
Abstract
Periodontitis is an infectious disease characterized by chronic inflammation and progressive destruction of periodontal tissues. Chronic inflammatory environment may affect immunomodulatory function of periodontal ligament stem cells (PDLSCs) and promote shift toward proinflammatory phenotype contributing to propagation of periodontitis. Therefore, suppression of inflammatory response in PDLSCs represents a novel therapeutic approach. Extracellular vesicles (EVs) have been shown to display anti-inflammatory and immunosuppressive actions in different tissues and could represent a potent therapeutic tools against chronic inflammation during periodontitis. In the present study, we investigated the effects of EVs on the basal and lipopolysaccharide (LPS)-induced activity of NFκB signaling pathway in PDLSCs. We also examined the impact of EVs on the osteogenic differentiation and expression of osteogenesis-related genes. EVs were purified by differential ultracentrifugation from PDLSCs grown on gelatin-coated alginate microcarriers in a bioreactor. NFκB reporter assays demonstrated that EVs permanently suppressed basal and LPS-induced activity of NFκB in PDLSCs. Combined treatment with EVs and anti-TLR4 antibody (Ab) resulted in attenuation of the inhibitory effect on the NFκB activity, suggesting a possible interference through a competition for TLR4 signaling pathway. EVs also increased phosphorylation of Akt and its downstream target GSK3β (Ser 9) indicating that PI3K/Akt signaling pathway may act as suppressor of NFκB activity. LPS stimulated osteogenic mineralization of PDLSCs. Unexpectedly, anti-TLR4 blocking Ab per se significantly decreased osteogenic mineralization of PDLSCs. EVs did not affect osteogenic mineralization, but partially suppressed inhibitory effect of anti-TLR4 blocking Ab. Gene expression studies revealed significant effects of EVs on osteogenesis-related genes and possible interference with TLR4 signaling in PDLSCs. In conclusion, our study demonstrates that EVs suppress basal and LPS-induced activity of NFκB signaling pathway in PDLSCs and could potentially be used for targeting of chronic inflammation during periodontitis.
Introduction
Periodontitis is a chronic infectious disease characterized by progressive destruction of periodontal tissues and leading to the tooth loss in adults. The development of periodontitis results from the complex interplay between dysbiotic oral microbiota and dysregulated host immune response causing chronic inflammation and progressive destruction of the tooth attachment structures of periodontal ligament (PDL) [1]. Chronic inflammation also leads to the exhaustion of endogenous pools of periodontal ligament stem cells (PDLSCs) slowing down remodeling and regeneration of periodontal tissues [2].
PDLSCs can differentiate into osteoblasts, cementoblast-like cells, and adipocytes in vitro [3] and are similar to the mesenchymal stem cells (MSCs) from other tissues. However, transplantation experiments using hydroxyapatite/tricalcium phosphate carriers demonstrated tissue-specific differentiation capacities of PDLSCs in vivo [4]. Thus, in contrast to the MSCs derived from bone marrow or dental pulp, PDLSCs generated cementum/PDL-like structures and contributed to the periodontal tissue repair [4].
Several studies also demonstrated that PDLSCs possess immunomodulatory activity with the ability to suppress immune reactions. PDLSCs exhibited a noncell contact-dependent suppression of peripheral blood mononuclear cell proliferation [5]. Another study showed that PDLSCs inhibited proliferation of allogeneic T cells through upregulation of cyclooxygenase-2 and prostaglandin E2 [6]. Similarly, to the MSCs from other tissues, PDLSCs also express indoleamine-2,3-dioxygenase 1 (IDO-1), which acts as a suppressor of local immune response by catalyzing oxidative degradation of
However, most of the in vitro studies have been performed with PDLSCs derived from healthy tissues, while actual immunomodulatory role of PDLSCs in the chronic inflammatory environment remains elusive. One study indirectly addressed this problem and found that PDLSCs from inflamed tissues exhibited impaired immunomodulatory properties [10]. PDLSCs also secreted proinflammatory cytokines in response to stimulation with LPS [11] or TLR2 agonist [9], suggesting that PDLSCs can also act as promoters of inflammation. It is known from other experimental models that tissue-specific fibroblasts can be reprogrammed to act as promoters of inflammation. For example, synovial fibroblasts [12] and cancer-associated fibroblasts [13] are important for the maintenance of chronic inflammation. We therefore suggest that impaired immunomodulatory function of PDLSCs and shift toward proinflammatory phenotype may contribute to the dysregulation of host immune response and propagation of periodontitis. Consequently, suppression of inflammatory response in PDLSCs could be a promising therapeutic approach against periodontitis.
Extracellular vesicles (EVs) provide a novel mechanism of intercellular communication via the transfer of biological information between cells [14]. Based on the mode of biogenesis and size, EVs could be classified as exosomes and microvesicles. Exosomes originate from multivesicular bodies and are 30–150 nm in diameter, while microvesicles (200–1,000 nm) are released by budding from the plasma membrane [15]. EVs are enclosed by phospholipid bilayer and contain many different proteins, nucleic acids, lipids, and metabolites that can be transferred to the distant sites of the organism. It is becoming increasingly evident that cargo content and therapeutic properties of EVs depend on parent cell type and its physiological state. Thus, EVs from healthy cells may have similar therapeutic properties as parent cells [16], while EVs from diseased tissues may act as facilitators of pathological processes [17,18]. Recent study used rat periodontal defect model to demonstrate that transplantation of conditioned medium from human PDLSCs enhanced periodontal tissue regeneration in a concentration-dependent manner [19]. EVs derived from the PDLSCs promoted bone regeneration in rats with calvarial defects [20]. Interestingly, EVs derived from human PDLSCs suppressed experimental autoimmune encephalomyelitis in mice [21]. A very recent report demonstrated that LPS-preconditioned PDLSCs induce M1 polarization of macrophages through EVs [22]. All these reports show that PDLSCs produce biologically active EVs that can be potentially exploited for therapeutic purposes.
In the present study, we demonstrate that EVs suppress basal and LPS-induced activity of NFκB signaling pathway in PDLSCs. These findings could be potentially exploited for the development of new therapeutic strategies targeting chronic inflammation during periodontitis.
Materials and Methods
Isolation and characterization of PDLSCs
Primary PDLSC cultures were isolated from healthy periodontal tissues of two donors using explant outgrowth method. Material was collected under the approval of the Lithuanian Bioethics Committee. Two intact premolars were obtained from two Caucasian females (18 and 21 years old) undergoing tooth extraction due to orthodontic reasons. PDL tissue was gently scraped from the middle third of the tooth root using surgical blades, minced and cut into 2 mm3 pieces, placed to the 35 mm diameter culture dishes, and cultured in a low glucose Dulbecco's modified Eagle's medium (Biochrom, Berlin, Germany) supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM
Microcarrier cell culture of PDLSCs
For the microcarrier culture of PDLSCs, we used BioLevitator™ (Hamilton Bonaduz AG) three-dimensional culturing platform and an alginate microcarrier cell culture system (Global Cell Solutions, Charlottesville, VA) [23]. The Global Eukaryotic Microcarriers (GEM™) (Global Cell Solutions) are composed of an alginate core embedded with paramagnetic particles and coated with adhesion molecules. In our experiments, we used gelatin-coated GEMs. Inoculation and culture of PDLSCs on GEMs in the BioLevitator was performed according to the manufacturer's recommendations with some modifications. In brief, 1.2 mL of prewashed, gelatin-coated GEMs were added to the LeviTube (Global Cell Solutions) containing 3.6 mL basal medium. Then, a single-cell suspension of PDLSCs (2.4 × 106 cells in 7.2 mL basal medium) was injected into the GEM/medium preparation in the LeviTube, and the inoculation program was initiated for 4 h. After inoculation, GEMs were checked via microscopy, and 30.0 mL of basal medium was added to the each LeviTube before the program for cell culture was initiated. The supernatants from PDLSCs grown on microcarriers were collected, and medium changes were performed twice a week. Imaging and cell counting was performed periodically by taking small amounts of cell-seeded microcarriers from the suspension cultures. PDLSCs on microcarriers were stained with 0.1 mg/mL Hoechst 33342 (Applichem, Darmstadt, Germany) and images were captured under UV light and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) filter by a Motic AE31 microscope equipped with a Moticam 2500 camera and Motic Images Plus 2.0 software.
Isolation of EVs
Isolation of EVs was performed using differential centrifugation according to the described protocol [24] with some modifications. All centrifugation steps were performed at 4°C. Supernatants were collected from PDLSC microcarrier cultures (basal medium supplemented with 10% of EV-depleted FCS) every 72 h and centrifuged successively at increasing speeds (300 g for 10 min, 2,000 g for 10 min, then at 20,000 g for 30 min). The final supernatants were ultracentrifuged at 100,000 g for 70 min in Sorvall LYNX 6000 ultracentrifuge, with rotor T29-8 × 50 in oak ridge centrifuge tubes with sealing caps (all from Thermo Fisher Scientific, Rochester, NY), then the pellets were washed in 40 mL PBS and ultracentrifuged again at 100,000 g for 70 min. Final pellets of EVs (exosomal fraction) were resuspended in sterile PBS and stored at −20°C.
Nanoparticle tracking analysis (NTA) was performed with NanoSight LM10 instrument (Malvern Panalytical).
All preparations of EVs were derived from the same PDLSC line. Before the experiment, all EVs preparations were pooled and divided into the single-dose aliquots. The yield of EVs prepared from the 8 mL of the supernatants derived from PDLSCs grown on microcarriers for 72 h was defined as single dose, or 1 activity unit (AU). According to the NTA measurements, single dose of EV contained 6.68 × 109 of EVs.
Transmission electron microscopy of EVs
EV samples for transmission electron microscopy (TEM) have been prepared according to the previously published protocol [24] with some modifications. In brief, EVs in PBS were fixed by adding 4% PFA to a final concentration of 2% and incubated for 40 min on ice. To absorb the sample, Formvar carbon-coated copper grid was floated on a 10 μL drop of EVs suspension for 20 min at room temperature. After adsorption, the grid was washed for 2 min at room temperature in a 100 μL drop of PBS. Then the grid was incubated in a 50 μL drop of 1% glutaraldehyde for 5 min at room temperature. Afterward, eight washing steps were performed (2 min for each) by transferring grid from one drop of distilled water to another. Samples were contrasted on a 50 μL drop of 2% neutral uranyl acetate for 5 min at room temperature in the dark. Thereafter, the grids were incubated on a 50 μL drop of 2% methylcellulose/0.4% uranyl acetate for 10 min on ice in the dark. Finally, grids were taken by stainless steel loop and excess of liquid removed by filter paper. Then, the grids on the same loop were air-dried for 5 min. All incubations were displayed on a Parafilm sheet with the coated side of grid facing the drop. Two grids were prepared under identical conditions for each EV sample. The sample was analyzed immediately with the transmission electron microscope JEOL JEM-2100F High Resolution EM-20023 at 80 kV. The images were captured with an Olympus Quemesa camera, using iTEM 5.2 software.
Confocal microscopy
Microcarrier cultures were washed twice with PBS, fixed with 4% PFA for 20 min at room temperature (RT), washed with PBS, permeabilized with 0.1% Triton X-100 (in PBS) for 15 min at RT, and treated with 1% bovine serum albumin (BSA) (in PBS) blocking buffer for 30 min at RT. After blocking, cells were incubated with a AlexaFluor®®647 phalloidin conjugate (Life Technologies) in 1% BSA for 1 h at RT. Finally, all sections were counterstained with 10 μg/mL DAPI (Applichem) for 5 min at RT in the dark, washed, and mounted in antifading fluorescent mounting medium (DakoCytomation, Huddinge, Sweden). Confocal images were acquired with confocal laser scanning microscope Leica TCS SP8 (Leica Microsystems).
Induction and quantification of osteogenic differentiation
For differentiation experiments, PDLSCs from the third passage were seeded at a density 5 × 103/cm2 in 6-well (35 mm diameter) culture dishes and grown in a basal medium until the cultures reached subconfluence. The osteogenic induction medium consisted of basal medium supplemented with 100 nM dexamethasone, 50 μg/mL ascorbic acid, and 10 mM β-glycerophosphate (all from Sigma). Cell cultures were treated with osteogenesis-inducing medium for 3 weeks, with medium change twice a week. For the assessment of osteogenic differentiation, cellular monolayers were washed with PBS, fixed for 20 min with 4% paraformaldehyde (Sigma) at room temperature, washed with deionized water, and stained with a 2% Alizarin Red S (Sigma) solution adjusted to pH 4.2 for 5 min. Control cultures without the differentiation stimuli were stained in the same manner.
For quantification of Alizarin Red staining, 800 μL of 10% (v/v) acetic acid was added to each well, and the plate was incubated at RT for 30 min with shaking. The monolayer, now loosely attached to the plate, was then scraped from the plate with a cell scraper and transferred with 10% (v/v) acetic acid to a 1.5 mL microcentrifuge tube. After vortexing for 30 s, microcentrifuge tubes were sealed with parafilm, heated to 85°C for 10 min, and kept on ice for 5 min. The slurry was then centrifuged at 20,000 g for 15 min, and 200 μL of the supernatant was removed and transferred to a new 1.5 mL microcentrifuge tube. Then, 75 μL of 10% ammonium hydroxide was added to neutralize the acidic environment. The pH was measured at this point to ensure that it was between 4.1 and 4.5. Aliquots (50 μL) of the supernatant were read in triplicate at 405 nm in 96-well format using opaque-walled, transparent-bottomed plates using a FLUOstar OPTIMA plate reader (BMG Labtech GmbH, Ortenberg, Germany). Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Bonferroni test using MaxStat Pro Statistics Software (Version 3.6). Statistical significance level was set at P < 0.05. Three independent experiments in triplicate were obtained to get statistically meaningful results indicated as significant (*P < 0.05), highly significant (**P < 0.01), and extremely significant (***P < 0.001) values.
Assessment of the effects of LPS and anti-TLR4 blocking antibody on the osteogenic differentiation of PDLSCs
PDLSCs from the third passage were seeded at a density 5 × 103/cm2 in 6-well (35 mm diameter) culture dishes and preincubated in EV-free basal medium for 72 h until the cultures reached subconfluence. Then, the PDLSCs were divided into five groups: PDLSCs cultured in the basal medium (control); PDLSCs treated with osteogenic induction/differentiation medium (OS); PDLSCs treated with 5 μg/mL of LPS derived from Escherichia coli (0127:B8) (Sigma) (OS+LPS). PDLSCs pretreated for 4 h with 0.5 μg/mL of anti-TLR4 blocking antibody (Ab) (HTA125; Abcam) and subjected to the fresh OS induction medium (OS+Ab); PDLSCs pretreated with anti-TLR4 blocking Ab and subjected to fresh OS induction medium plus 5 μg/mL of LPS (OS+Ab+LPS).
In total, PDLSCs received three treatments with LPS and anti-TLR4 blocking Ab—on 4th, 7th, and 10th day after induction of osteogenic differentiation. Every time basal or OS medium was changed in all experimental groups.
Quantification of osteogenic differentiation was performed on 14th and 21st day after induction of osteogenic differentiation.
The effects of EVs on the osteogenic differentiation of PDLSCs
PDLSCs were cultured as described in previous section and then divided into seven experimental groups (Fig. 6B):
PDLSCs cultured in the basal medium (Control);
PDLSCs treated with osteogenic induction medium (OS);
PDLSCs pretreated for 4 h with 0.5 μg/mL of anti-TLR4 blocking Ab and subjected to the fresh OS induction medium (OS+Ab);
PDLSCs in OS treated with 5 AU of EVs (OS+EVs);
PDLSCs pretreated with Ab, then subjected to OS induction medium and treated with 5 AU of EVs (OS+Ab+EVs);
PDLSCs treated with 5 μg/mL of LPS and 5 AU of EVs (OS+LPS+EVs);
PDLSCs pretreated with Ab, then subjected to OS induction medium and treated with 5 μg/mL of LPS plus 5 AU of EVs (OS+LPS+Ab+EVs).
PDLSCs were treated with EVs, LPS, and anti-TLR4 blocking Ab—on 4th, 7th and 10th day after induction of osteogenic differentiation. During every treatment, basal or OS medium was changed in all experimental groups (Fig. 6B).
Quantification of osteogenic differentiation and isolation of RNA were performed on 7th, 10th, and 14th day after induction of osteogenic differentiation.
Monitoring of NFκB activity in human PDLSCs
PDLSCs were transiently transfected with NFκB reporter plasmid pNiFty2-SEAP (InvivoGen) containing endothelial cell-leukocyte adhesion molecule promoter, five NFκB repeated transcription factor binding sites and secreted alkaline phosphatase (SEAP) reporter gene. Nucleofection was performed with 4D-Nucleofector instrument (Lonza) in 100 μL Nucleocuvettes™ (0.3 × 106 cells +2 μg of pNiFty2-SEAP; impulse code FF104).
After 12 h postnucleofection, when necessary, cells were washed and pretreated for 12 h with 0.5 μg/mL of anti-TLR4 blocking Ab (HTA125; Abcam), then cells were washed again and subjected to one of the following treatments: 5 μg/mL of LPS (Sigma); 1 AU of EVs; and LPS +1 AU of EVs.
In total, there were eight experimental points (Fig. 1): control; LPS; EVs; LPS+EVs; Ab; LPS+Ab; EVs+Ab; and LPS+EVs+Ab. SEAP activity in supernatants was determined after 12, 24, 36, 48, 60, and 72 h using colorimetric assay (OZ Biosciences) according to the manufacturer's instructions.
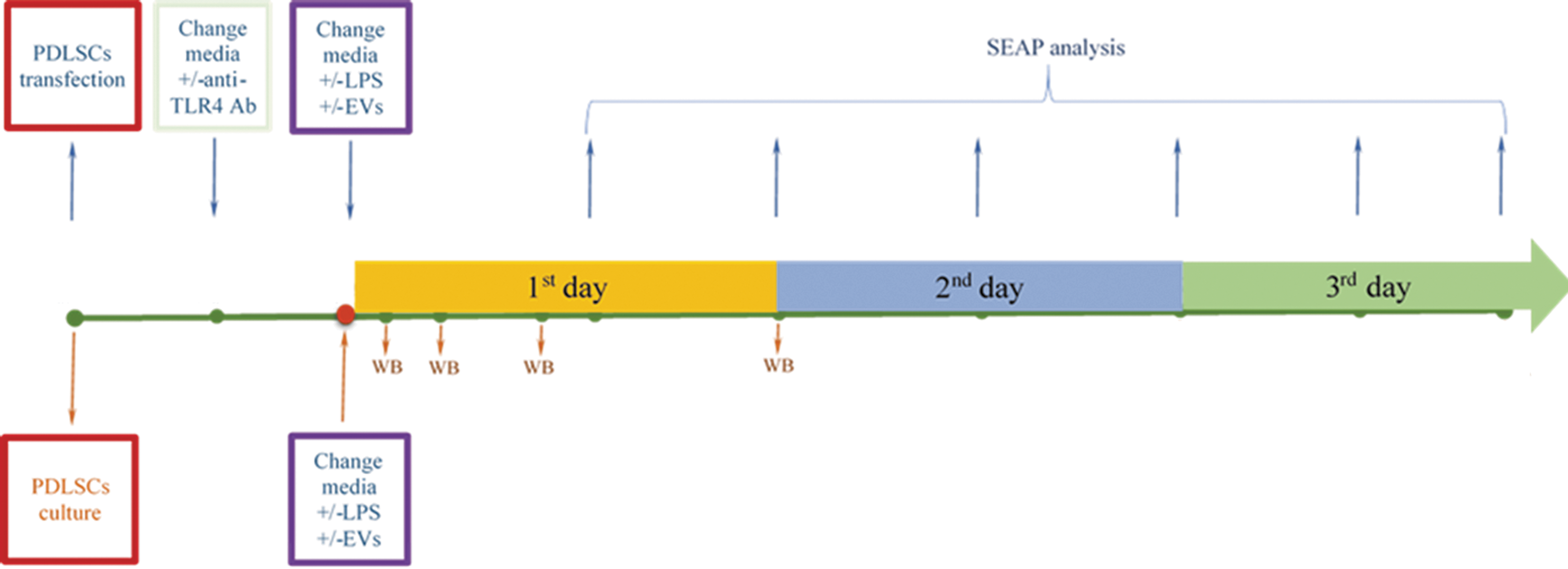
Monitoring of NFκB activity in human PDLSCs. PDLSCs, periodontal ligament stem cells.
Data represent measurements from at least four independent experiments. Data represent mean ± standard error of the mean (SEM) values. Groups were compared by two-way ANOVA followed by Bonferroni's posttest. GraphPad Prism® software version 5.0 (GraphPad Software, Inc.) was used for all data analysis. ***P < 0.001; **P < 0.01; *P < 0.05.
Protein isolation and western blot analysis
For preparation of total cell lysates, cell monolayers were washed twice with cold PBS, pH 7.3, and lysed in Pierce radioimmunoprecipitation assay (RIPA) buffer supplemented with 1× Halt protease inhibitor cocktail for 15 min on ice. Samples were centrifuged at 14,000 g for 30 min at 4°C. Supernatants derived after centrifugation of cellular lysates were aliquoted and kept at −20°C until analyzed. EVs were incubated in RIPA buffer for 30 min on ice and then dissolved in Laemmli sample buffer, boiled, and kept at −20°C until analyzed. Protein concentrations were measured with the NanoPhotometer Pearl (Implen). For western blot analysis, cell and EV lysates diluted in a Laemmli sample buffer were heated for 5 min at 95°C. The same amounts of proteins from EVs and cellular lysates were loaded on Mini-PROTEAN TGX precast gels (Bio-Rad), subjected to electrophoresis in Mini-PROTEAN Tetra cell apparatus (Bio-Rad), then blotted onto a polyvinylidene fluoride (PVDF) membrane in a semidry Trans-Blot Turbo transfer system (Bio-Rad) and blocked for 1 h at room temperature with 5% BSA in PBS containing 0.18% Tween-20 (PBS-Tw). The membranes were then probed with primary Abs against CD63 (Thermo Fisher Scientific) and MFG-E8 (Santa Cruz) for 1 h at room temperature. Alternatively, membranes were incubated overnight at 4°C with Abs against phospho Akt (Ser 473), phospho p38 (Thr180/Tyr182), phospho GSK3β (Ser 9), iIDO-1, β actin (all from Cell Signaling), or phospho p65 (Ser 536) (Thermo Fisher Scientific). After incubation with primary Abs, membranes were washed three times in PBS-Tw. After washing, membranes were incubated further with horseradish peroxidase-conjugated secondary Ab for 1 h at room temperature. Washing procedure was repeated and immunoreactive bands were detected with Clarity ECL western blotting substrate (Bio-Rad) using ChemiDoc MP system (Bio-Rad).
RNA extraction, cDNA synthesis, and real-time polymerase chain reaction
Total RNA was isolated from cultured PDLSCs using the RNeasy Mini kit (QIAGEN, Venlo, Netherlands) according to the manufacturer's instructions with minor changes. Two micrograms of total RNA was converted into cDNA in 35 μL reaction mixture using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific). Quantitative polymerase chain reaction (qPCR) was performed using Maxima SYBR Green qPCR master mix (Thermo Scientific). For qPCRs, we used 0.25 μL of cDNA samples and 0.2 μM of primers per 25 μL of reaction volume. The PCR cycling started from initial 10 min denaturation step at 95°C and was followed by 40 cycles of 15 s denaturation at 95°C, 30 s annealing at 55°C and 30 s elongation at 72°C. Specificity of PCR products was confirmed by the melting curve analysis (95°C for 30 s and melting analysis from 55°C to 95°C with increment 0.5°C for 5 s). For normalization of cDNA levels, the housekeeping gene hypoxanthine guanine phosphoribosyltransferase was used. Primer sequences are listed in the Supplementary Table S2. qPCR experiments were performed in triplicates repeated at least three times.
A statistical analysis of the data was performed using a one-way ANOVA followed by a Dunnett's test using MaxStat Pro Statistics Software (Version 3.6). Statistical significance was set at P < 0.05.
Results
Characterization of EVs derived from microcarrier cell cultures of PDLSCs
NTA analysis revealed that the majority of particles were distributed in the range between 112 and 182 nm (Fig. 3C). EV preparations were enriched with exosomal markers CD63 and MFG-E8 (Fig. 3A). Transmission electron microscopy of whole-mount EVs showed the typical cup-shape appearance of majority of particles, indicating pure EV preparations (Fig. 3B).
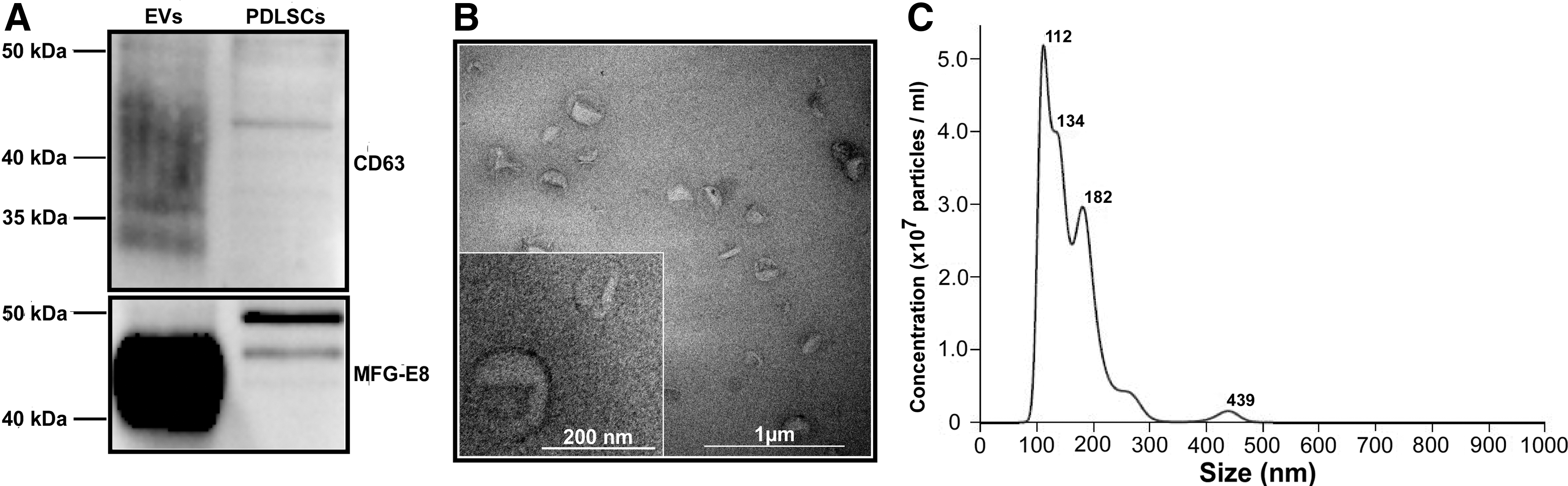
Characterization of EVs isolated from human PDLSCs.
EVs suppress basal and LPS-induced activity of NFκB signaling pathway in PDLSCs
NFκB signaling pathway is a key regulator of the inflammatory response in periodontal tissues.
We therefore investigated whether EVs derived from the microcarrier cultures of PDLSCs may affect LPS-induced activation of NFκB in standard PDLSC cultures. PDLSCs were transiently transfected with NFκB reporter plasmid pNiFty2-SEAP (InvivoGen), and SEAP activity was continuously monitored in the supernatants after 12, 24, 48, 60, and 72 h of treatment with different combinations of LPS, EVs, and anti-TLR4 blocking Ab (Fig. 1). We found that LPS induced moderate upregulation of NFκB activity in PDLSCs after 12, 24, 36, and 48 h (by 25%, 13%, 25%, and 31%, respectively). As expected, anti-TLR4 Ab markedly suppressed LPS-induced NFκB activation. Unexpectedly, treatment with anti-TLR4 Ab alone resulted in a permanent downregulation (by about 34%) of basal NFκB activity in PDLSCs (Fig. 4). EVs also permanently downregulated basal NFκB activity in PDLSCs and have similar effects to anti-TLR4 Ab (Fig. 4). Interestingly, combined treatment with EVs and anti-TLR4 Ab resulted in attenuation of the inhibitory effect on the NFκB activity in PDLSCs. EVs moderately suppressed NFκB signaling pathway in LPS-treated PDLSCs after 12, 36, and 48 h. Combined treatment using LPS and EVs in PDLSCs pretreated with anti-TLR4 blocking Ab potentiated inhibitory effect on the NFκB activity (Fig. 4). These findings demonstrate that EVs suppress basal and LPS-induced activity of NFκB signaling pathway in PDLSCs.
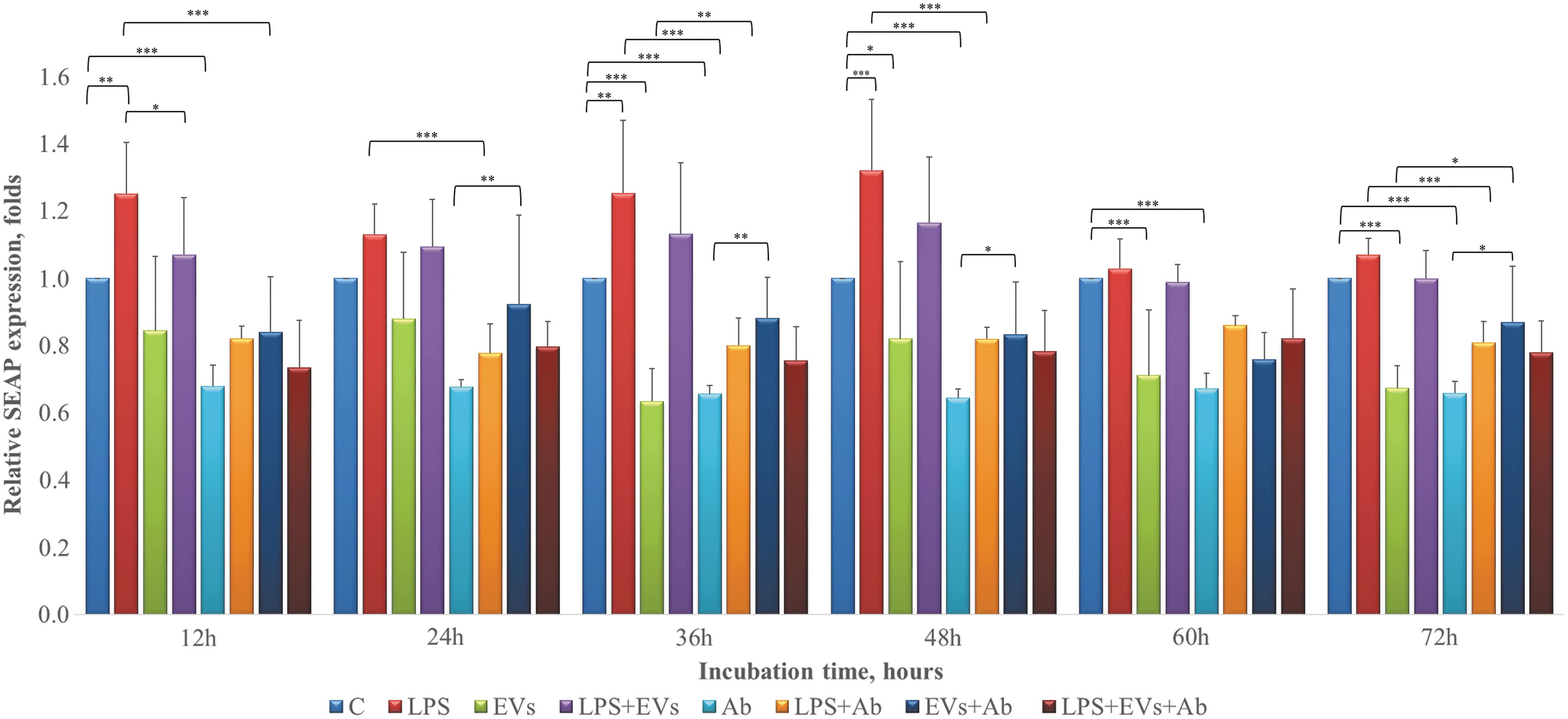
EVs suppress basal and LPS-induced activation of NFκB signaling pathway in human PDLSCs. PDLSCs were transiently transfected with NFκB reporter plasmid pNiFty2-SEAP (InvivoGen). Nucleofection was performed with 4D-Nucleofector instrument (Lonza). After 12 h post nucleofection, when necessary, cells were washed and pretreated for 12 h with 0.5 μg/mL of anti-TLR4 blocking Ab and then subjected to one of the following treatments: 5 μg/mL of LPS, EVs (1 AU), and LPS+EVs. SEAP activity in supernatants was determined after 12, 24, 48, 60, and 72 h using colorimetric assay (OZ Biosciences). Data represent measurements from at least four independent experiments. Data represent mean ± SEM values. Groups were compared by two-way ANOVA followed by Bonferroni's posttest. GraphPad Prism® software version 5.0 (GraphPad Software, Inc.) was used for all data analysis. ***P < 0.001; **P < 0.01; *P < 0.05. ANOVA, analysis of variance; AU, activity unit; LPS, lipopolysaccharide; SEAP, secreted alkaline phosphatase; SEM, standard error of the mean..
Intracellular signaling events in PDLSCs upon exposure to EVs and LPS
We next tested how EVs and (or) LPS affect selected intracellular signaling pathways in PDLSCs (Fig. 5). We found, that EVs significantly increased phosphorylation of Akt (Ser 473) after 30 min, 2 h, and 24 h of incubation, whereas combined treatment with LPS slightly decreased Akt phosphorylation after 2 and 24 h (Fig. 5A). GSK3β represents one of the numerous downstream targets of Akt, and we found that EVs increased inhibitory phosphorylation of GSK3β (Ser 9) after 30 min and 24 h (Fig. 5B). Treatment with LPS alone increased phosphorylation of GSK3β after 30 min and 2 h. Combined treatment with EVs slightly increased phosphorylation of GSK3β after 30 min and 8 h, but decreased after 2 and 24 h (Fig. 5B). EVs also induced phosphorylation of p38 (Thr180/Tyr182) after 30 min of incubation. LPS slightly increased phosphorylation of p38 kinase only after 2 h (Fig. 5C). LPS induced phosphorylation of p65 subunit at Ser 536, which is critical for activation of the canonical NFκB pathway [25]. LPS increased phosphorylation of p65 after 30 min, 2 h, and 8 h (Fig. 5D). After 8 h of incubation, EVs slightly reduced LPS-induced phosphorylation of p65 (Fig. 5D). Neither LPS nor EVs induced expression of iIDO-1 in PDLSCs (Fig. 5E). However, positive controls treated with IFN-γ demonstrated robust IDO-1 expression after 8 h of incubation (Fig. 5E).
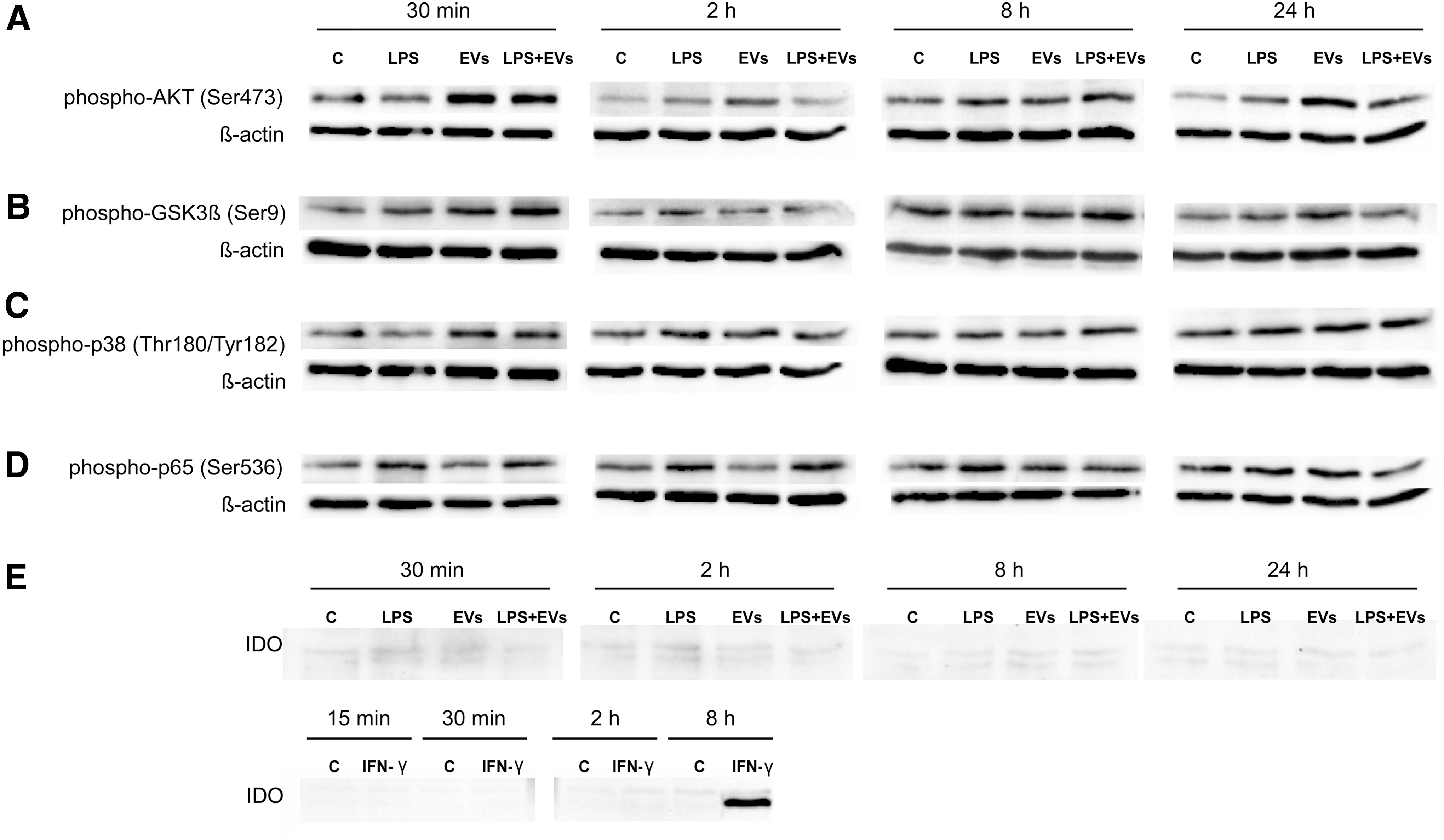
Intracellular signaling events in human PDLSCs upon exposure to EVs and LPS. PDLSCs were treated with EVs (1 AU), 5 μg/mL of LPS, and LPS+EVs for 30 min, 2 h, 8 h, and 24 h, then cell lysates were subjected to electrophoresis, blotted, and the membrane was probed with Abs against
LPS stimulates osteogenic differentiation of PDLSCs
We tested how LPS derived from E. coli (0127:B8; Sigma) affects osteogenic differentiation of PDLSCs. After induction of osteogenic differentiation, PDLSCs were treated with 5 μg/mL of LPS and (or) 0.5 μg/mL of anti-TLR4 blocking Ab. Staining by Alizarin Red S revealed robust calcium depositions in PDLSC cultures after 2 and 3 weeks of osteogenic differentiation (Fig. 6A). Quantification of the Alizarin Red-stained samples demonstrated that after 3 weeks LPS treatment increased osteogenic differentiation of PDLSC cultures by 24.8% (Fig. 6A). Surprisingly, treatment with anti-TLR4 blocking Ab alone significantly decreased mineralization in differentiating PDLSC cultures. Thus, when compared to differentiating controls, treatment with anti-TLR4 blocking Ab decreased Alizarin Red S staining by 26% and 19.6% after 2 and 3 weeks, respectively (Fig. 6A). Anti-TLR4 blocking Ab also slightly suppressed osteogenic differentiation in the LPS-treated PDLSCs (Fig. 6A).
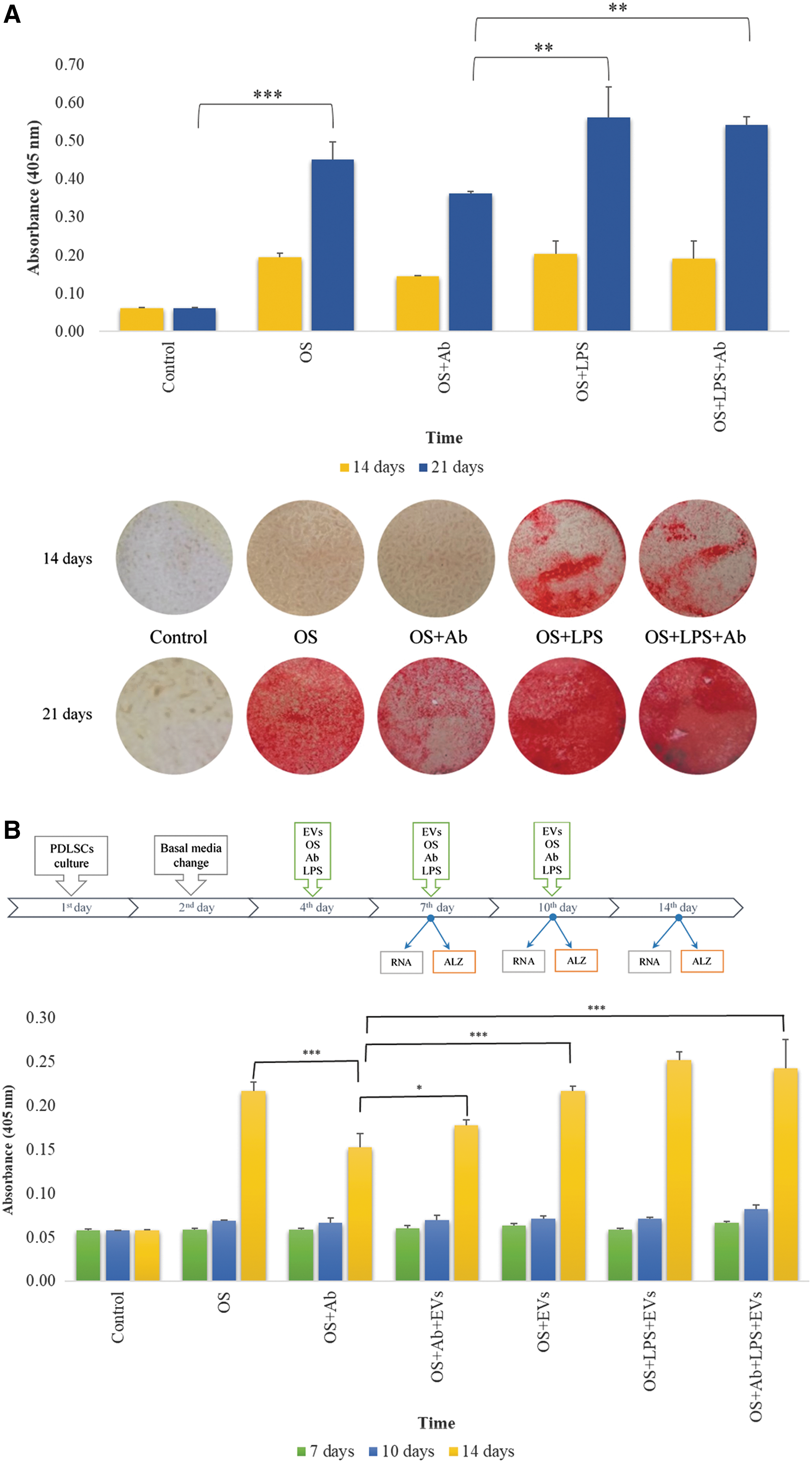
The effects of EVs and LPS on the osteogenic differentiation of PDLSCs.
EVs partially neutralize TLR4 blocking effect on the osteogenic differentiation of PDLSCs
We next tested how EVs affect osteogenic differentiation of PDLSCs. In total, PDLSCs received three treatments with EVs (5 AU each) and anti-TLR4 blocking Ab—on 4th, 7th, and 10th day after induction of osteogenic differentiation (Fig. 6B). Our data demonstrate that EVs did not affect osteogenic mineralization of PDLSC cultures (Fig. 6B). Interestingly, EVs partially suppressed inhibitory effect of anti-TLR4 blocking Ab. Thus, when compared to Ab treatment alone, combined treatment with EVs increased mineralization of PDLSC cultures by 16.3% (Fig. 6B). These results suggest a possible interference between EVs and TLR4 signaling during osteogenic differentiation of PDLSCs.
The effects of EVs on the expression of osteogenesis-related genes in PDLSCs
We used real-time PCR to compare expression levels of some genes important for osteogenesis during osteogenic differentiation of PDLSCs treated with EVs and (or) anti-TLR4 blocking Ab.
Gene expression analyses were performed 7, 10, and 14 days after induction of osteogenic differentiation (Figs. 6B and 7). We found that EVs and anti-TLR4 blocking Ab induced similar effects on the expression of alkaline phosphatase (ALP) and osteocalcin (OCN). Results show downregulation after 7 days followed by upregulation on 10th and 14th day of osteogenic differentiation (Fig. 7). Interestingly, combined treatment with EVs and anti-TLR4 blocking Ab resulted in opposite effects (Fig. 7). Both EVs and anti-TLR4 blocking Ab suppressed BMP2 gene expression on 10th day of osteogenic differentiation, whereas combined treatment resulted in significant upregulation of BMP2 expression (Fig. 7). EVs also upregulated the expression of genes coding bone extracellular matrix proteins osteopontin (OPN), bone sialoprotein (BSP), and cementum protein 23 (CP23) after 10 days of osteogenic differentiation (Fig. 7). Our results show that EVs may act as a potent regulator of genes important for osteogenesis and also interfere with TLR4 signaling in PDLSCs.
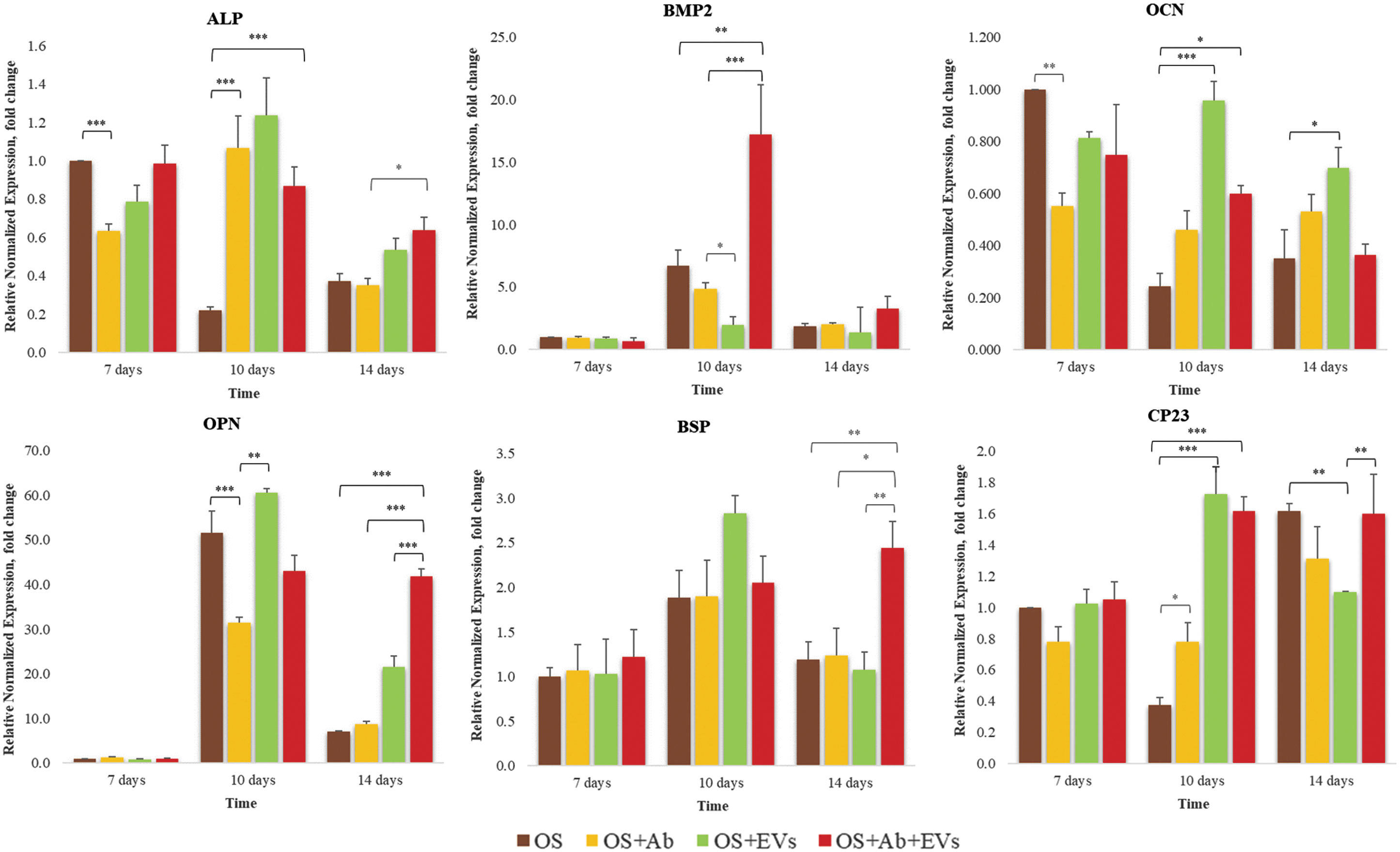
The effects of EVs on the expression of selected osteogenesis-related genes in the PDLSCs. Gene expression levels of ALP, BMP2, OCN, OPN, BSP, and CP23 were analyzed in differentiating (OS); differentiating PDLSCs treated with of anti-TLR4 blocking Ab (HTA125; Abcam) (OS+Ab); differentiating PDLSCs treated with 5 AU of EVs (OS+EVs); differentiating PDLSCs treated with of anti-TLR4 blocking Ab and 5 AU of EVs (OS+Ab+EVs). RNA was extracted after 7, 10, and 14 days of induction of osteogenic differentiation. qPCR experiments were performed in triplicates and repeated at least three times. A statistical analysis of the data was performed using a one-way ANOVA followed by a Dunnett's test using MaxStat Pro Statistics Software (Version 3.6). Statistical significance was set at P < 0.05. qPCR, quantitative polymerase chain reaction.
Discussion
Several lines of evidence indicate that chronic inflammatory environment may affect immunomodulatory function of PDLSCs and promote shift toward proinflammatory phenotype contributing to propagation of periodontitis [9 –11]. Accordingly, suppression of inflammatory response in PDLSCs may represent a promising therapeutic approach. EVs have been shown to display anti-inflammatory [26] and immunosuppressive actions [27] in different tissues and therefore could represent a potent therapeutic tool against chronic inflammation during periodontitis. Currently tooth extraction is needed for the establishment of PDLSC cultures making autologous transplantation impractical for patients who do not need tooth extraction. In addition, PDLSCs derived from the inflamed tissues may have impaired functional properties [10]. In this respect, use of EVs derived from “healthy” allogeneic PDLSC cultures may represent a promising therapeutic approach. In contrast to the cells, EVs represent less risk associated with immune rejection. EVs are also relatively simple and stable systems and therefore are more suitable for the large scale clinical manufacturing [28]. In the present study, we used a commercially available three-dimensional culturing platform and alginate microcarrier cell culture system for the propagation of human PDLSCs. This system allows scaling-up of cell production in a relatively small volume of medium. We have also previously demonstrated that microcarrier culture allows rapid generation of large numbers of PDLSCs prespecified toward an osteogenic-like phenotype [23]. In this study, EVs have been isolated from the supernatants of the microcarrier PDLSC cultures (Fig. 2). According to the size, morphology and characteristic markers of EV fractions contained vesicles that could be classified as exosomes (Fig. 3).
Morphology of PDLSCs grown in bioreactor on gelatin-coated microcarriers. Immunocytochemical staining of PDLSCs grown in bioreactor on gelatin-coated microcarriers (F-actin-red, DAPI-blue). DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride.
NFκB signaling pathway regulates inflammatory response by controlling the expression of pro- and anti-inflammatory genes in many tissues [29]. In this study, we demonstrate that EVs suppress basal and LPS-induced activity of NFκB in PDLSCs (Fig. 4). Interestingly, EVs treatment strongly and permanently suppressed basal NFκB activity, while LPS induced rather moderate response of NFκB signaling in the PDLSCs. We suggest that these effects may be due to the high basal activity of NFκB signaling in PDLSCs. The low reactivity of PDLSCs to bacterial LPS could also be explained by the lack of membrane bound CD14, which is an important component of TLR4 multireceptor complex [30].
We demonstrate that EVs activate PI3K/Akt signaling pathway in PDLSCs (Fig. 5A). Furthermore, LPS suppressed EV-induced Akt phosphorylation after 2 and 24 h of incubation (Fig. 5A). EVs also increased inhibitory phosphorylation of GSK3β (Ser 9), a downstream target of Akt, after 30 min and 24 h of incubation (Fig. 5B). It is known that PI3K/Akt negatively regulates NFκB p65 transactivation via the inactivation of GSK-3β [31]. Our findings are in agreement with reports showing that PI3K/Akt pathway acts as a negative regulator of the inflammatory responses induced by TLRs [31 –33]. We therefore suggest that EVs suppress NFκB signaling by activating PI3K/Akt pathway. However, further studies using pathway-specific inhibitors are needed to confirm these findings. LPS induced phosphorylation of p65 subunit at Ser 536, which is critical for translocation and transactivation of NFκB [25] (Fig. 5A, D). However, EVs only slightly inhibited LPS-induced p65 phosphorylation after 8 h of incubation, suggesting that other signaling pathways are involved in the suppression of NFκB. Interestingly, neither LPS nor EVs induced expression of IDO-1 in PDLSCs (Fig. 5E). This result is in contrast with recent observation that IDO-1 expression in PDLSCs can be upregulated by LPS [8].
Another interesting finding of our study is that anti-TLR4 Ab per se permanently downregulated basal NFκB activity in PDLSCs (Fig. 4). Furthermore, combined treatment with EVs and anti-TLR4 blocking Ab resulted in the attenuation of the inhibitory effect on the NFκB activity suggesting a possible interference through a competition for TLR4 signaling pathway. Indeed, several lines of evidence indicate that TLR4/NFκB signaling pathway is a potential target of EVs. Exosomes derived from human MSCs induced Myd88-dependent response in THP-1 cell line with NFκB-SEAP reporter [34]. The same study showed specific interaction of exosomes with TLR4, but not TLR2 receptors. EVs derived from different types of body fluids induced NFκB- and STAT3-mediated secretion of inflammatory cytokines in TLR-dependent manner [35]. EVs can also act as carriers for the endogenous TLR4 ligands. For example, HSP72 and HSP105 on the tumor-derived EVs surface induced IL-6 secretion of dendritic cells in a TLR2- and TLR4-dependent manner [36]. EVs can carry fibronectin 1 and HSP70 that also can serve as endogenous ligands of TLR4 [34,37]. Furthermore, murine MSC-derived myristoylated tomato tagged EVs colocalized with TLR4 receptors [38]. Although the exact mechanisms by which EVs affect TLR4/NFκB signaling are presently unclear, it is likely that EVs may directly interact with components of TLR4 multireceptor complex and affect signal transduction from the plasma membrane. Our observation that combined treatment with EVs and anti-TLR4 blocking Ab attenuates inhibitory effect on NFκB indirectly supports this possibility. On the contrary, EVs induced long-term effects (up to 72 h), indicating that other regulatory mechanism may be involved. Further studies are needed to clarify the mechanisms underlying inhibitory effect of EVs on the NFκB signaling pathway in the PDLSCs.
There have been contradictory reports in the literature about the effects of LPS on the osteogenic differentiation of PDLSCs. Thus, LPS from E. coli (O55:B5) decreased osteogenic differentiation of human PDLSCs through TLR4 regulated NFκB pathway [39], while another recent report demonstrated that LPS from E. coli (O55:B5) stimulated osteogenic differentiation of human PDLSCs through Wnt/beta-catenin-induced transcriptional activator with a PDZ motif (TAZ) elevation [40]. These discrepancies may reflect differences in the experimental design and use of different concentrations of LPS. We found that treatment with 5 μg/mL of LPS from E. coli (0127:B8) stimulated osteogenic differentiation of PDLSCs. Interestingly, treatment with anti-TLR4 blocking Ab alone significantly decreased mineralization in differentiating PDLSC cultures (Fig. 6A). Since anti-TLR4 Ab also suppresses basal NFκB activity in PDLSCs, we speculate that this mechanism could be also involved in the suppression of osteogenic differentiation. Our results show that EVs did not significantly affect osteogenic mineralization of PDLSC cultures (Fig. 6B). By contrast, it has been demonstrated that NFκB inhibits osteogenic differentiation of MSCs [41]. In this study, authors used selective small molecule inhibitor of IκB kinase (IKKVI) to suppress NFκB signaling pathway in MSCs. By contrast, EVs have complex cargo composition and induce pleiotropic effects in target cells. We therefore speculate that in addition to the suppression of NFκB activity, EVs also affected other signaling pathways important for the osteogenic mineralization. Our observation that treatment with EVs significantly increased expression of ALP, OCN, BSP, and CP23 gene expression, but downregulated BMP2 expression on the 10 days of osteogenic differentiation (Fig. 7) indirectly supports this possibility. Interestingly, EVs partially suppressed inhibitory effect of anti-TLR4 blocking Ab on the osteogenic mineralization of PDLSC (Fig. 6B), suggesting possible interference between EVs and TLR4 signaling during osteogenic differentiation of PDLSCs. Although EVs did not affect osteogenic mineralization of PDLSCs, gene expression studies revealed significant effects on osteogenesis-related genes (Fig. 7). Importantly, in most cases, EVs and anti-TLR4 blocking Ab displayed similar effects on gene expression of selected genes, while combined treatment resulted in opposite effects (Fig. 7). For instance, EVs and anti-TLR4 blocking Ab suppressed expression of BMP2 gene on the 10 days of osteogenic differentiation, whereas combined treatment induced significant upregulation of BMP2 expression (Fig. 7). These results provide further support for possible interference of EVs and TLR4 signaling in PDLSCs.
In summary, our study demonstrates that EVs suppress basal and LPS-induced activity of NFκB signaling pathway in PDLSCs and could potentially be used for targeting of chronic inflammation during periodontitis.
Footnotes
Acknowledgment
This work was partially supported by National Research Programme “Healthy ageing” (grant no. SEN-15090) from Research Council of Lithuania.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.