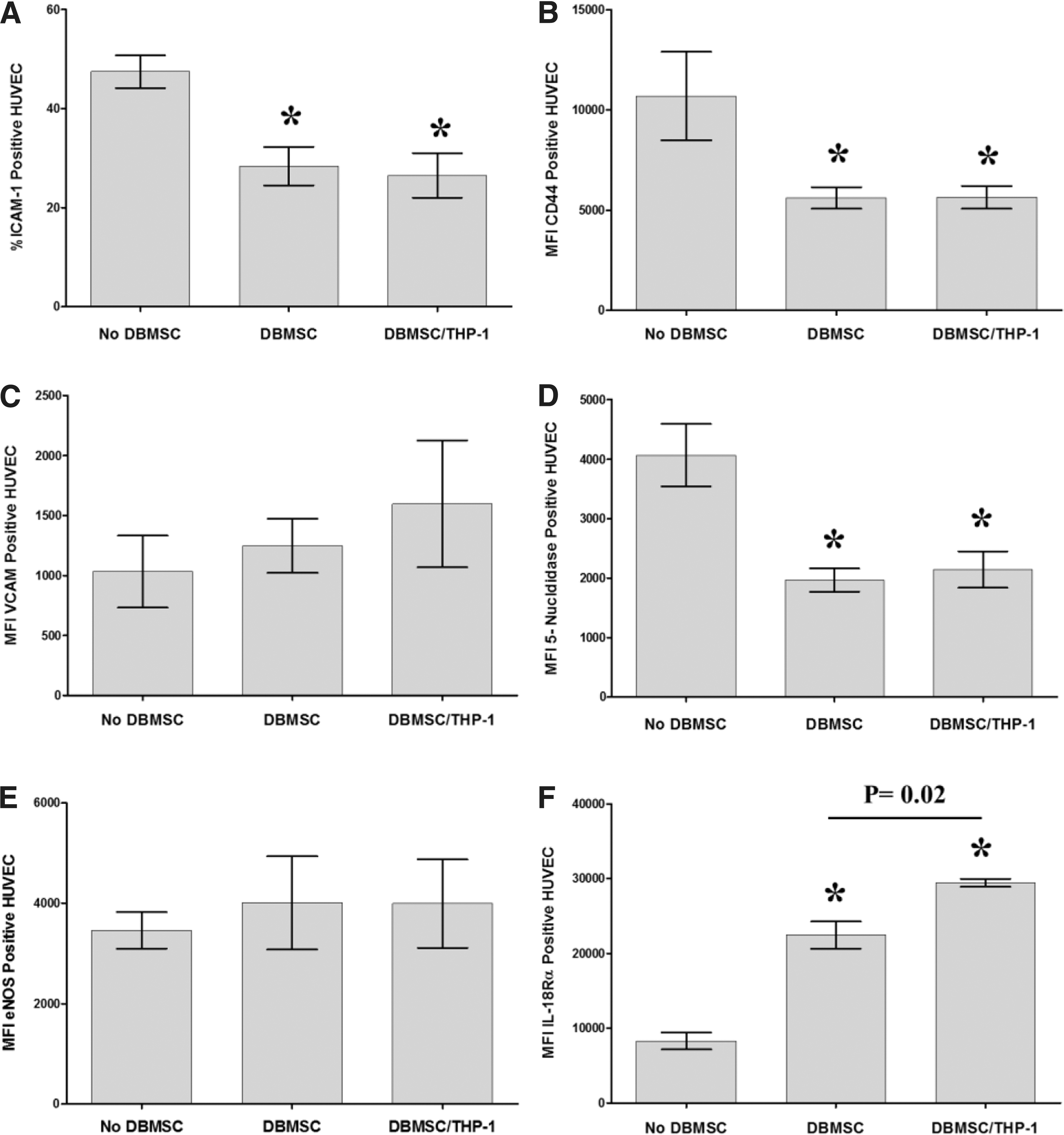
In this study, we also found that the interaction between DBMSCs and monocytes reduced monocyte adhesion to endothelial cells (Fig. 3). This provides further evidence of the protective effects of DBMSCs on endothelial cells. Activated endothelial cells at atherosclerotic lesions within a blood vessel wall express high levels of adhesion molecules and cytokines, both of which mediate the recruitment of additional monocytes. Accumulation of monocytes and monocyte-derived macrophages in the vessel wall results in chronic inflammation and leads to the development and progression of atherosclerosis. Monocyte activation of endothelial cells induces endothelial cell expression of inflammatory cytokines, such as TNF-α and IL-1β [27]. In this study, we also showed for the first time that direct contact between monocytes and endothelial cells increases endothelial cell expression of other inflammatory cytokines and receptors (ie, IL-6, IL-8, and IL18Rβ, respectively) (Fig. 6). The consequences of high levels of these cytokines in inflammatory diseases are reported by many studies [39 –42]. These cytokines contribute to increased proliferation of endothelial cells in atherosclerosis, and to the firm binding of monocytes to endothelial cells through enhanced expression of adhesion molecules, such as ICAM-1, VCAM-1, and CD44 [15,43]. In this study, we showed that DBMSCs reduced endothelial cell expression of inflammatory proteins. In a cell–cell contact assay, DBMSCs directly reduced endothelial cell expression of TNF-α (Fig. 5) while through their interaction with monocytes, endothelial cell expression of IL18Rβ, TNF-α, IL-6, and IL-8 were also reduced (Fig. 5). However, the CM of DBMSCs, monocytes, and DBMSC/THP-1 had no effect on endothelial cell expression of these inflammatory proteins. These data demonstrate the ability of DBMSCs to protect endothelial cells from inflammation through direct contact with the endothelial cells or by inhibiting the inflammatory effect of monocytes on endothelial cells, as previously reported [15,43]. DBMSCs or DBMSCs pretreated with monocytes induced endothelial cell expression of IL-18Rα but CM from DBMSCs or DBMSC/THP-1 had no effect on endothelial cell expression of IL-18Rα. This may indicate that the protective role of DBMSC on endothelial cells does not involve the IL-18 pathway. Further studies are needed to better understand the protective role of DBMSCs on endothelial cells via the IL-18 pathway.
We also found that in a cell–cell contact assay, DBMSCs can reduce endothelial cell expression of ICAM-1 and CD44 through direct contact, or after being exposed to monocytes (Fig. 5). There was no difference between the effects of DBMSCs and DBMSC/THP-1 on endothelial expression of ICAM-1 and CD44. Furthermore, the increased expression of ICAM-1 expression in endothelial cells by monocytes was significantly reduced if the monocytes were previously exposed to DBMSCs (Fig. 4). Moreover, DBMSCs reduced the ability of monocytes to enhance the expression of VCAM-1 in endothelial cells, but statistically this was not significant. These data suggest that DBMSCs can regulate the permeability of the endothelium through modulating the expression of specific endothelial cell adhesion molecules, such as ICAM-1 and CD44. This is a significant finding since the permeability of endothelium is enhanced in atherosclerosis [44]. Atherosclerosis is characterized by increased expression of ICAM-1, VCAM-1, and CD44 in activated endothelium and this strengthens the adhesion of monocytes to the endothelium while simultaneously enhancing endothelium permeability, which subsequently permits the transmigration of monocytes into the underlying stroma [44 –47]. Thus, reducing endothelium permeability controls the adhesion and transmigration of monocytes and this prevents inflammation triggered by inflammatory cells in atherosclerosis [44]. Therefore, our data indicate that DBMSCs could be useful in treating atherosclerosis by reducing endothelial cell activation through a mechanism that involves a decreased expression of adhesion molecules, and the adhesion of monocytes to endothelium, which would reduce the permeability of endothelial cells, and consequently reduce the influx of monocytes and thereby lower inflammation.
We also assessed the effects of DBMSCs on endothelial cell migration. The migration of cells plays a central role in a variety of physiological and pathological processes [48]. Cell migration involves a complex, tightly regulated multistep process [48]. Endothelial cells migrate during vasculogenesis and angiogenesis but also in a damaged vessel to restore vessel integrity [48]. Depending on the environment, endothelial cells can migrate in response to intercellular and environmental signals, which modulate the process [48]. In this study, we report that endothelial cell migration is increased in response to the CM of DBMSCs, monocytes, and monocytes pretreated with DBMSCs. As previously discussed, DBMSCs secrete many cytokines, chemokines, and growth factors with various biological functions. For example, they secrete vascular endothelial growth factor (VEGF), which induces endothelial cell migration [10,49]. VEGF is also secreted by monocytes [50]. This may explain the enhanced migration of endothelial cells in this study, and suggests that DBMSCs can also influence endothelial cell migration: a critical step in angiogenesis that is triggered in atherosclerosis [51]. Although CM of DBMSCs, monocytes, and monocytes pretreated with DBMSCs promoted endothelial cell migration, under other conditions DBMSCs appear to inhibit angiogenesis. We found that endothelial cell expression of a proangiogenic factor known as ecto-5′-nucleotidase (CD73) was significantly reduced by DBMSCs either in direct contact, or through their interaction with monocytes (Fig. 5). In contrast, monocytes significantly increased endothelial cell expression of CD73, a promoter of angiogenesis [52] (Fig. 6). Studies reported that angiogenesis is inhibited in cancer cells when the activity of CD73 is decreased [53]. Another study revealed that more capillary-like structures were formed in CD73+/+ endothelial cells than CD73−/− endothelial cells [54]. Thus, CD73 can promote endothelial cells to form new blood vessels.
As previously discussed, DBMSCs also reduced endothelial cell expression of CD44, which may account for the observation that DBMSCs inhibit endothelial capillary-like structures since CD44 is a promoter of angiogenesis [55]. Antibodies against CD44 inhibited in vitro endothelial tube formation [55], therefore DBMSCs may inhibit angiogenesis, or inhibit monocyte induction of endothelial new blood vessel formation [56], by reducing the activity of CD73 and CD44. Future in vitro and in vivo studies are necessary to elucidate the role of DBMSCs in angiogenesis.
In conclusion, our study shows for the first time a protective role of DBMSCs on endothelial cells, at least in part through their modulatory effects on several early events key to the development of inflammatory diseases, such as atherosclerosis; the adhesion of monocytes to the endothelium, which in turn increases endothelium permeability, and induces endothelial cell proliferation. This is a significant finding because these events activate endothelial cells, and enhance their expression of adhesion, and inflammatory markers. Therefore, DBMSCs may be a promising therapeutic strategy for atherosclerosis.
Footnotes
Acknowledgments
The authors thank the staff and patients of the Delivery Unit, King Abdul Aziz Medical City for their help in obtaining placentae. This study was supported by grants from King Abdulla International Medical Research Centre (grant no. RC12/133).
Author Disclosure Statement
No competing financial interests exist.