
Abstract
The secretome of mesenchymal stromal cells (MSCs) is enriched for biotherapeutic effectors contained within and independent of extracellular vesicles (EVs) that may support tissue regeneration as an injectable agent. We have demonstrated that the intrapancreatic injection of concentrated conditioned media (CM) produced by bone marrow MSC supports islet regeneration and restored glycemic control in hyperglycemic mice, ultimately providing a platform to elucidate components of the MSC secretome. Herein, we extend these findings using human pancreas-derived MSC (Panc-MSC) as “biofactories” to enrich for tissue regenerative stimuli housed within distinct compartments of the secretome. Specifically, we utilized 100 kDa ultrafiltration as a simple method to debulk protein mass and to enrich for EVs while concentrating the MSC secretome into an injectable volume for preclinical assessments in murine models of blood vessel and islet regeneration. EV enrichment (EV+) was validated using nanoscale flow cytometry and atomic force microscopy, in addition to the detection of classical EV markers CD9, CD81, and CD63 using label-free mass spectrometry. EV+ CM was predominately enriched with mediators of wound healing and epithelial-to-mesenchymal transition that supported functional regeneration in mesenchymal and nonmesenchymal tissues. For example, EV+ CM supported human microvascular endothelial cell tubule formation in vitro and enhanced the recovery of blood perfusion following intramuscular injection in nonobese diabetic/severe combined immunodeficiency mice with unilateral hind limb ischemia. Furthermore, EV+ CM increased islet number and β cell mass, elevated circulating insulin, and improved glycemic control following intrapancreatic injection in streptozotocin-treated mice. Collectively, this study provides foundational evidence that Panc-MSC, readily propagated from the subculture of human islets, may be utilized for regenerative medicine applications.
Introduction
The therapeutic capacity of mesenchymal stromal cells (MSCs) has been demonstrated in preclinical models for decades [1,2]; however, clinical translation after MSC transplantation is often underwhelming [3]. Limited recapitulation of preclinical efficacy is complicated by transient cell engraftment [4], limited biodistribution [5 –7], and/or the loss of a pro-regenerative secretome [8 –11] during ex vivo expansion. We have previously shown that bone marrow (BM) MSC can be utilized as directable “biofactories” to generate conditioned media (CM) capable of stimulating islet regeneration and normalize glycemic control following intrapancreatic (iPan) injection [12,13]. Although cell-free biotherapeutics mitigate compatibility and cell survival issues associated with cellular therapies [14], additional investigation remains warranted to maximize the potential efficacy of MSC-derived biotherapeutics [15].
Deciphering the phenotypic segregation between regenerative MSC and MSC progeny, such as fibroblasts, is under continuous scrutiny [16 –18]. Indeed, a tissue regenerative secretome may delineate MSC from related nontherapeutic cells [19,20]. We recently reported that MSC derived from human pancreatic tissue preparations [pancreas-derived MSC (Panc-MSC)] demonstrates classical MSC characteristics of plastic adherence, stromal marker (CD73, CD90, CD105) expression, and multipotent differentiation; however, a distinct secretory “fingerprint” was observed in comparison to BM-MSC after proteomic analyses [21]. Despite these inherent differences, both MSC sources generate a secretome that has demonstrated biotherapeutic activity in vivo, supporting that the tissue regenerative secretome of MSC may be similar within distinct tissues [14,22]. As cell-free biotherapeutics develop in early translational studies or clinical trials, it remains necessary to (1) understand mechanisms of cellular communication driving tissue regeneration and (2) to reduce heterogeneity during the production of cell-free biotherapeutics.
The secretome of MSC is enriched with bioactive stimuli derived from amino acids, lipid or nucleic acid moieties that mediate regenerative processes within target cell populations [14,23]. Notably, extracellular vesicles (EVs) encapsulate a portion of these stimuli within a lipid bi-layer protected from enzymatic degradation, thus permitting delivery of cargo to both local and distant cell populations [24 –26]. Classification of EVs from MSC is primarily performed by EV size and/or subcellular origin [27 –29], although the method of EV purification will ultimately determine final composition. Exosomes (50–100 nm) are derived through endosomal biogenesis and are released following fusion of multivesicular bodies with the plasma membrane. Alternatively, microvesicles (100 nm–1 μm) are generated from blebbing of cytoplasmic compartments and localized “pinching” of the plasma membrane [30]. Although interest in the enrichment of therapeutic EVs is increasing, it remains undetermined as to what degree of EV purity will translate to therapeutic efficacy. Because the complexity of tissue regenerative stimuli represents the culmination of many components within [27,31 –33], characterization of the MSC secretome using highly sensitive “-omic” analyses would benefit from simple strategies to deplete or enrich for EVs to improve resolution and protein identification [34]. However, an ideal enrichment strategy should also permit parallel in vitro and in vivo analyses to advance ongoing preclinical efforts to identify therapeutic MSC [12]. Indeed, collaborative research efforts to standardize the MSC secretome and develop novel biotherapeutics will advance the forefront of regenerative medicine research.
In this study, we present an adaptable workflow to validate the production of a MSC-generated biotherapeutic enriched with EVs and bioactive stimuli. Ultrafiltration (UF) was used to concentrate serum-free CM generated by human Panc-MSC, producing an injectable cocktail with minimal manipulation. UF was chosen as a cost-effective method to support the identification and comparative analyses of individual components within the MSC secretome. Within this study, we use multidisciplinary efforts to validate the enrichment of therapeutic stimuli using nanoscale flow cytometry, atomic force microscopy (AFM), and label-free mass spectrometry, combined with a series of in vitro and in vivo functional investigations. Collectively, this study supports ongoing efforts to develop ready-made biotherapeutics for regenerative medicine applications.
Materials and Methods
All studies were approved by the Human Ethics Research Committee at Western University (REB#12935).
UF of MSC-generated CM
Panc-MSC was used to generate serum-free CM, as previously described. Briefly, human islets were obtained through the Integrative Islet Distribution Program and initially established as plastic-adherent colonies in AmnioMAX C-100 for 7 days before nonadherent populations were removed. Panc-MSC was cultured in supplemented AmnioMAX C-100 until 80% confluency and was either harvested for passage or switched to basal AmnioMAX C-100 to generate CM (Supplementary Fig. S1). CM was subsequently processed in one of three ways. (1) CM was concentrated by UF in 3 kDa centrifuge filter units for 45 min at 2,800g. This fraction (bulk) contained both EVs and soluble proteins >3 kDa. (2) CM was concentrated in 100 kDa centrifuge filter units for 20 min at 2,800g. This fraction (EV+) was enriched with EVs, liable proteins, or macromolecular complexes. (3) CM which passed through the 100 kDa filter was subsequently centrifuged in 3 kDa centrifuge filter units for 40 min at 2,800g to produce a subfraction depleted for EVs (EV−). CM was concentrated in 10 mL batches to produce ∼250–300 μL of bulk or EV− CM fractions, whereas 120 μL of the EV+ CM fraction was typically generated from 20 mL of Panc-MSC CM. Protein concentration of each fraction varied between 0.3 and 0.6 μg/μL, by absorbance at 660 nm.
Nanoscale flow cytometry
Bulk, EV+, and EV− CM generated from Panc-MSC were analyzed for the number of microparticles/μL by nanoscale flow cytometry. Serial injections (2, 5, 10, or 20 μL) of each concentrate were diluted to 300 μL with 0.22 μm-filtered phosphate-buffered saline (PBS) within low-attachment 96-well plates and stored at 4°C before analysis at room temperature (RT). EVs were enumerated in duplicate on the Apogee A-60 nanoscale flow cytometer (nFC) with autosampler, capable of EV resolution between 150 and 1,000 nm [35]. One hundred microliters of diluted CM was injected and analyzed at 10.5 μL/min for 1 min. The size of secreted microparticles was estimated using silica beads ranging 110–1,300 nm using properties of large-angle light scatter and small-angle light scatter, as previously reported [35] (Fig. 1A). Silica beads provide a refractive index (λ = 1.42) that is closer to cells (λ = 1.35–1.39) than commonly used polystyrene beads (λ = 1.59). The resolution of exosomes (<100 nm) from background noise was unattainable based on the properties of the A-60 nFC used during this study. To assess protein expression within EVs, EV− or EV+ CM was incubated with conjugated antibodies in a 1:1 ratio that was diluted fivefold with PBS before incubation overnight at 4°C. Following incubation, samples were diluted to 300 μL and analyzed by nFC as described above. Parent cells were stained in parallel and analyzed by flow cytometry using a LSRII flow cytometer at London Regional Flow Cytometry Facility. Conventional flow cytometry and nFC data were analyzed using FlowJo v10.2. Antibodies used are listed in Supplementary Table S1.
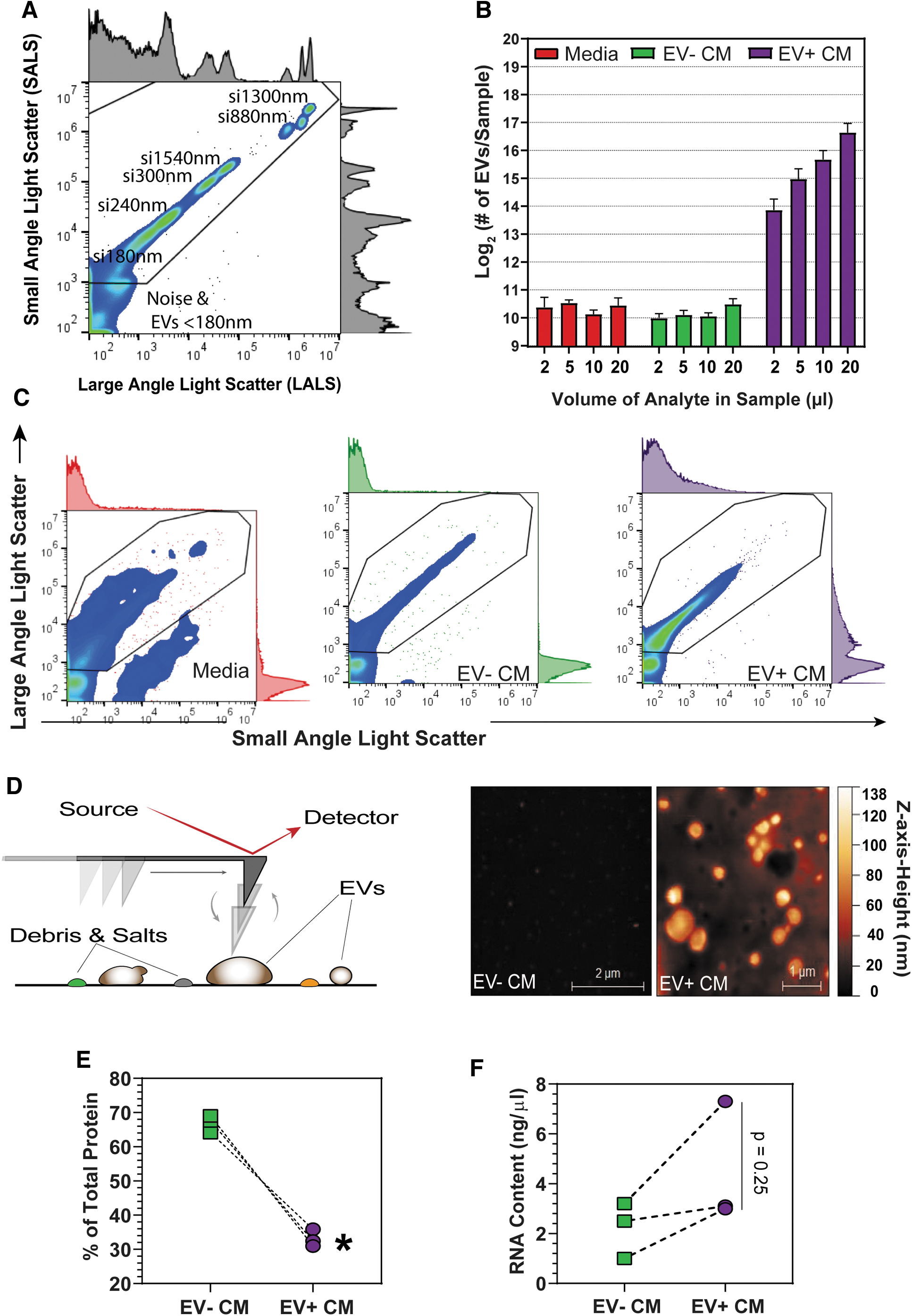
EVs are enriched from the secretome of Panc-MSC by 100-kDa ultrafiltration.
Protein and RNA quantification of Panc-MSC EV− and EV+ CM
CM fractions from MSC populations were analyzed for total protein content with a urea-based lysis buffer [36] containing ammonium bicarbonate, dithiothreitol, and 20% sodium dodecyl sulfate at a 1:1 ratio followed by tip probe sonication. Lysed CM was diluted ∼200-fold with an ionic detergent compatibility reagent (Thermo Fisher) and incubated for 5 min. RNA was extracted from a normalized 50 μg of EV+ or EV− CM using an RNeasy Plus Mini Kit (Qiagen), according to manufacturer's instructions. RNA was eluted in RNAase-free ddH2O and stored at −80°C before quantification using a ND 1000 NanoDrop spectrophotometer (ThermoFisher).
Atomic force microscopy
EV+ and EV− CM fractions were washed twice and diluted 1:10 with 0.22 μm filtered PBS. CM fractions were pipetted as 10 μL microdroplets onto sterile glass coverslips and dried at RT in preparation for AFM imaging. AFM measurements were performed using a BioScope Catalyst AFM (Bruker) equipped with NCL tips (NanoWorld) using Nanoscope software. Images were recorded in noncontact mode in air at a line rate of 1 Hz and processed using the postacquisition software Gwyddion.
Label-free liquid chromatography–tandem mass spectrometry
Methods of protein precipitation, digestion, liquid chromatography–tandem mass spectrometry (LC-MS/MS), and database analyses have been described in detail in previous studies [12,36]. LC-MS/MS settings are outlined in Supplementary Table S2. Annotated enrichment analyses were performed using open-source Metascape [37] (
Cell tracker CMTPX labeling of Panc-MSC EVs
Panc-MSC CM was incubated for 40 min with Cell Tracker CMTPX, a fluorescent dye that becomes a membrane impermeable by intra-EV esterase activity, at final concentration of 1 μM. CMTPX was added to CM and subsequently processed by UF to generate CMPTX-labeled EV+ and EV− CM subfractions. EV− CM was devoid of EVs retaining CMTPX metabolites. Both EV+ and EV− CM subfractions were washed with PBS to remove residual dye before analyses. CMPTX+ EVs were analyzed by nFC (see above) and visualized using oil immersion confocal imaging at 63 × with 4 × digital zoom (Leica TSP8).
Human microvascular endothelial cell uptake of Panc-MSC EVs
Human microvascular endothelial cells (HMVECs) were seeded at 9.40 × 103 cells/cm2 in complete endothelial growth media, EGM-2 (EBM-2 + 5% fetal bovine serum, insulin growth factor, basic fibroblast growth factor, epidermal growth factor, and vascular endothelial growth factor) for 24 h. HMVECs were washed twice with PBS and cultured in serum/growth factor-deprived EBM-2 for up to 48 h. To quantify the rate of uptake of EVs by HMVEC using flow cytometry, CMTPX+ EV+ or EV− CM was added to serum-free HMVEC cultures for up to 12 h. Geometric mean fluorescence intensity of CMTPX was measured at 0.5, 1, and 12 h in harvested HMVEC. Confocal images were acquired at 1 or 12 h after CMFDA-labeled HMVEC was fixed using 10% formalin and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Alternatively, saponin-permeabilized HMVEC was stained for cytoplasmic actin using phalloidin ifluor 488 counterstained with DAPI. EV internalization was visualized through the Z-plane at 0.1 μm increments under 63 × oil immersion and 2 × or 5 × digital zoom (Leica TSP8). Line scan analyses were collected using Fiji software (ImageJ), and three-dimensional (3D) volume projections were performed using LASX software (Leica).
Assessment of tubule formation
To understand the functional relevance of EV-uptake in HMVEC, we assessed HMVEC tubule formation under serum starved conditions in the presence and absence of bulk, EV+ or EV− CM. A measure of 1.2 × 105 HMVECs was cultured on growth factor-reduced Geltrex (LifeTechnologies) in EBM-2 or EBM-2 supplemented with 30 μg of bulk, EV+ or EV− CM generated by BM-MSC or Panc-MSC. At 24 h, four photomicrographs were taken per well, and complete tube formation was enumerated by manual counting in a blinded manner using Fiji software (ImageJ).
Femoral artery ligation and intramuscular injection
Animal procedures to assess vascular regeneration were approved by the Animal Care Committee at the University of Western Ontario according to guidelines of the Animal Use Protocol (2015-012).Unilateral hind limb ischemia was induced in anesthetized (100 mg/kg ketamine/xylazine, maintained with 2% isoflurane, 0.8 L/min) nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice through surgical ligation and cauterization of the femoral artery and vein ligation, as previously described [39]. NOD/SCID mice with unilateral hind-limb ischemia (perfusion ratio <0.1) were injected in a blinded manner 24 h after surgery (day 0) with a total of 80 μg by intramuscular (i.m) injections (20 μL/injection) at three sites in the thigh muscles and one injection in the calf muscle. To serve as a vehicle control, mice received i.m.-injections of basal AmnioMAX C100. Anesthetized NOD/SCID mice were warmed to 37°C for 5 min, and hind limb blood perfusion was measured using Laser Doppler perfusion imaging (LDPI) in a blinded manner. LDPI was performed on 1-, 3-, 7-, 14-, 21-, and 28-days post-transplantation and quantified by comparing the perfusion ratio (ischemic/nonischemic). Endothelial density within thigh muscle tissue cryosections, >100 μm apart, was assessed by CD31 using immunohistochemical detection of 3,3′-diaminobenzidine counterstained with hematoxylin. Immunohistochemical images were acquired using a Leica SP5, and insulin area measurements were performed using ImageJ software.
Streptozotocin-induced hyperglycemia and iPan injection of CM
Animal procedures to assess islet regeneration were approved by the Animal Care Committee at the University of Western Ontario according to guidelines of the Animal Use Protocol (2015-033). Pancreatic β cell ablation and nonfasted hyperglycemia were induced in NOD/SCID mice aged 8- to 10-week old (Jackson Laboratories, Bar Harbor, ME) by intraperitoneal injection of streptozotocin (STZ, 35 mg/kg/day) for five consecutive days, as previously described [13]. On day 10, hyperglycemic (15–25 mmol/L) mice received iPan injection [13] of basal AmnioMAX C-100 or 8 μg total protein of concentrated EV+ or EV− CM subfractions. Injections were performed in a blinded manner. Nonfasted systemic blood glucose levels were monitored weekly for up to 42 days. Stabilization of chronic hyperglycemia was determined by two consecutive nonfasted blood glucose measurements (2 weeks total) below day 10 measurement. At euthanasia, blood was collected through cardiac puncture, and serum was isolated by centrifugation following 15-min incubation at RT to allow for platelet coagulation. Twenty microliters of serum/mouse was analyzed in duplicate for insulin quantitated against a standard curve using absorbance at 450 and 650 nm, according to manufacturer's instructions (ALPCO, Salem, NH). Islet histology using colorimetric insulin staining and β cell mass quantification was performed as previously described [13]. Immunohistochemical images were acquired using a Leica Aperio TS2, and insulin area measurements were performed using Leica software.
Statistical analyses
Analyses of significance were performed by Welch's student's t-test or analysis of variance (ANOVA) for in vitro or in vivo experiments to account for unequal variance between control and treatment conditions. A two-way ANOVA with post hoc Tukey's t-test was used to assess significance of average resting blood glucose levels over time. Rate of hyperglycemia stabilization was determined by Kaplan–Meier Curve and Log-rank analysis with Mantel–Cox test. Outliers were detected with Grubb's test with confidence level at P = 0.0001. GraphPad (Prism) software was used for statistical analysis of in vitro and in vivo experiments. Statistical analyses of proteomic data between EV+ and EV− CM were performed using Perseus software. Proteins were considered significantly enriched (>2-fold) using permutation false discovery (P < 0.05). Bioinformatic analyses were determined by Metascape, FunRich, or Enrichr annotated enrichment using default settings.
Results
UF enriched for EVs secreted by Panc-MSC
Enrichment of EVs from CM [34] or plasma [35] using UF has been previously described; however, UF-based enrichment of EVs from Panc-MSC CM has not been reported. We recently analyzed EV-like structures, isolated from Panc-MSC, using custom fabricated nanohole trapping and surface enhanced Raman spectrometry (SERS) [23]. Thus, we hypothesized that EV+ CM, produced by 100 kDa-UF (Supplementary Fig. S1), would consist of a heterogenous mixture enriched for EVs, soluble proteins, and macromolecular complexes with undetermined therapeutic potential. As an internal control, we concentrated flow through of 100 kDA-UF units in 3 kDa-UF units to produce EV-depleted CM (EV− CM). We validated the enrichment of EVs within EV+ CM by measuring EV content (170–1,300 nm) using nanoscale flow cytometry analyses (Fig. 1A). Positive linear detection of EVs (170 nm–1 μm) was observed with increasing concentrations of EV+ CM analyzed at a fixed acquisition volume, whereas the number EV− CM events did not surpass background measurements of basal media (Fig. 1B, C). The majority of EVs in EV+ CM was estimated to be smaller than 300 nm, relative to silica bead standards (Supplementary Fig. S2A, B). Indeed, AFM validated the enrichment of 3D vesicle structures in EV+ CM (Fig. 1D) with cup or dome-shaped architecture, consistent with previous studies [40,41]. Although parallel EV-like structures were not observed in EV− CM, the majority of total protein mass (∼70%) was contained within EV− CM (Fig. 1E). In contrast, EV+ CM demonstrated a trending increase of enriched RNA content compared to EV− CM (Fig. 1F). Collectively, these data indicate that 100 kDa-UF generated an EV-enriched subfraction of CM containing both protein and RNA content.
Classical EV surface proteins and bioactive stimuli were found exclusively within EV+ CM
We used label-free LC-MS/MS to characterize protein cargo within EV− or EV+ CM. Although EV− CM contained ∼70% of total protein mass (Fig. 1E), the diversity of proteomic cargo was distinct within EV+ CM compared to EV− CM (Supplementary Fig. S2C). For example, 1,458 ± 63.02 versus 877.30 ± 78.19 unique proteins were detected in at least one EV+ CM or EV− CM sample, respectively (Fig. 2A). EV+ CM was enriched with ∼342 proteins not detected in any EV− CM samples, whereas only 18 proteins were exclusively identified in EV− CM (Fig. 2A and Supplementary Tables S3 and S4). EV+ CM was enriched with proteins associated with vesicle lumen, exosomes, or ribosomal components described in previous EV studies [42,43] (Supplementary Fig. S2D). Classical EV markers [43] such as CD9, CD81, and CD63 were exclusively detected within EV+ CM (Fig. 2B). We validated CD81 and CD63 expression on the surface of EVs in EV+ CM and parental cells using nanoscale and conventional flow cytometry (Fig. 2C), respectively. We also detected THY1/CD90 and ITGA6/CD49f expression on secreted EVs and on the plasma membrane of parental cells (Fig. 2C). Proteins enriched in EV+ CM were associated with epithelial to mesenchymal transition (EMT), extracellular matrix (ECM) organization, and interactions at the vascular wall (Fig. 2D and Supplementary Fig. S2E, F). For example, EV+ CM was enriched for several potent cytokines (eg, HGF [44], ANGPT1 [45], Wnt5A [46]), ECM macromolecules (COL16A1/8A1), and integrins (ITGA5/6, ITGB6). In addition to an enrichment in RNA cargo, EV+ CM was also enriched with ribosomal machinery previously described in EVs from adipose-derived MSC [42]. Collectively, these data indicate an enrichment of EV-specific stimuli and extracellular matrix components using 100 kDa-UF.
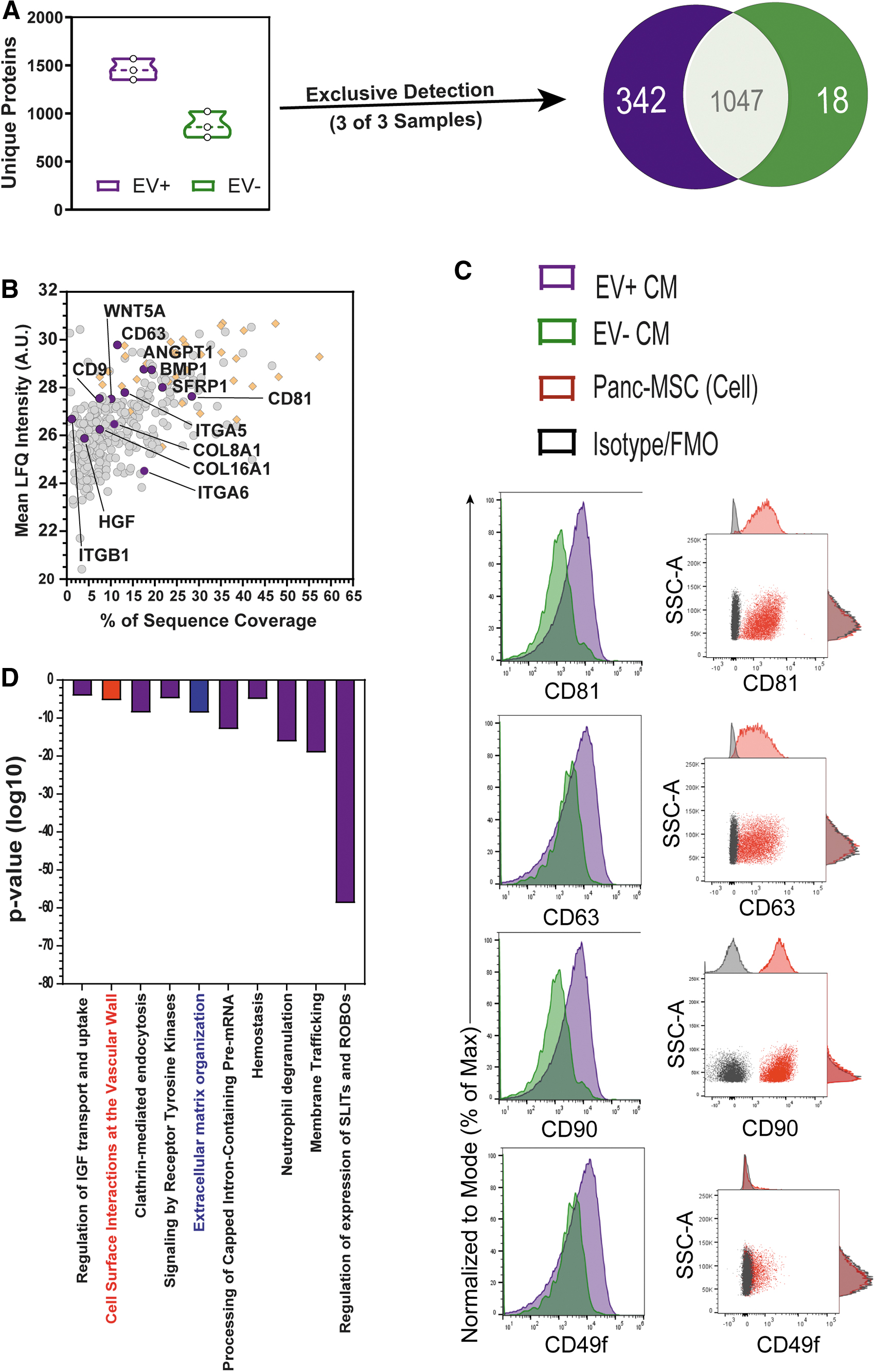
Classical EV membrane markers CD9, CD81, CD63 were exclusively detected in EV+ CM. Despite containing less total protein by mass (Fig. 1E),
Panc-MSC EVs internalized by endothelial cells and support vascular regeneration
Vascular supportive and matrix remodeling mural cells represent the progeny of MSC [1,2,7,47,48]. Indeed, EV+ CM was highly associated with annotations of ECM organization and cell surface interactions at the vascular wall and included several mediators of cell–cell and/or cell–matrix interactions during regenerative processes. EVs express membrane bound integrins and/or integrin ligands [43] to facilitate the docking of EVs to the exterior surface of cell populations expressing complementary machinery [49], exemplified by the capacity of MSC to stimulate regeneration within distant tissues [50]. To visualize EV uptake within endothelial cells, labeling of EVs was accomplished with Cell TrackerTM CMTPX supplementation into cell-free CM before UF (Supplementary Fig. S3A). This approach was used because the enzymatic activity necessary to metabolize CMTPX into a membrane-impermeable product would need to occur within the lumenized core of EVs, as opposed to lumen-deficient lipoproteins. Increased linear detection of CMPTX+ EVs was exclusively observed in EV+ CM using both nanoscale flow cytometry and confocal analyses (Supplementary Fig. S3B–E). EV internalization occurs through several mechanisms, such as phagocytosis [51], macropinocytosis [52], and clathrin- or caveola-mediated endocytic processes [53,54]. To visualize EV internalization, CMTPX+ EVs were exposed to CMFDA-labeled HMVEC in culture and assessed for fluorescence at 12 h using both flow cytometry and confocal microscopy (Fig. 3A–F). CMPTX+ EV–like structures were identified at the z-plane cytoplasmic compartments within HMVEC using phalloidin to mark actin cytoskeletal network (Fig. 3E, F). As expected, CMTPX internalization was not observed within HMVEC supplemented with EV− CM.
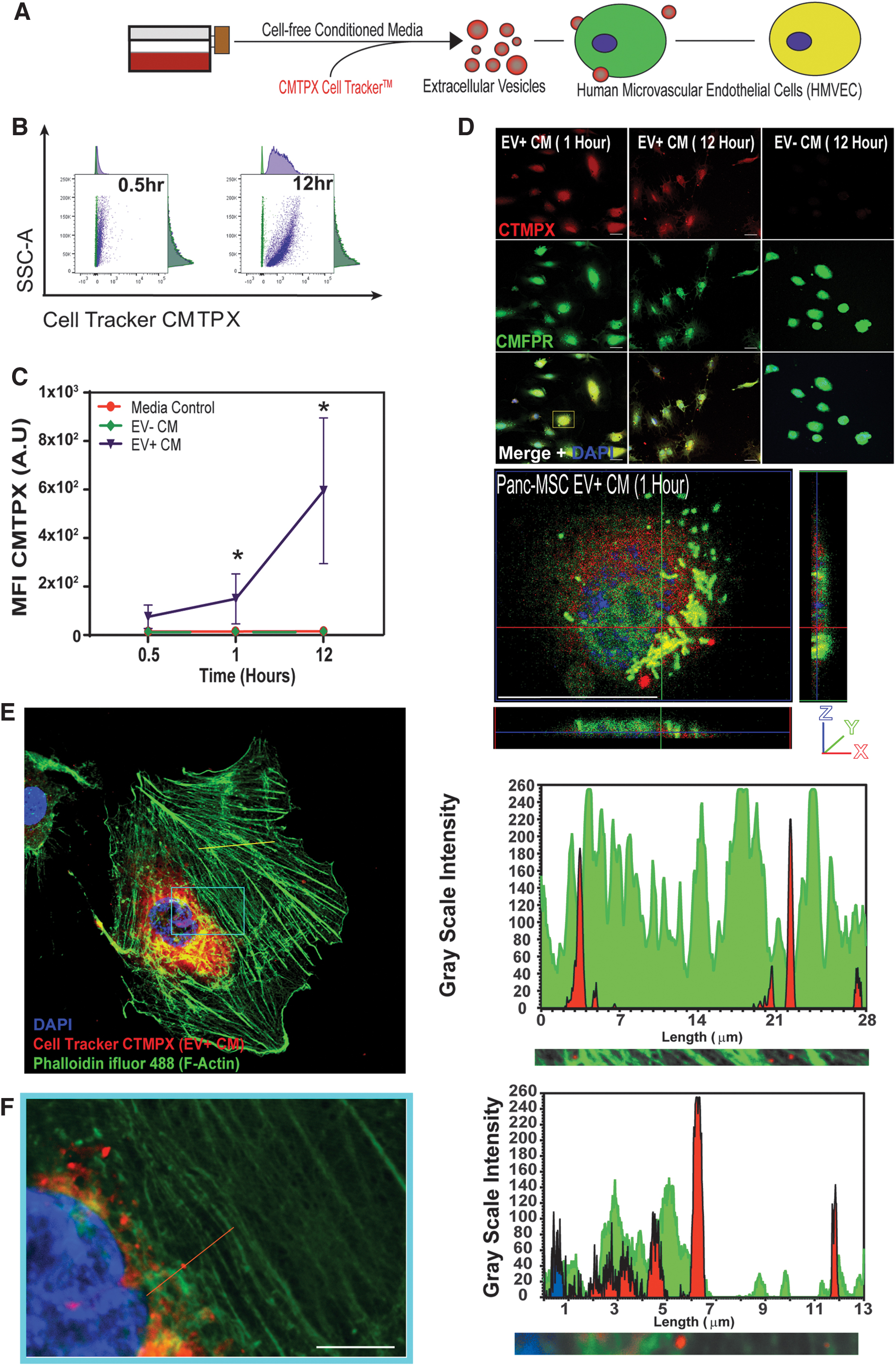
HMVEC uptake of EVs secreted by human Panc-MSC.
EV+ CM demonstrated vascular regenerative properties
High-throughput in vitro assays provide a platform to screen for therapeutic drug [55] or biological agents [56,57] before testing in preclinical models in vivo. We performed endothelial tubule formation assays, under serum conditions in growth factor-free GeltrexTM media, to assess whether EV+ or EV− CM enhanced tubule formation in a suboptimal microenvironment. At 24 h, HMVEC formed comparable number of tubules when exposed to EV−, EV+, or bulk CM (Fig. 4A, B). Notably, all three conditions enhanced tubule formation compared to vehicle controls. At this point, we speculated that both EV cargo and EV-independent stimuli in the secretome of Panc-MSC mediate functional alterations in target cells. Accordingly, we investigated whether subfractions from Panc-MSC could be injected as a biotherapeutic to enhance vascular regeneration in a murine model of surgically-induced unilateral hind limb ischemia (Fig. 4C–E). Compared to vehicle control, i.m. injection of bulk and EV+ CM enhanced blood perfusion above endogenous recovery up to 28 days postinjection (Fig. 4F and Supplementary Fig. S4), as determined by area under the curve (AUC) analyses (*P < 0.05; Fig. 4G). Several mice that received i.m. injections of EV− CM demonstrated modest blood perfusion recovery. However, the perfusion ratio AUC remained statistically comparable to vehicle control. Histological analyses of abductor muscles in ischemic thighs demonstrated a trending increase in CD31 area in mice injected with EV+ CM, although this was found to be statistically comparable to vehicle control mice (Fig. 4H, I). Histological analyses throughout the injected hind limb are necessary to identify vasculogenic mechanisms; however, these studies were considered out of the scope of this study. Ultimately, we considered these data as early evidence of the vascular regenerative potential of EV+ CM.
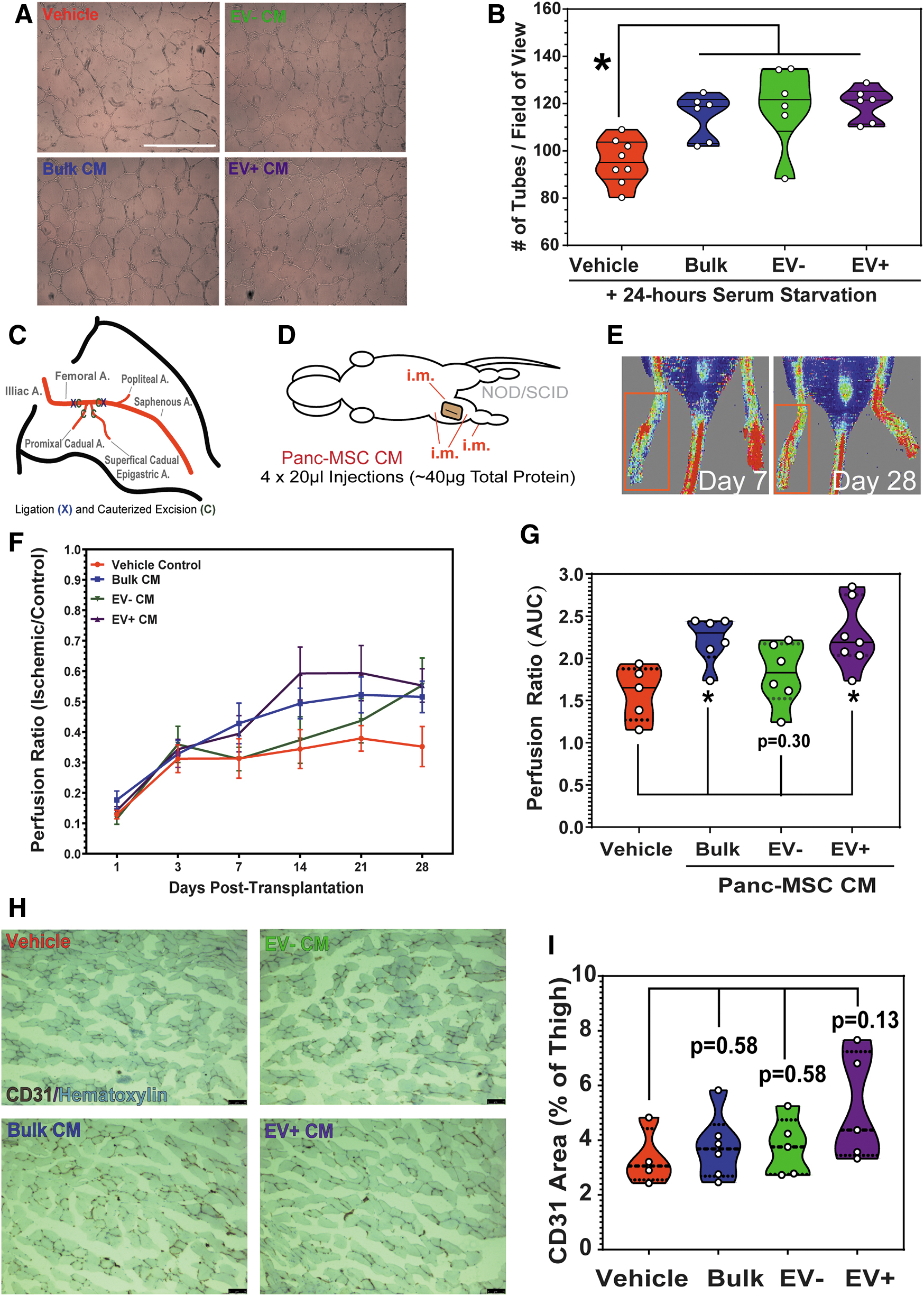
Panc-MSC CM enhances endothelial function in vitro and in vivo.
EV− and EV+ CM were enriched with effectors involved in wound response and tissue regeneration
Considering that in vitro tubule formation was equally supported by both EV− and EV+ CM, we hypothesized that biotherapeutic stimuli were contained within both CM subfractions. Seven hundred forty-three of 1,047 proteins were commonly detected in ≥66% of EV+ and EV− CM samples (Fig. 5A and Supplementary Table S7). To perform statistical comparisons [58], missing value imputation was performed on common proteins without compromising data integrity (Fig. 5B). Permutation-based false discovery rate (P > 0.05) determined that 201 proteins were significantly enriched (≥2-fold) in the EV+ CM subfraction, whereas 153 proteins were significantly enriched within the EV− CM subfraction (Fig. 5C and Supplementary Tables S5 and S6). MSCs support homeostasis and facilitate tissue regeneration through modulation of the immune system, promotion of angiogenesis, and remodeling of the tissue microenvironment [1,2]. With these properties in mind, we speculated that serum starvation in the microenvironment of two-dimensional culture MSC likely mimics acute tissue injury and leads to a “wounding-like response” from cultured MSC [59 –61]. Indeed, 389 proteins detected at comparative levels in EV− and EV+ CM were associated with EMT transitions and angiogenesis (Fig. 5D). Likewise, enriched proteins within EV− and EV+ CM were also associated with EMT or complementary mechanisms of tissue regeneration (Fig. 5E and Supplementary Fig. S5A). For example, we identified several known mediators of the wound response enriched within EV+ CM, including Periostin, TGF-B1, EGFR, and FN1 (Supplementary Fig. S5B). Unexpectedly, EV− CM was enriched for membrane proteins, including PROCR (Supplementary Fig. S5C). It remains unclear whether some proteins can be secreted within or independent of EVs or if the overlap of detected proteins may be an artifact of UF and EV rupture. Alternative methods of EV enrichment and experimentation will be required to elucidate the significance of individual biological effectors.
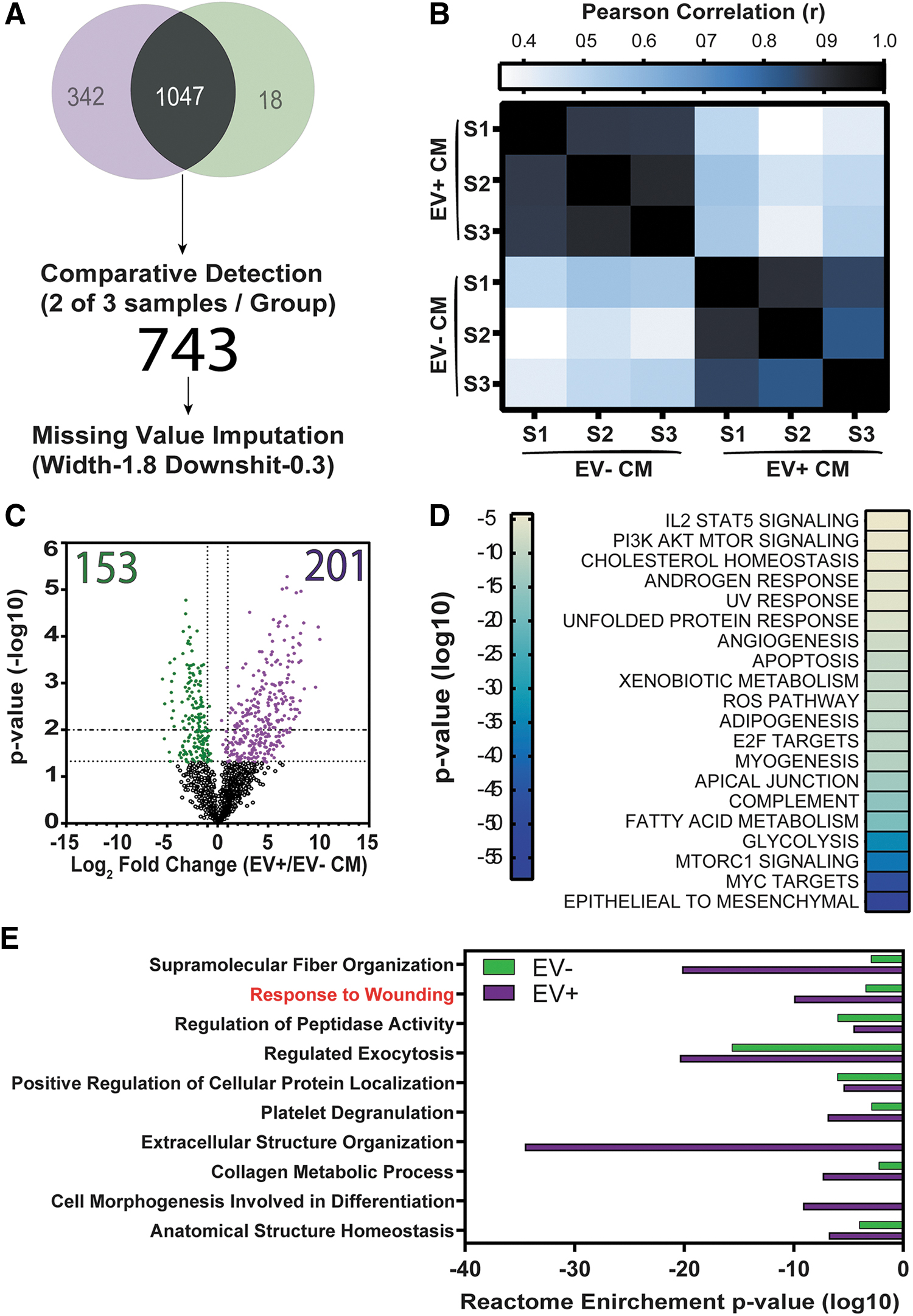
The secretome of Panc-MSC was enriched with mediators of epithelial to mesenchymal transition and response to wound healing annotations.
Intrapancreatic injection of EV+ CM stimulated islet regeneration and stabilized hyperglycemia in STZ-treated NOD/SCID mice
We sought to assess whether EV− or EV+ CM can stimulate complex tissue regeneration in a model where endogenous recovery was minimal or absent without intervention. STZ-induced β cell ablation induces chronic hyperglycemia, with limited regeneration of insulin-secreting β cells [13,62]. Importantly, islet regeneration requires vasculogenic processes to establish intra- and inter-islet vascular networks for functional glycemic control [13,63]. Accordingly, we monitored nonfasted resting and glucose-challenged glycemic levels as a surrogate to assess the recovery of endogenous β cell function and release of insulin into systemic circulation (Fig. 6A). To this end, a single iPan injection of bulk, EV+, or EV− CM reduced nonfasted resting blood glucose within 11 days, stabilized glycemia for up to 32 days postinjection (Fig. 6B and Supplementary Fig. S6), and significantly reduced blood glucose concentration compared to injection of vehicle control, as determined by AUC (Fig. 6C). Furthermore, islet number and β cell mass were significantly increased in the pancreas of mice injected with EV+ CM (Fig. 7A–C). Mice demonstrated an improved physiologic response to glucose bolus injection (Supplementary Fig. S7), and a significant increase in circulating insulin at 32 days postinjection was detected in nonfasted serum from mice injected with bulk, EV−, or EV+ CM (Fig. 7D). Serum insulin levels significantly correlated with resting blood glucose in nonfasted mice at day 32 postinjection (Supplementary Fig. S8A). To compare efficacy across treatment groups, we defined stabilized glycemia as two consecutive weeks with a nonfasted blood glucose measurement less than the day 10 measurement (Supplementary Fig. S8B). These data suggested that bulk CM and both EV− and EV+ CM subfractions were capable of mitigating chronic hyperglycemia. Specifically, iPan injection of EV−, EV+, or bulk CM led to stabilized glycemia in ∼50% of hyperglycemic mice after 11–18 days post-transplantation. Failure to stabilize glycemia may be due to variable retention of CM within the pancreatic microenvironment following iPan injection. Regardless, total β cell mass was increased in every mouse injected with EV+ Panc-MSC CM compared to vehicle controls (Fig. 7C). These data provide evidence that the secretome of Panc-MSC contains bioactive stimuli that support endogenous islet regeneration.
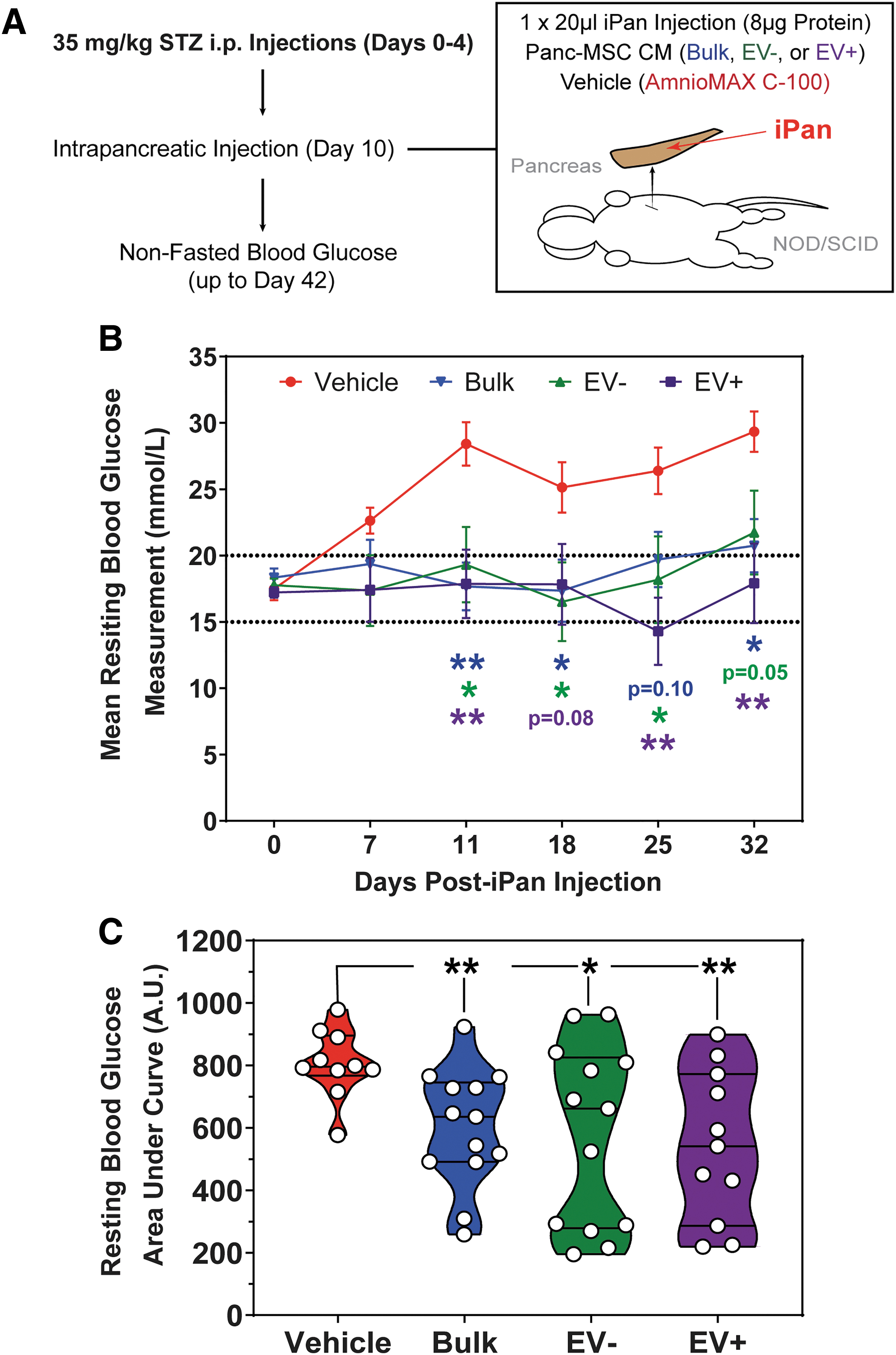
Intrapancreatic injection of Panc-MSC EV+ or EV− CM stabilized blood glucose levels in STZ-treated hyperglycemic mice.
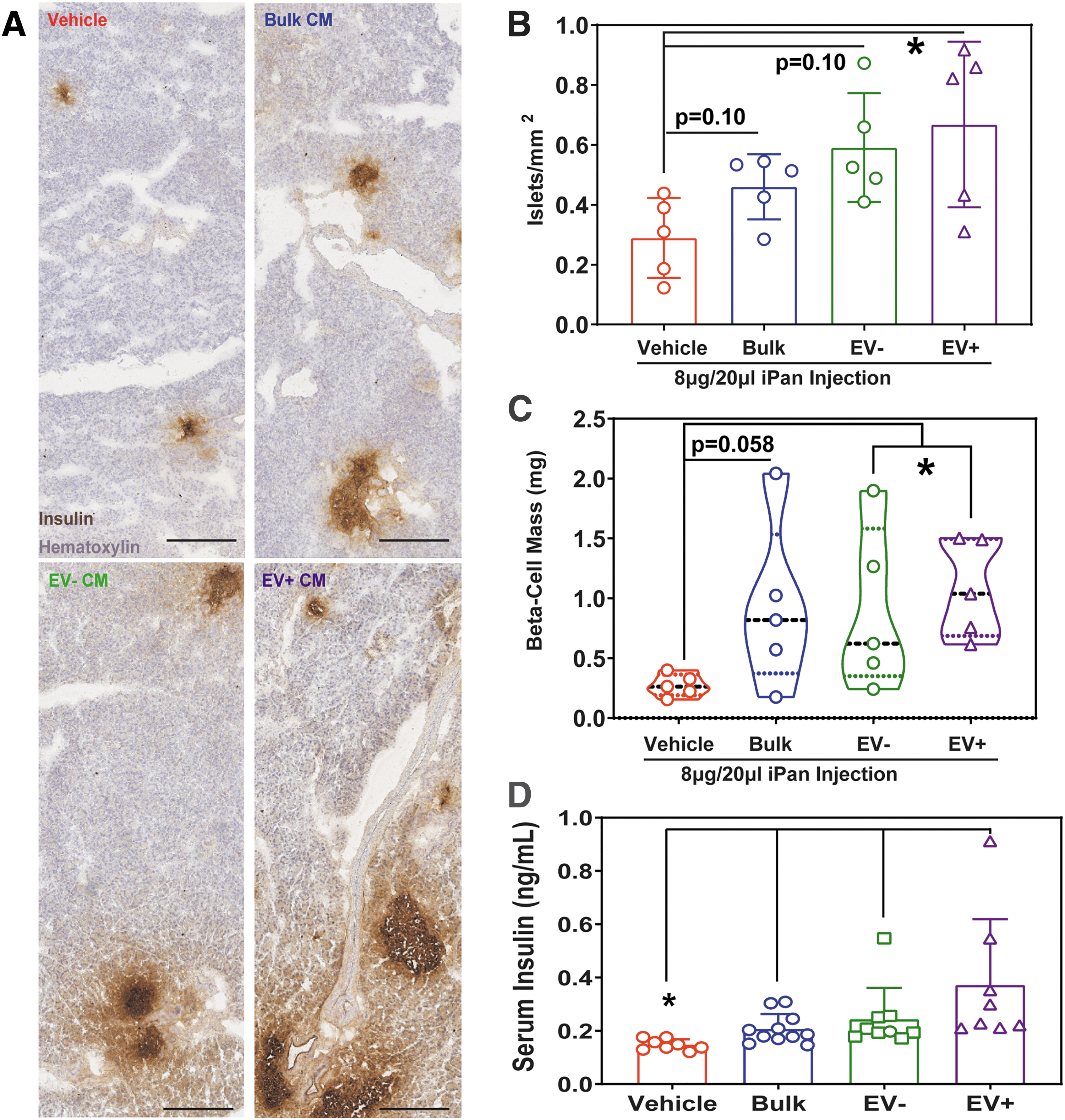
Intrapancreatic injection of Panc-MSC EV+ CM increased islet number, β cell mass, and serum insulin in STZ-treated hyperglycemic mice.
Discussion
This study used a multidisciplinary approach to characterize a potential biotherapeutic CM generated from the secretome of human Panc-MSC. Specifically, UF was used to concentrate the secretome into injectable EV-enriched and EV-depleted subfractions. This method was validated by a series of in vitro analyses using AFM, confocal microscopy, nFC, and conventional flow cytometric analyses. In addition to augmenting endothelial tubule formation in vitro, i.m. injection of EV+ CM enhanced recovery of blood perfusion in mice with hindlimb limb ischemia, supporting previous reports of pro-angiogenic stimuli in the MSC secretome [21,64]. Furthermore, iPan injection of EV− or EV+ CM improved glucose homeostasis, increased circulating insulin, and stimulated islet regeneration in hyperglycemic mice. Nonetheless, using a simple and adaptable workflow, label-free LC-MS/MS was used to characterize therapeutic components of the MSC secretome with vascular and islet regenerative properties.
The therapeutic activity of human MSC has recently been attributed to the release of EVs [27,65] and traditional mechanisms of secreted protein transfer [66,67]. Despite these studies demonstrating a clear role for EVs toward the therapeutic functions of MSC, it remains unclear if increased EV purity enhances biotherapeutic applications [14,22,33,68,69]. Witwer et al. recently highlighted the collective and international focus to improve guidelines for classifying MSC and MSC-generated EVs designed for therapeutic applications [27], specifically acknowledging that the microenvironment of culture [70] and/or source of origin [21,71 –73] can lead to variable phenotypic and functional characteristics from expanded MSC [21,59]. Indeed, unwanted differentiation of MSC ex vivo can lead to a loss of therapeutic activity [14,20,74], such as the acquisition of a senescent secretome and increased production of pro-inflammatory cytokines [20,75]. EVs may also be enriched from the MSC secretome by techniques, such as ultracentrifugation [76], density/size-based fractionation [77,78], and/or proprietary commercial separation kits [79]. Each of these methods obtains the purity necessary to elucidate biotherapeutic stimuli that are truly exclusive or independent of EVs [80]; logistic considerations may limit scalability required for high-throughput screens. While our parallel efforts continue to develop individual EV capture for high-throughput SERS analyses [23], herein we demonstrate the utility of 100 kDa-UF as a robust method to enrich for soluble and EV-contained bioactive stimuli from MSC CM. The unavoidable caveat to this method is the copurification of proteins or protein complexes independent of EVs. Moreover, the nature of UF may rupture unstable EVs, as indicated by the detection of vesicle proteins and surface markers (eg, CD201) in EV− CM. In agreement with Whittaker et al., our data suggest that therapeutic stimuli are likely not exclusive to MSC-derived EVs [69].
The attractiveness of EVs for therapeutic applications is, in part, due to the expression of surface proteins that mediate targeted uptake by recipient cells [27,81]. EV proteins interact with the extracellular matrix or recipient cells [81]. For example, integrins commonly expressed on MSC [82] can facilitate EV docking and uptake in recipient cells [81,83 –85]. Our proteomic and nFC data suggest that EVs generated by Panc-MSC expressed integrins in addition to the classical EV-markers CD81 and CD63. Exposure to EV+ CM showed accumulation of CMPTX-labeled EVs at the plasma membrane and within the cytoplasm of endothelial cells in vitro. This uptake correlated with augmented tubule-forming function in a “serum-starved” environment in vitro and enhanced recovery of blood perfusion following vascular injury in vivo. Although we demonstrated tissue regenerative functions of injected EV+ CM, the contribution of specific EV-bound cargo toward functional response(s) observed in recipient cells (ie, endothelial or pancreatic β cells) remains to be specified. Nonetheless, our data suggest that effectors independent of EVs likely contribute to tissue regenerative mechanisms in vivo. This phenomena has been exemplified in previous studies, such as EV-independent induction of tumor progression [86] and Argonaut 2 complex shuttling of microRNAs in human plasma [87].
The secretory action of tissue-resident MSC populations can influence the molecular and cellular make-up of the microenvironment, including the infiltration hematopoietic cells following tissue injury, such as macrophage polarization following the lateral transfer of MSC cargo [88 –90]. Indeed, our previous study demonstrated a strong chemotactic activity of Panc-MSC CM using the directed in vivo angiogenesis assay [21]. Thus, future studies are required to determine how the secretome of Panc-MSC influences the tissue regenerative properties of infiltrating hematopoietic, stromal, or progenitor cells. At this time, we also cannot specify whether the therapeutic activity demonstrated by injection of EV+ CM was also impacted by nucleic acid cargo; bioactive lipids [91] or metabolites [92] may also contribute to the therapeutic functions of EVs. As the field of cell-free biotherapeutics advances, we envision the requirement of multiplatform analyses to elucidate the biotherapeutic cargo within MSC EVs.
A collection of previous studies has demonstrated the potential of engineering the secretome of MSC to meet therapeutic needs [13,36,74]. Several stimuli with potent tissue regenerative functions were exclusively detected in the EV+ CM. For example, Wnt5a signals through both the canonical and noncanonical signaling pathways to activate pro-angiogenic pathways in vivo through endothelial cells [46] or through the polarization of infiltrating monocytes [46,93]. Furthermore, impaired neovascularization during diabetes can be reversed by supplementation of HGF [44]; and VEGF-A, ANGPT1, and POSTN have each demonstrated pro-regenerative function in previous studies [94 –96]. Taken together, an opportunity to refine the production of cell-free biotherapeutics exists. We have recently demonstrated that supplementation of the Wnt-pathway inhibitor (CHIR99021) to BM-MSC can enhance islet regenerative functions of injected bulk CM [13], whereas other studies have demonstrated that 3D bioreactor culture [74] or hypoxic priming [97] may enhance the secretome of BM-MSC. In addition, directed engineering of MSC toward molecular targets in recipient cells has demonstrated applicability. For example, a recent clinical trial (NCT03608631) has engineered BM-MSC to produce EVs with shRNA against pancreatic cancer cells with KRASG12D mutations [98]. Certainly, the development of cell-free biotherapeutics will require consideration of the therapeutic cargo within and independent of EVs and elucidate biological mechanisms activated in recipient cells.
Conclusion
Collectively, the knowledge obtained in this study will lay the foundation to elucidating the role of human pancreatic mesenchymal cells during islet regeneration. Specifically, this work demonstrates that Panc-MSC generates a secretome with islet regenerative properties, and bioactive stimuli are distributed throughout the Pan-MSC secretome. In the murine pancreas, progenitor-like cells with mesenchymal characteristics have been recently described in both embryonic [99] and adult [100] pancreatic tissues that contribute to islet regeneration through mechanisms of cell–cell communication. Given that our Panc-MSCs are derived from pancreatic islet preps and express putative progenitor markers (eg, CD201), further research is warranted to elucidate the identity of these cells in situ.
Footnotes
Acknowledgments
This work was supported by operating grants from the Canadian Institute of Health Research (CIHR MOP# 378189 and 426890) and Juvenile Diabetes Research Foundation (2-SRA-2015-60-Q-R) awarded to D.A.H. and a NSERC Discovery grant and Canadian Foundation for Innovation grants awarded to G.A.L.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by operating grants from the Canadian Institute of Health Research (CIHR MOP# 378189 and 426890) and Juvenile Diabetes Research Foundation (2-SRA-2015-60-Q-R) awarded to DAH, and a NSERC Discovery grant and Canadian Foundation for Innovation grants awarded to GAJ.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.