
Abstract
Lung fibrosis is a progressive fatal disease, and the underlying mechanisms remain unclear. These involve a combination of altered fibroblasts, excessive accumulation of extracellular matrix, inflammation, and aberrant activation of epithelial cells. Previously, we showed that high-fat diet (HFD) induces lung inflammation, aberrant activation of stem cells, and lung mitochondria impairment. Therefore, we hypothesized that HFD-induced changes would influence lung fibrosis. Mice were fed standard diet (SD) or HFD, administered bleomycin, then examined for fibrosis severity and the start of repair 3 weeks after injury, and for fibrosis repair/resolution 6–9 weeks after injury. At 3 weeks, no significant differences in inflammation and fibrosis severity were observed between SD- and HFD-fed mice. However, infiltration of alveolar type (AT)-2 cells and bronchioalveolar stem cells (BASCs) into the fibrotic areas (the start of repair) was impaired in HFD-fed mice. At 6 weeks, SD-fed mice showed near-complete resolution/repair of fibrosis and inflammation, while HFD-fed mice still showed residual fibrosis and inflammation. Infiltration of the fibrotic areas with AT2 cells was observed, but very few BASCs were detectable. At 9 weeks, mice from both groups showed complete resolution/repair of fibrosis and inflammation, indicating that HFD induced delayed, rather than failed, resolution of fibrosis and alveolar repair. To further confirm the direct role of enhanced fatty-acid oxidation (FAO) in delayed resolution/repair, we administered etomoxir, a FAO inhibitor, to HFD-fed mice for 3–6 weeks after bleomycin injury. Inhibition of FAO abolished the HFD-induced delay in alveolar repair and fibrosis resolution at both time points. In conclusion, after a fibrosis-inducing injury, HFD slows resolution of fibrosis/inflammation and delays alveolar repair by slowing the contribution of AT2 stem cells and abolishing the contribution of BASCs in the repair process. FAO activation appears to be involved in this delay mechanism; thus, inhibiting FAO may be useful in the treatment of lung injury and fibrosis.
Introduction
A
It is now widely believed that part of the various disadvantages of HFD is due to an effect on stem cells and their niches. For example, the effect of HFD on the intestine is mediated through the activation of intestinal stem cells, which enhances the risk of intestinal tumor formation [4]. Exposing adult mice to several weeks of HFD also resulted in a decreased number of skeletal muscle stem cells and promoted muscle fibrogenic transformation [5]. With the increasing change to a “western diet pattern” worldwide and its high fat content, it is essential to understand the various influences of HFD on body organs and cells and its interaction with various diseases.
Recently, we showed that HFD induces aberrant activation of lung stem cells, low-grade inflammation, and impairment of lung mitochondria, a phenotype similar to aging-induced changes in the lung [6]. Stem cell dysfunction, lung inflammation, and mitochondria are all implicated in the development of lung fibrosis, and “exaggerated” lung aging may promote aberrant lung epithelium development in fibrotic lung diseases [7]. Ge et al. previously examined the effect of HFD on the lungs in the context of allergen-induced asthma [8]. They found that HFD induced airway hyper-responsiveness and TGF-β1 levels, but reduced allergen challenge-induced lung inflammation and cytokine, chemokine, and eicosanoid levels [8]. The source of TGF-β1 induction was in the epithelial cells, rather than the inflammatory cells, indicating a direct effect of HFD on lung epithelium independent of its effect on inflammation. TGF-β1 is a potent inducer of collagen expression and promotes subepithelial fibrosis.
Accordingly, in this study, we hypothesized that HFD will increase the severity and/or delay the repair of the lung after a fibrosis-inducing injury. We examined the HFD-induced changes in the response of the distal lung epithelial stem cells that are known to be the major contributors to lung repair/aberrant repair after a fibrosis-inducing bleomycin injury. These stem cells are the alveolar type (AT)-2 cells, which repair the alveoli; the club cells, which repair the airways but might overproliferate and descend to the lung parenchyma, causing aberrant repair and forming honeycombs and bronchiolization; and the bronchioalveolar stem cells (BASCs), which contribute to the repair of both terminal bronchioles and alveoli.
Materials and Methods
Fibrosis induction and diet protocols
Animal experiments were approved by the Institutional Animal Care and Use Committee of Keio University. C57BL6J mice were used in all experiments. All mice (n = 75) were kept on standard chow (CE-2; Clea Japan) diet [standard diet (SD)] ad libitum from weaning until 8 weeks of age. They were then split into two groups: an HFD group, which was switched to a high-fat chow (HFD32; Clea Japan) ad libitum (group 3; n = 15), and a group that continued on standard chow (n = 60). Four weeks later, half of the SD-fed mice continued on an SD (groups 1 and 4; n = 30, 15/group), while the other half was switched to HFD (groups 2 and 5; n = 30, 15/group). All mice in groups 1, 2, and 3 were administered bleomycin (1 μg/g in 60 μL of water; Nippon Kayaku), while mice in groups 4 and 5 received vehicle. Five mice from each group were sacrificed at 3, 6, and 9 weeks (Fig. 1A and Supplementary Fig. S1). Two ad hoc groups (n = 10/groups) were added later. They were similar to group 2, but one was treated with etomoxir (25 μg/g every other day starting on the same day as bleomycin administration; CSN Pharma) or vehicle for 3 and 6 weeks (Supplementary Fig. S1). The body weights of all the mice were measured and recorded weekly. More detailed description of the mice management and diet contents is given in the Supplementary Data.
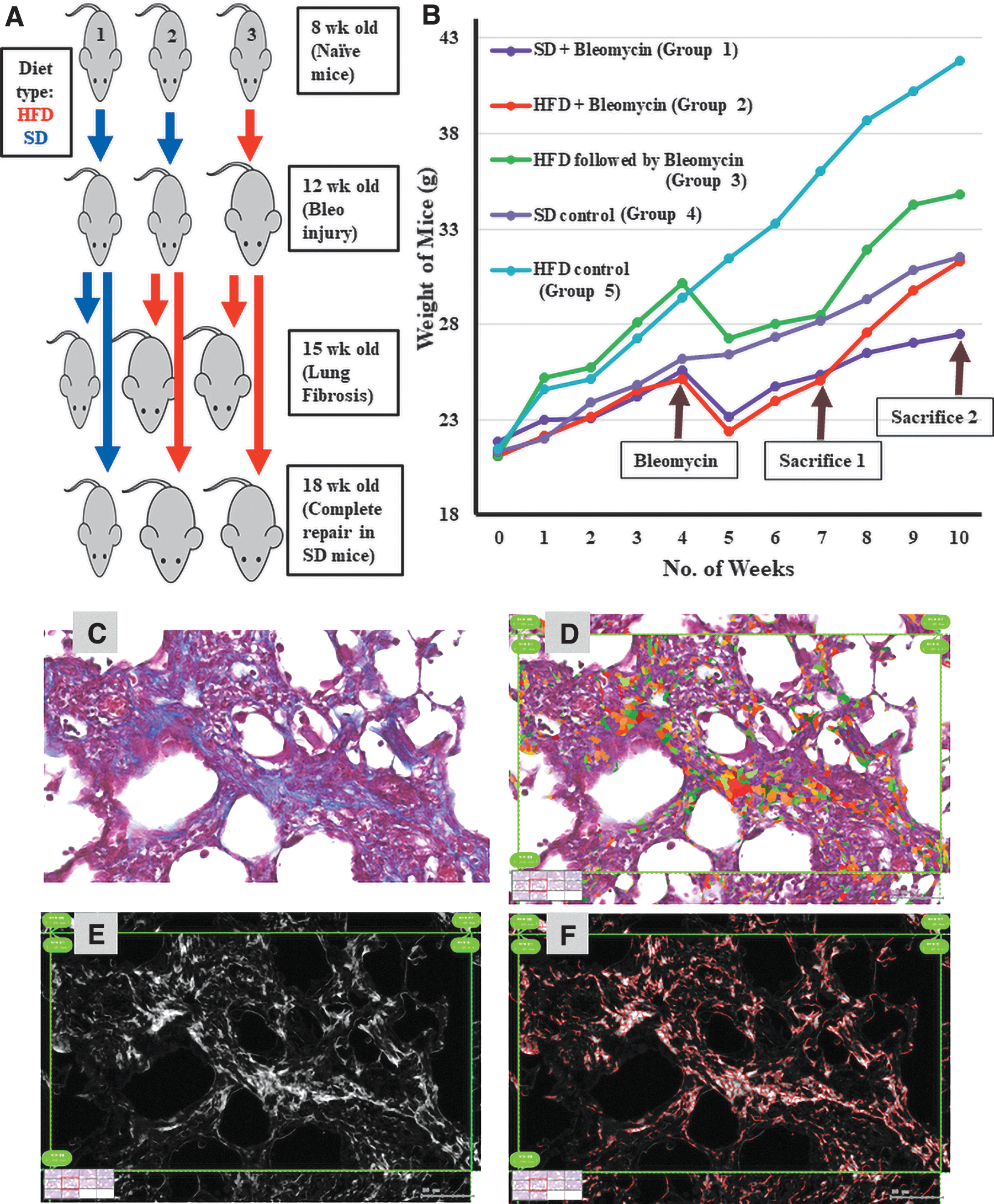
An animal model for the study of the effect of HFD on lung fibrosis severity and repair.
Lung collection and tissue histology
Mice were anesthetized and exsanguinated as previously described [6,9]. A brief description is given in the Supplementary Data.
Polymerase chain reaction and quantitative real-time polymerase chain reaction
Total RNA was extracted from whole lung tissues using the RNeasy Kit (Qiagen) according to the manufacturer's protocol. Complementary DNA (cDNA) was synthesized from total RNA using the High Capacity RNA to cDNA Kit (Thermo Fisher Scientific). The real time polymerase chain reaction was prepared using the SYBR FAST ABI Prism quantitative polymerase chain reaction Kit (Kapa Biosystems) according to the manufacturer's protocol. Gene expression levels were analyzed using the StepOnePlus Real-Time PCR System (Applied Biosystems) and the 7500 Fast Real-Time PCR System (Applied Biosystems). Mouse β-actin was used as an endogenous control for normalization. The primers used are shown in Supplementary Table S1.
Assessment of fibrosis severity and scoring systems for fibrosis resolution and accumulation of alveolar macrophages
Lung sections were stained with hematoxylin and eosin (H&E) and Masson's trichrome stain, where the fibrotic tissue stains blue. Digital images were captured from all slides using a NanoZoomer digital imaging system (Hamamatsu Photonics). All digital images were processed using the HistoQuest software (Tissue Gnostics). First, all nonblue (nonfibrotic) areas were extracted from the images. The remaining blue (fibrotic) areas were then converted into black and white shades based on their density. The area and density of these shades were then quantified and analyzed to reflect variations in the severity of fibrosis. A detailed description of the fibrosis resolution scoring method is given in the Supplementary Data and Supplementary Fig. S2.
Quantification of BASC
Cells that were double positive for CC10 and Sftpc and located within a circle with a diameter of 400 μm with bronchioalveolar duct junctions (BADJs) at its center were counted.
Quantification of AT2 and club cell infiltration of fibrotic areas
When more than 20 Sftpc+/CC10− cells were observed within the fibrotic area using a high-power field (HPF; × 400), the area was scored as positive for AT2 infiltration. When more than 10 Sftpc−/CC10+ cells were observed within a fibrotic area using HPF, the area was scored as positive for club cell infiltration.
Statistical analysis
Data were quantified from the abovementioned animals/samples/images, and averages and standard deviations were calculated and compared using the two-tailed Student's t-test. Statistical significance was set at P < 0.05.
Results
Establishing the HFD protocols and the lung fibrosis-induced injury and repair model
Instilling bleomycin (and less commonly silica or asbestos) into mouse lungs through the trachea causes alveolar epithelial cell injury, inflammation, and fibrosis that reach its peak 3 weeks after injury. In most cases, inflammation clears and alveoli are repaired after a further 3 weeks and fibrosis resolves. However, in a few cases, the alveolar epithelium fails to regenerate, and instead, airway (club) cells expand, leading to bronchiolization of the lung parenchyma and the formation of honeycomb cysts [10].
We used this animal model of alveolar injury, fibrosis, and repair to identify the influence of HFD on the extent of fibrosis and the ability of the alveolar stem cells to repair the lung after an injury. After weaning, all mice were fed a SD until they were 8 weeks old (Fig. 1A, B), and then groups 3 and 5 were switched to HFD for 4 weeks. When all mice were 12 weeks old, bleomycin (1 mg/kg) was administered to all mice in groups 1, 2, and 3, while groups 4 and 5 received vehicle. Group 2 was switched to HFD, while groups 1 and 4 were continued on the SD. Three weeks later (15 weeks old, peak fibrosis time point), half of the mice in each group were sacrificed, the right lungs were collected for histological assessment, and the left lungs were processed for RNA and protein isolation. After a further 3 weeks (18 weeks old, repair time point), all remaining mice in all groups were sacrificed and similarly processed (Fig. 1A, B). Groups 2 and 3 will allow us to differentiate between the effects of “starting” a HFD concomitantly with the lung injury, and the effect of receiving the injury, while “already obese,” which means that the influences of HFD on naive lungs, which we reported in our previous study, are already in place [6].
To verify the efficiency of the diet protocols and the success of the bleomycin injury (which is known to induce 5%–15% weight loss in the first week), we recorded the body weight of all mice in all groups weekly (Fig. 1B). As expected, mice fed HFD (groups 3 and 5) gained more weight than mice in the SD-fed groups. After the initial bleomycin injury-induced weight loss, mice in groups 1, 2, and 3 started to gain weight again, with HFD-fed groups (2 and 3) gaining weight faster than SD-fed group 1 (Fig. 1B).
Examining the effect of HFD on fibrosis severity, inflammation, and epithelial cells 3 weeks after bleomycin injury (maximum fibrosis/inflammation and early epithelial repair time point)
Several methods for assessing fibrosis severity in the bleomycin animal model have been described, including histopathological analysis, microcomputed tomography, and measurement of collagen content. The current gold standard for assessing lung fibrosis is histopathological quantification. It is performed visually on Masson's trichrome-stained paraffin lung sections [11]. However, this method is semiquantitative, and identification of the various degrees of severity, ranging from 0 (normal lung) to 8 (total fibrous obliteration), is subjective (due to the patchy distribution of fibrosis within the same lung) and can vary markedly among researchers. Several digital imaging analysis methods have also been proposed to overcome the limitations of visual scoring systems [12].
In this study, we used HistoQuest image analysis software to automate the quantification of fibrosis severity in lung sections. Lung sections from all the mice sacrificed at the “peak fibrosis time point” were stained with H&E and Masson's trichrome stain and captured digitally on the NanoZoomer, where the severity of fibrosis was quantified (Fig. 1C–F). As shown in Supplementary Table S2, quantification of the images from all samples showed that a HFD, whether started with the injury or before it, had no significant influence on the severity of fibrosis, compared to the SD.
As both M1 and M2 macrophages are known to play critical roles during the development and control of fibrosis [13], we also examined histological sections from all groups for variations in macrophage phenotype and density of infiltration of the fibrotic areas and their neighboring nonfibrotic areas. As shown in Supplementary Fig. S3, regardless of diet type, bleomycin-treated lungs (groups 1, 2, and 3) had a significantly higher infiltration of macrophages, both in the fibrotic (Supplementary Fig. S3A–C) and the neighboring nonfibrotic areas (Supplementary Fig. S3D–F), compared to the vehicle-treated lungs (groups 4 and 5) (Supplementary Fig. S3G, H). In the bleomycin-treated lungs, 30%–60% of the macrophages were arginase-1 positive (M2 macrophages), and no significant difference was detected among the groups based on the diet protocol. Vehicle-treated lungs showed only 1%–3% arginase-1 positive cells (Supplementary Fig. S4A–I).
Gene expression of the fibrosis and inflammatory markers Tgf-b1, Col1a1, Col3a1, Tgf-b2, Tnf-a, IFN-g, GM-CSF, RANTES, IL-1a, IL-2, IL-6, IL-12, HIF-1a, CTGF, MMP2, and MMP9 was also examined in all lung samples. Although many of them showed a trend toward higher expression in bleomycin-treated groups, as expected, no significant differences were detected among the bleomycin-injected lungs of groups 3, 4, and 5 (Supplementary Fig. S4).
We then looked for signs that showed the start of the regeneration of damaged alveolar epithelial cells. Initial repair of the alveolar epithelium typically requires AT2 stem cells to proliferate and “invade/infiltrate” fibrotic foci. In contrast, aberrant repair will appear in the form of fewer AT2 cells within the fibrotic foci, accompanied by enhanced proliferation of the nonepithelial mesenchymal cells (ie, fibroblasts and inflammatory cells), and the airway club cells, which will descend into the lung parenchyma, causing bronchiolization of the lung parenchyma and honeycomb cyst formation.
Quantification of the presence of AT2 cells within fibrotic areas showed that 70% (14 out of 20 foci examined) of the SD-fed mice fibrotic foci were invaded with AT2 cells (more than 20 cells/HPF) versus 40%–45% (8 in group 2 and 9 in group 3; out of 20 foci examined in each group, P value <0.05 between group 1 vs. group 2 or 3) of the HFD-fed mice foci, indicating a delayed alveolar repair process (Fig. 2A–G and Table 1). Quantification of the presence of club-like cells within fibrotic areas showed that only 30% (6 out of 20 foci examined) of the SD-fed mice fibrotic foci were invaded with club-like cells (more than 10 cells/HPF) versus 80%–85% (16 in group 2 and 17 in group 3; out of the 20 foci examined in each group, P value <0.05 between group 1 vs. group 2 or 3) of the HFD-fed mice foci, indicating an aberrant repair process (Fig. 2A–G and Table 1).
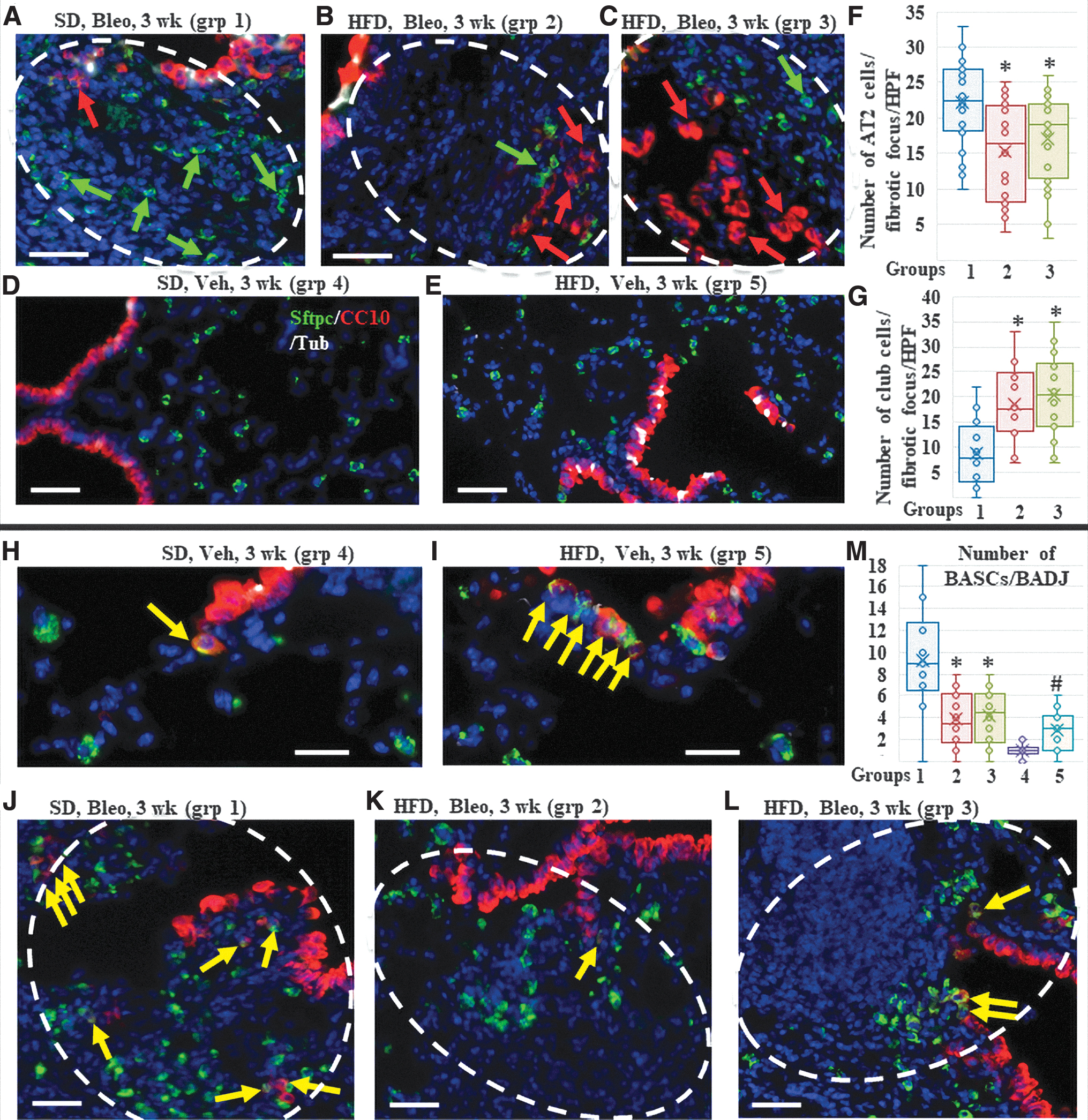
Assessment of the lungs 3 weeks after bleomycin (peak fibrosis time point).
Summary of the Histological Changes Observed in the Lung Parenchyma at the Different Time Points
Fibrosis: +++: extensive (more than 40% of the area), ++: moderate (25%–40% of area), +: mild: (10%–25% of area), (−): absent (less than 10%). AT2 cells: infiltration into the fibrotic areas (start of repair). BASCs: presence at the bronchioalveolar duct junction or infiltration into nearby fibrotic areas (contributing to the repair process). Club cells: descent into fibrotic and/or repaired parenchymal areas (bronchiolization of the lung parenchyma, ie, aberrant repair). PCNA+: proliferating cells. aSMA+: myofibroblasts (indicative of ongoing fibrosis).
AT, alveolar type; BASC, bronchioalveolar stem cell; Eto, etomoxir; HFD, high-fat diet; SD, standard diet; ttt, treatment; Veh, vehicle.
Recently, stem cells that coexpress markers of club and AT2 cells (CC10 and Sftpc) and reside in the vicinity of BADJ, called BASCs, have been shown to be a major contributor to the repair of bleomycin-induced injury [14,15]. To identify whether HFD influences the contribution of BASCs at the start of regeneration of the damaged alveolar epithelial cells, we examined and quantified cells that were double positive for CC10 and Sftpc at BADJ and their nearby fibrotic foci. We found that in the vehicle-treated control mice, HFD resulted in a small but significant increase in the number of BASCs (0–2 cells/BADJ, average: 1 ± 0.76 in the SD-fed mice vs. 1–6 cells/BADJ, average: 2.9 ± 1.8, P < 0.05). Conversely, in the bleomycin-injured fibrotic mice, SD-fed mice showed a significantly larger number of CC10+/Sftpc+ cells at BADJs and within the parenchymal fibrotic foci, compared to a few or no cells in the HFD-fed mice [0–18 cells/BADJ, average: 9.3 ± 4.31 in SD-fed mice vs. 0–8 cells/BADJ, average: 3.9 ± 2.5 (group 2) and 4.2 ± 2.42 (group 3), P value <0.05 (groups 1 vs. 2), <0.05 (groups 1 vs. 3), and 0.36 (groups 2 vs. 3)] (Fig. 2H–M).
To further confirm the involvement of each of these cells (AT2, club, and BASCs) in the ongoing repair/remodeling process, proliferating (PCNA+) cells around and within the fibrotic foci were also quantified in triple-stained slides. Significantly higher numbers of proliferating AT2 and BASCs were detected in SD-fed mice than in HFD-fed mice. Conversely, quantification of proliferating (PCNA+) club cells in terminal bronchioles and nearby parenchymal fibrotic areas revealed significantly higher cell numbers in both HFD-fed mice groups than in the lungs from the SD-fed mice group. In contrast, quantification of the proliferating nonepithelial cells (ie, fibroblasts/myofibroblasts or inflammatory cells) showed no significant difference among groups 1–3, indicating a similar ongoing fibrotic process (Fig. 3A–G) (Table 2).
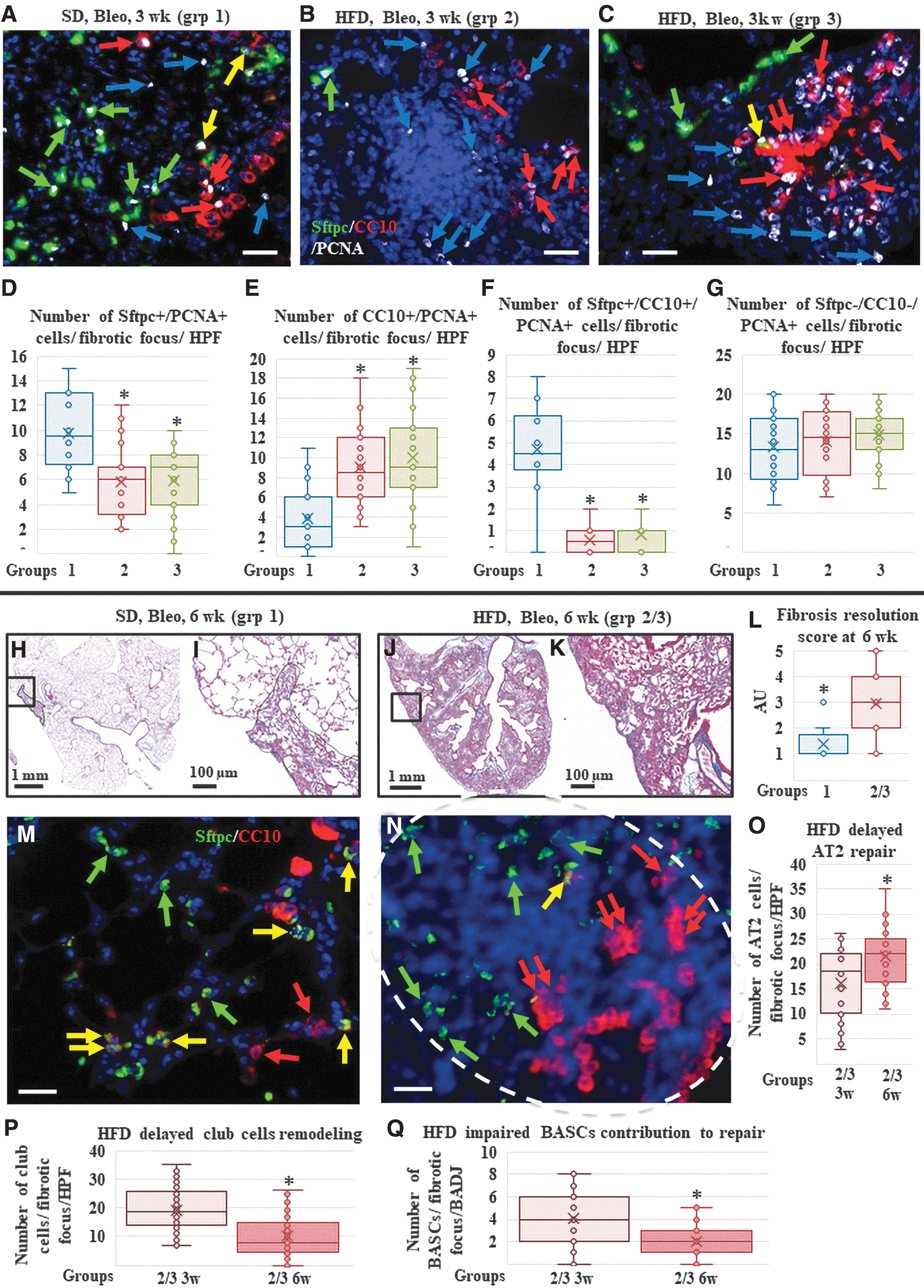
Assessment of the lungs 3 and 6 weeks after bleomycin (fibrosis and repair time points).
Proliferating Cell Types at the Different Time Points
Proliferating AT2 cells: ongoing repair of the parenchyma. Proliferating BASCs: contributing to the ongoing repair of the parenchyma and airways. Proliferating club cells in the parenchymal areas, not in the airways: ongoing remodeling and bronchiolization of the lung parenchyma, that is, aberrant repair. In the airways: ongoing repair of the airways. Nonepithelial cells: inflammatory cells, fibroblasts/myofibroblasts, indicative of ongoing fibrosis and inflammation.
Collectively, these data indicate that HFD does not influence fibrosis severity, number and phenotype of macrophages, or the expression of fibrosis and inflammatory markers, but significantly impaired AT2 cell proliferation and infiltration of the fibrotic foci and reduced the number of BASCs near and within the fibrotic foci, while increasing aberrant repair by club cells. As no significant differences were found between mice groups 2 and 3 (HFD started with fibrosis induction or before it), for simplicity, data from these two groups were pooled together and presented as HFD-fed mice versus SD-fed mice.
Examining the effect of HFD on the resolution of fibrosis and inflammation and on alveolar repair 6 weeks after bleomycin injury (minimal residual fibrosis/inflammation and epithelial repair time point)
At the 6-week time point, lung histological examination by H&E and Masson's trichrome staining showed near-complete resolution of inflammation, fibrosis, and return of the airways and alveolar compartments into near-normal histology in the SD-fed bleomycin-injured mice. In contrast, lungs from bleomycin-injured HFD-fed mice groups showed significantly higher residual inflammation, fibrosis, and areas with aberrant epithelial repair in the form of bronchiolization and honeycomb cyst formation (Fig. 3H–L). Immunofluorescent staining showed that the density of lung macrophages returned to levels that were comparable to those of the vehicle-treated lungs in most of the bleomycin-injured SD-fed mice with very few arginase1+ cells (group 1), while in the HFD-fed mice, many macrophages were still seen in and around the residual fibrotic foci, with 10%–40% of the inflammatory cells showing arginase1+ staining (M2 macrophages) (Supplementary Fig. S5).
Epithelial repair was also explored by immunostaining lung sections with epithelial markers. The SD-fed mice lungs had near-normal histological appearance and distribution of AT2 cells (Sftpc+/CC10−) in the alveoli. Club cells (Sftpc−/CC10+) were seen lining apparently normal airways and were rarely seen in the lung parenchyma. Importantly, many BASCs (Sftpc+/CC10+) were detectable at the BADJ, in the apparently normal (repaired) lung parenchyma, and within a few areas with residual fibrosis (Fig. 3M and Supplementary Fig. S6). In contrast, HFD-fed mice lungs showed increased “invasion/infiltration” of the fibrotic foci with AT2 cells (Sftpc+/CC10−; similar to what was seen in the SD-fed mice at 3 weeks, and significantly higher than the HFD-fed mice at 3 weeks). Club cells (Sftpc−/CC10+) were seen lining the apparently normal airways, but several were also still seen in lung parenchyma forming typical honeycomb cysts and “bronchiolization.”
Quantification showed a significant decrease in parenchymal club cells compared to that observed in the HFD-fed mice at 3 weeks, indicating ongoing remodeling to remove the “unnecessary” club cells from the parenchyma (Fig. 3M–Q and Supplementary Fig. S6). Interestingly, significantly fewer BASCs (Sftpc+/CC10+) were seen in the BADJ, both in the apparently normal (repaired) lung parenchyma and in the areas with residual fibrosis (Fig. 3M–Q and Supplementary Fig. S6).
PCNA staining revealed that in SD-fed mice, the overall number of PCNA+ cells was lower than that at the 3-week time point. Most PCNA+ cells were AT2 and club cells. Fewer SPC-CC10-PCNA+ cells (mesenchymal, ie, fibroblasts and inflammatory cells) were observed, suggesting a further resolution of fibrosis. Proliferation of BASCs was also low. In the HFD-fed mice, the number of PCNA+ cells was still abundant, similar to the 3-week time point. PCNA+ cells not only included AT2 cells in the areas of repair but also several club cells (in areas with bronchiolization and honeycombing) and SPC-CC10-PCNA+ cells (mesenchymal, ie, fibroblasts and inflammatory cells) suggestive of continued fibrosis/inflammation. PCNA+ BASCs were almost absent (Fig. 4A–F, Supplementary Fig. S7, and Supplementary Table S2).
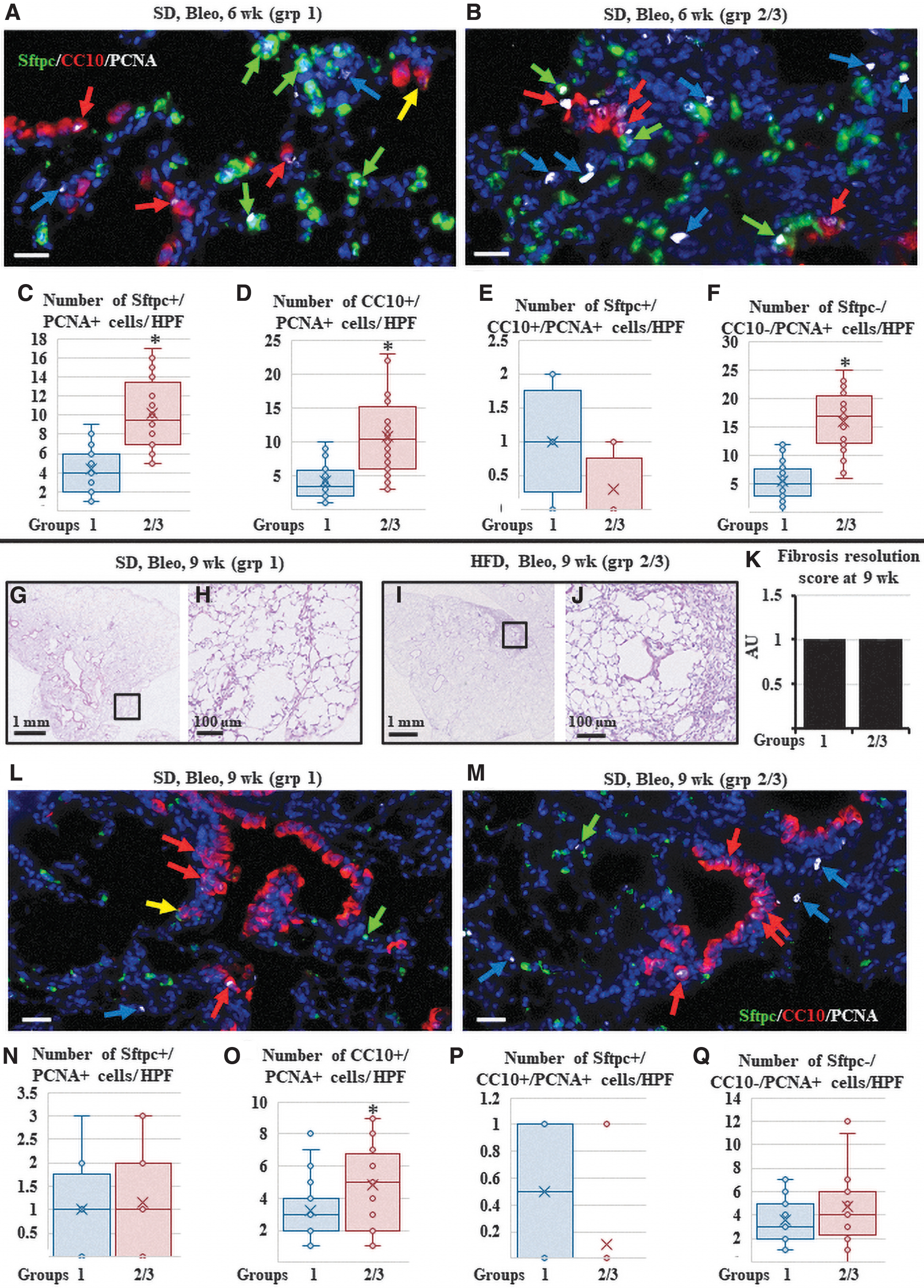
Assessment of the lungs 6 and 9 weeks after bleomycin (delayed repair and complete repair time points).
These data indicate that HFD causes significant delay/failure of fibrosis and inflammation resolution, significantly delays AT2 cell proliferation and infiltration of fibrotic foci, and impairs the contribution of BASCs to epithelial repair.
Examining the effect of HFD on the resolution of fibrosis and inflammation and on the alveolar repair 9 weeks after bleomycin injury (complete repair time point)
To identify whether HFD-induced delay in the repair and resolution of fibrosis, which was observed 6 weeks after fibrosis induction, are enduring effects, or whether the lung will eventually repair and resolve fibrosis, bleomycin-injured SD and HFD mice were examined 9 weeks after injury. At the 9-week time point, lung histological examination by H&E and Masson's trichrome staining showed near-complete resolution of inflammation and fibrosis in both SD and HFD-fed mice. However, areas with aberrant epithelial repair in the form of bronchiolization and honeycomb cyst formation were still detected in the HFD-fed mice (Fig. 4G–K).
Immunofluorescent staining showed the return of lung macrophage density levels that were comparable to the vehicle-treated lungs in most of the bleomycin-injured HFD-fed mice with very few arginase1+ cells (groups 2 and 3) and thus became indistinguishable from the SD-fed mice (data not shown). Immunostaining for epithelial markers revealed near-normal histological appearance and distribution of AT2 (Sftpc+/CC10−) cells in the alveoli of both SD-fed and HFD-fed mice lungs. Club cells (Sftpc−/CC10+) were seen lining apparently normal airways. However, in the HFD-fed mice, some parenchymal areas still showed aberrant epithelial repair in the form of club cell-lined bronchiolization and honeycomb cyst formation. Notably, BASCs (Sftpc+/CC10+) were rarely detected in the lung parenchyma (Fig. 4L–Q and Supplementary Fig. S8).
PCNA staining revealed fewer overall PCNA+ cells in comparison to that seen at 6 weeks in both diet groups, further confirming a near-complete repair of the lungs and return to homeostasis. Very few Sftpc+/PCNA+ cells were observed, indicating the completion of AT2 cell repair. The number of proliferating CC10/PCNA cells was also lower than the numbers observed at 6 weeks in both diet groups, but they were still significantly higher in HFD-fed lungs than in SD-fed lungs, suggesting that airway repair/remodeling is slower than alveolar repair, especially under the influence of a HFD. PCNA+ mesenchymal cells were also lower than their levels at 6 weeks, and no significant difference was detected between the SD and HFD-fed lungs. Proliferating BASCs were almost absent in both groups (Fig. 4L–Q and Supplementary Fig. S8). These data confirm that the HFD-induced delay of fibrosis and inflammation resolution observed at 6 weeks was not detectable at 9 weeks, although some aberrantly repaired areas persisted (in the form of enduring bronchiolization and honeycombs).
Examining the effect of inhibiting fatty-acid oxidation on the HFD-induced delayed/impaired resolution of fibrosis and inflammation and on the aberrant repair of alveoli at 3 and 6 weeks after bleomycin injury
HFD is associated with dysregulated glucose and lipid metabolism, resulting in enhanced fatty-acid oxidation (FAO) [16]. The functions of several adult stem cells have been shown to be influenced by changes in FAO and lipid metabolism [17]. To identify whether the HFD-induced delay/disturbance in lung repair after bleomycin injury was mediated through the induction of FAO, we administered the FAO blocker Etomoxir to the HFD-fed mice in a “repeat” of group 2 for 3 and 6 weeks after bleomycin injury (Supplementary Fig. S1).
At 3 weeks (peak fibrosis time point), inhibiting FAO showed no significant effect on fibrosis severity (Fig. 5A–D), infiltration of macrophages, or percentage of arginase-1 positive (M2) macrophages (Supplementary Fig. S9). Immunostaining for epithelial markers showed the presence of AT2 cells within fibrotic foci in 75% (15 out of 20 foci examined) of the “HFD-fed etomoxir-treated” mice lungs, while the “HFD-fed, vehicle-treated” mice lungs showed this in only 40% (8 out of 20 foci examined) of the group (P < 0.05; Fig. 5I, J, N and Table 1). Quantification of the club cells within the fibrotic areas showed that only 25% (5 out of 20 foci examined) of the “HFD-fed etomoxir-treated” mice lungs were invaded with club-like cells, while the “HFD-fed, vehicle-treated” mice showed invasion in 80% (16 out of 20 foci examined) of the group (P < 0.05; Fig. 5I, J, O and Table 1).
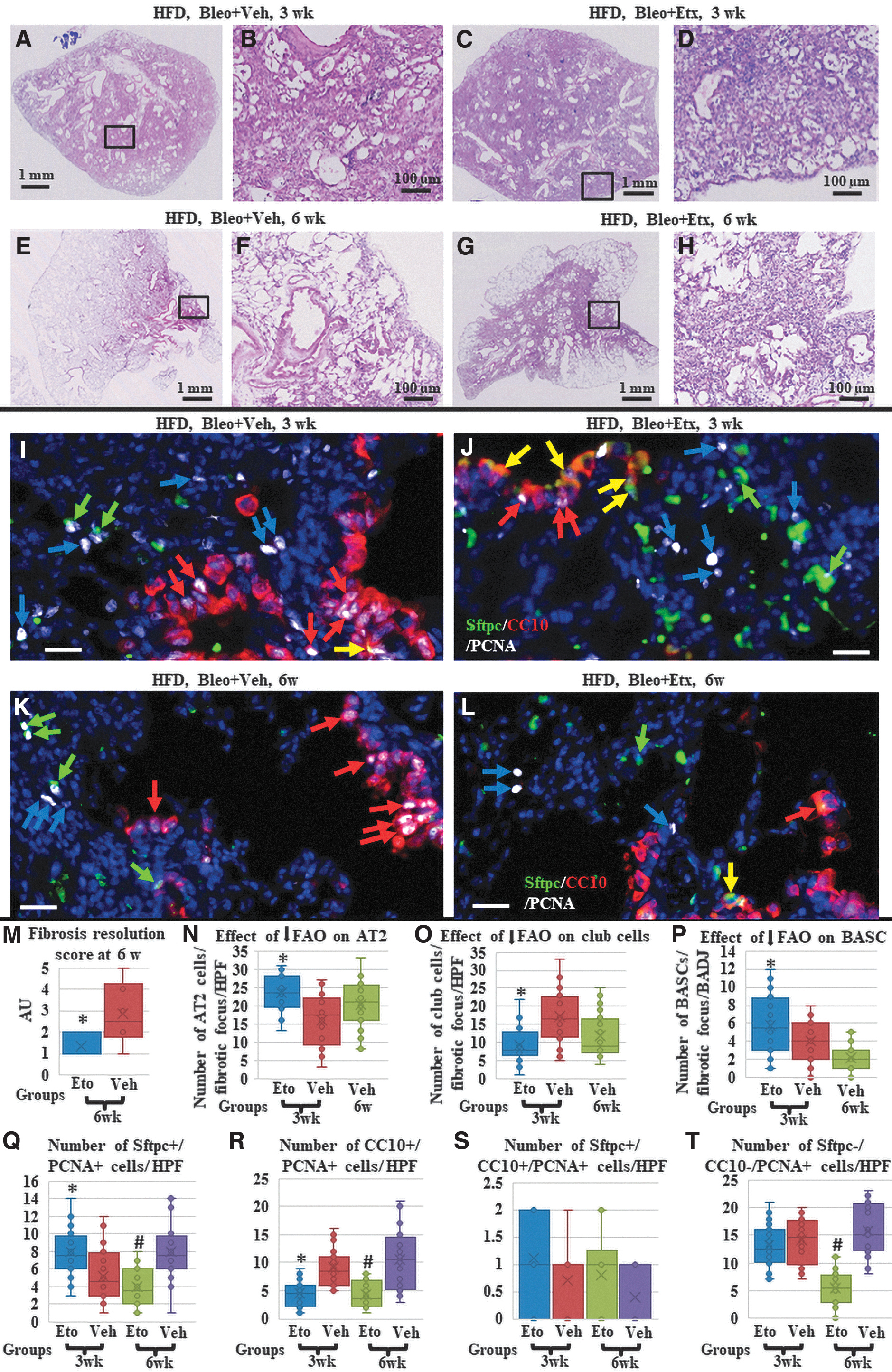
Effect of blocking FAO on the HFD-induced impaired and delayed repair.
Regarding BASCs, the “HFD-fed etomoxir-treated” mice lungs showed a small but significant increase in the number of CC10+/Sftpc+ cells at BADJ, and within the parenchymal fibrotic foci compared to the “HFD-fed, vehicle-treated” mice lungs, which showed a few BASCs. However, the number of BASCs in the “HFD-fed etomoxir-treated” mice lungs was still significantly lower than the number observed in group 1, SD-fed mice lungs at 3 weeks (indicating that blocking FAO could rescue the HFD-induced impairment of BASC contribution in repair; only partially) (1–12 cell/BADJ, average: 5.85 + 3.3 in HFD-fed etomoxir-treated mice lungs versus 0–7 cells/BADJ, average: 4.05 + 6.36 in “HFD-fed, vehicle-treated” mice lungs, P values <0.05, and <0.05 versus the 1–18 cell/BADJ observed in group 1 (Fig. 5I, J, P).
Quantification of the proliferating (PCNA+) cells further confirmed the overall improvement in the start of the repair process in response to blocking FAO. Significantly higher numbers of proliferating AT2 cells (but not BASCs) were detected in the lungs of the HFD-fed etomoxir-treated mice than in the lungs of the HFD-fed, vehicle-treated mice. In contrast, proliferating club cells in terminal bronchioles and nearby parenchymal fibrotic areas were significantly lower in HFD-fed etomoxir-treated mice lungs than in lungs from HFD-fed, vehicle-treated mice. Proliferating nonepithelial cells (fibroblasts/myofibroblasts or inflammatory cells) were not influenced by FAO inhibition, confirming a similar ongoing fibrotic process (Fig. 5I, J, Q–T and Table 2).
In contrast, at 6 weeks (delayed repair time point), lungs from the “HFD-fed etomoxir-treated” mice showed a significant resolution of fibrosis (Fig. 5E–H, M) reduction in the number of macrophages and percentage of arginase-1 positive (M2) macrophages (Supplementary Fig. S8), while the “HFD-fed, vehicle-treated” mice showed a typical delayed repair as described above for groups 2/3. Immunostaining for epithelial markers showed near-normal histological appearance and distribution of AT2 cells in the alveoli, and few club cells in the lung parenchyma in the “HFD-fed etomoxir-treated” mice lungs, similar to what was seen in group 1 at 6 weeks. However, BASCs were rarely detectable at the BADJ of the apparently normal (repaired) lung parenchyma, and within the few areas with residual fibrosis, so BASC numbers were significantly lower than those observed in group 1. The “HFD-fed, vehicle-treated” mice showed typical delayed repair as described above for groups 2/3 (Fig. 5K, L, N–P).
PCNA staining in “HFD-fed etomoxir-treated” mice confirmed a profile similar to that seen in the SD-fed mice of group 1, where the overall number of PCNA+ cells was significantly lower than its 3-week time point. The majority of the PCNA+ cells were AT2 and club cells. Significantly fewer SPC-CC10-PCNA+ cells (mesenchymal, ie, fibroblasts and inflammatory cells) were observed, suggesting a further resolution of fibrosis. Proliferating BASCs were almost absent. In “HFD-fed, vehicle-treated” mice, the numbers of CC10+ PCNA+ and SPC-CC10-PCNA+ cells were still abundant, similar to what was seen in groups 2/3, confirming HFD-induced aberrant repair and delayed fibrosis resolution (Fig. 5J, L, Q–T and Table 2). These data indicate that blocking FAO in HFD-fed mice alleviated the HFD-induced delay in the repair of bleomycin-induced lung fibrosis.
Discussion
Diet has a critical influence on body physiology and health. Several recent studies have shown that variations in the calorie and fat content of the diet exert a wide variety of effects on adult tissue stem cells [18]. Previous studies have observed that HFD can trigger a tendency toward fibrosis development, but the mechanisms involved remain elusive [8,19]. In this study, we show for the first time that a HFD causes a delay in lung fibrosis resolution after a fibrosis-inducing injury, a delay in AT2 stem cell proliferation, and a reduction in the contribution of BASCs in the repair process. However, HFD did not result in more severe fibrosis or increased mortality.
Bleomycin is known to cause severe damage to AT1, AT2, and airway club cells and to induce fibroblasts to proliferate and differentiate into extracellular matrix (ECM)-depositing myofibroblasts, which results in fibrosis of the lung tissues [20]. Resolution of this fibrosis requires removal of the myofibroblasts, clearance of the ECM, and replacement of the damaged alveolar and airway epithelium. Although the mechanism of lung fibrosis resolution is complex and involves a dynamic interplay among multiple cells in the lungs, the role played by AT2 cells and other epithelial stem cells in fibrosis development, resolution, and aberrant resolution has recently attracted a lot of interest [21,22].
AT2 and club cells are well-established stem cells of the alveolar compartments and airways, respectively. They proliferate and differentiate into AT2 and AT1 or club and ciliated cells during the repair and resolution of bleomycin-induced injury and fibrosis [23,24]. Accumulating evidence indicates that impaired AT2 cell function (caused by genetic factors, aging, mechanical tension, environmental stress, etc.) is a major contributor to fibrogenesis [22]. In contrast, club cells seem to be involved in the development of lung injury and fibrosis [25].
As we have shown previously that HFD causes several potentially harmful changes in the lung in homeostasis by inducing changes in lung stem cells [6], we expanded the spectrum of the deleterious effects caused by HFD by documenting its harmful influence on fibrosis resolution and epithelial repair. In contrast, AT2 and club cells are also known to have secretory functions (and thus termed facultative stem cells). Previous studies showed minimal effect of HFD on AT2 secretory function; the secretion of surfactant [26,27], while it seemed to modulate the pro-inflammatory secretions of the club cell [28].
FAO plays a critical regulatory role during maintenance and repair after injury in several adult stem cell populations. The precise mechanisms by which FAO affects stem cells may differ across stem cells, such as quiescent versus highly proliferative stem cells, and type and severity of injury. Adult stem cells are negatively affected by pharmacological or genetic ablation of the components of the FAO machinery [17]. As HFDs seemed to negatively influence lung repair after fibrosis-inducing injury, we expected that pharmacological blocking of FAO would reverse the delaying effect of HFD, although some previous reports showed that blocking FAO seemed to exert harmful effects on adult hematopoietic stem cells and neural stem/progenitor cells [29,30]. Indeed, we found that inhibiting FAO with etomoxir reversed most of the HFD-induced delay in epithelial cell repair and fibrosis resolution.
Previously, Li et al. showed that after lung injury with bleomycin (under SD conditions), glycolytic and pentose phosphate pathways are promoted, whereas fatty acid synthesis is repressed in mouse AT2 cells, which eventually results in fibrosis resolution and epithelial repair [31]. Therefore, we propose that in the presence of the HFD-induced excess in fatty acids, AT2 stem cells are pushed toward FAO as an energy source, rather than the classic glycolysis, thus impairing its repair capacity. In contrast, Li et al. showed that, under SD conditions, autophagy and glucose metabolism were essential for the maintenance of airway epithelium through an effect on the club (progenitor) cells during steady state or allergic inflammation [32]. Further studies are needed to elucidate the exact metabolic pathways used by AT2 and other lung stem cells during homeostasis and after injury.
Conclusion
It seems that calorie restriction and fasting are generally beneficial to adult stem cell function, while HFDs impair stem cell function or create opportunities for tumorigenesis. However, the effects of each diet (or even variations in the component of the diet that does not influence its calorie content) on stem cell biology are complex and vary greatly between tissues and during different injuries/diseases. Given the ongoing interest in using dietary interventions and developing dietary mimetics as treatments for various diseases and disorders, further research is required to better delineate the specific response of adult (lung) stem cells and diet-induced changes in metabolic and energy pathways.
Footnotes
Acknowledgments
The authors thank Sachiko Wada for technical support and the Collaborative Research Resources, Keio University School of Medicine for technical support and reagents.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Japan Society for Promotion of Science JSPS Grant-in-Aid for Scientific Research (A.E.H. 20K08548, M.O. 18K15934).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.