
Abstract
The extracellular vesicles (EVs) secreted by mesenchymal stromal cells (MSC-EVs) exhibit immunoregulatory functions dependent on their parent cells. MSC-EVs are promising candidates for treating neuroinflammation in neurological diseases due to their acellular nature and their ability to reach the central nervous system. However, the conditions of MSCs for producing EVs with the highest anti-inflammatory efficacy are still unknown. Therefore, the first objective was to study the characteristics of the EVs produced by MSCs cultured in different conditions. The second objective was to evaluate the in vitro anti-inflammatory properties of those EVs in feline stimulated mixed glia. Umbilical cord-derived MSCs were treated with serum-free (SF) media, inflammatory (IF) media, or media supplemented with 5% EV-depleted fetal bovine serum (FBS). The isolated MSC-EVs were characterized by particle size and yield, and their anti-inflammatory ability was evaluated in lipopolysaccharide (LPS) stimulated feline mixed glia. All EV isolates were <160 nm, and the primary mixed glia consisted of microglia, astrocytes, neurons, and endothelial cells. Our results indicate that IF-EVs statistically significantly decreased the production of interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α) and downregulated the transcription of the, nuclear factor kappa B p65 subunit in inflammatory mixed glia after 48 hours. In addition, SF- and FBS-EVs significantly reduced in vitro the secretion of IL-6 after 48 hours, but only SF-EVs achieved a significant effect on inhibiting the expression of p65 at 48 hours. Moreover, messenger RNA (mRNA) levels of inducible nitric oxide synthase (iNOS) were significantly decreased following treatment with SF-EV for 24 hours. This study demonstrates that MSC culture conditions affect the therapeutic potential of the secreted EVs in feline mixed glia.
Introduction
Mesenchymal stromal cells (MSCs) are multipotent stem cells that can repair tissue damage, promote angiogenesis, and regulate the immune responses while exhibiting low immunogenicity. 1,2 Their therapeutic effect is due to their secretome 3 consisted of soluble components such as cytokines, growth factors, and extracellular vesicles (EVs). 4 Specifically, EVs are double-layered membrane particles that are being released from all types of cells under normal and abnormal conditions to sustain cellular communication and homeostasis. 4 Various cellular processes are responsible for generating different types of EVs, which can vary in size (30 nm–5 μm) and content. 4,5 Moreover, EVs are acellular particles that exhibit long half-life circulation, can be easily collected from tissues and biological fluids, and can cross the blood–brain barrier (BBB). 6,7
Research shows that EVs secreted by MSCs (MSC-EVs) exhibit immunoregulatory properties similar to their parent cells, 6,8 which include increasing the circulation and function of T-regulatory cells, inhibiting adaptive immune responses and antigen presentation, decreasing antibody production and T-cell activation, reducing macrophage recruitment, and polarizing macrophages into the M2 anti-inflammatory state. 3,9 In addition, issues related to immunogenicity/immunotoxicity have not been encountered following the in vivo administration of EVs derived from normal cells 10,11 and MSCs. 8 All of the above support the superiority of EV-therapy over their cellular components, 12 given that EV administration can also resolve safety issues associated with cellular transplantation. 5,6
The therapeutic effect of stem cell-derived EVs has been explored against a variety of neurological diseases, including Alzheimer’s disease, 13 traumatic brain injury, 14,15 graft-versus-host disease, 16 thromboembolic stroke, 17 ischemic stroke, 11 multiple sclerosis, 18 Parkinson’s disease, 19 and nerve ligation-induced injury. 20 Those neuropathologies are characterized by neuroinflammation, which is a complex and heterogeneous immune process in the brain and spinal cord. Neuroinflammation is accompanied by dysfunction in glial cells responsible for tissue homeostasis and neuronal health and infiltration of peripheral immune cells. 21,22 Prolonged glia overreaction, known as microgliosis and astrogliosis, results in neurodegeneration, which in turn exacerbates the neurological disease progression. 23,24 Anti-inflammatory drugs such as nonsteroidal anti-inflammatory drugs are the current treatment against neuroinflammation, but they often have minimum clinical effect 25,26 and their long-term administration is related to specific adverse reactions and risks. 27 Therefore, new therapeutic approaches are currently being investigated. 20 The anti-inflammatory properties and the high brain-homing ability of stem cell-derived EVs make them potential candidates to treat neuroinflammation as they appear to target all the main components of neuroinflammation. 17 Specifically, previous studies have shown that stem cell-derived EVs can suppress microglia activation 13,28 and reduce oxidative stress 29 and the circulation of pro-inflammatory mediators in the central nervous system (CNS) 30 among other functions. 3,31,32
It is known that the condition of the parent cells can affect the biological action of EVs due to differences in their cargo and morphological characteristics. 33 We previously proved that the source of MSCs alters the immunoregulatory potential and the characteristics of the secreted EVs. 34 Specifically, we found that EVs from umbilical cord-derived MSCs (UC-MSCs) exhibited improved anti-inflammatory properties in comparison with those isolated from bone marrow MSCs. 34 Moreover, we found that cell culture conditions of MSCs affect the functionality and the morphology of the produced EVs. 34 Specifically, EVs isolated from serum-free (SF) or supplemented with EV-depleted fetal bovine serum (FBS) culture conditions induce stress and influence the physiological effects of the MSCs. 34 Cell culture conditions affect the EV cargo too, since Pei et al. showed that the miR-146a-5p microRNA delivered by human SF-EVs from UC-MSCs exerted an anti-inflammatory effect in lung chemical toxicity, by inhibiting the formation of nuclear factor kappa B (NF-κB), a key factor in inflammation. 35 In contrast, priming MSCs with pro-inflammatory mediators has been shown to enhance the immunosuppressive functions of the secreted EVs. 14,36 Therefore, EV release methods from the parent cells have to be taken into consideration when conceptualizing EV-based therapeutics, and literature is still inconclusive in identifying the optimal conditions for the production of MSC-EVs.
Researchers who investigate the development of novel therapeutics that target neurodegenerative diseases often use rodent mixed glia cell cultures as a reliable in vitro platform. 37 However, rodent glia substantially differ from humans' in morphology, function, complexity, secretion, and gene expression profiles, which complicates translational neuroscience. 37,38 Therefore, EV studies performed in humans or evolutionally similar species are needed to draw appropriate conclusions that can impact future clinical decisions. In contrast, small animals such as feline suffer from naturally occurring neurodegenerative diseases identical to those that occur in humans, allowing the conduction of long-term studies testing the efficacy of novel therapeutics. 39 Specifically, the feline species serve as a valuable model for translational research in neurodegenerative diseases since cats have similar pathology, physiological properties, and brain size to humans. 40
Our goal is to identify whether culture conditions (SF, inflammatory [IF], FBS) of MSCs affect the functionality of the generated EVs when added to feline-stimulated mixed glia. Therefore, our objectives are to (1) evaluate whether the characteristics and functionality of EVs are affected by various MSCs’ culture conditions and (2) to investigate whether stem cell-derived EVs can alleviate in vitro the neuroinfammatory response produced by feline-stimulated mixed glia. Our hypothesis is that the cell culture conditions of the parent cells will affect the anti-inflammatory potential of their secreted EVs when added in lipopolysaccharide (LPS)-stimulated mixed glia from normal cats.
Materials and Methods
Isolation and characterization of EVs from UC-MSCs
EVs from human UC-MSCs were isolated following previously described protocols. 34 Briefly, commercially available human UC-MSCs were purchased and cultured up to passage 5 according to manufacturer’s instructions. At 70% confluency, cells were washed twice with phosphate-buffered saline without calcium and magnesium (PBS −/−, VWR, USA) and then cultured in different MSC culture conditions as follows: (1) 24 h in SF with alfa minimum essential medium (VWR, USA) with 2 mml/L GlutaMAX (Gibco, USA), (2) 24 h in media supplemented with 5% EV-depleted FBS (Gibco, USA), or (3) 48 h in IF media with 15 ng/mL tumor necrosis factor alpha (TNF-α) and 20 ng/mL interferon gamma (IFN-γ) (Thermo Scientific, USA) and then 24 h incubation in SF conditions. Media from each condition were collected, filtered with 0.22 μm (VWR Life Sciences, USA), and subsequently ultrafiltrated with 100 kDa molecular weight cutoff (Amicon; Millipore-Sigma, USA) at 4,000 g for 10 min. The EVs on top of the filter were washed twice using PBS with calcium and magnesium (PBS +/+) and were collected and stored at −20°C. Later, the various EV isolates were titrated using the EXOCET enzymatic assay (System Bioscience, USA), whereas the total protein concentration was evaluated via Micro BCA™ Protein Assay Kit (Thermo Fisher Scientific, USA). Transmission electron microscopy (TEM) was utilized to confirm the proper EV morphology, and flow cytometry (FC) (antibody-coated MACSPlex Exosome Capture Beads & Detection reagents, Millipore Sigma) was performed to phenotypically characterize the hUC-MSC-EV products as previously described. 34 In addition, EV samples were analyzed by RoosterBio® (Maryland, USA) for particle concentration, size, and distribution via nanoparticle tracking analysis (NTA).
Isolation of feline mixed glia
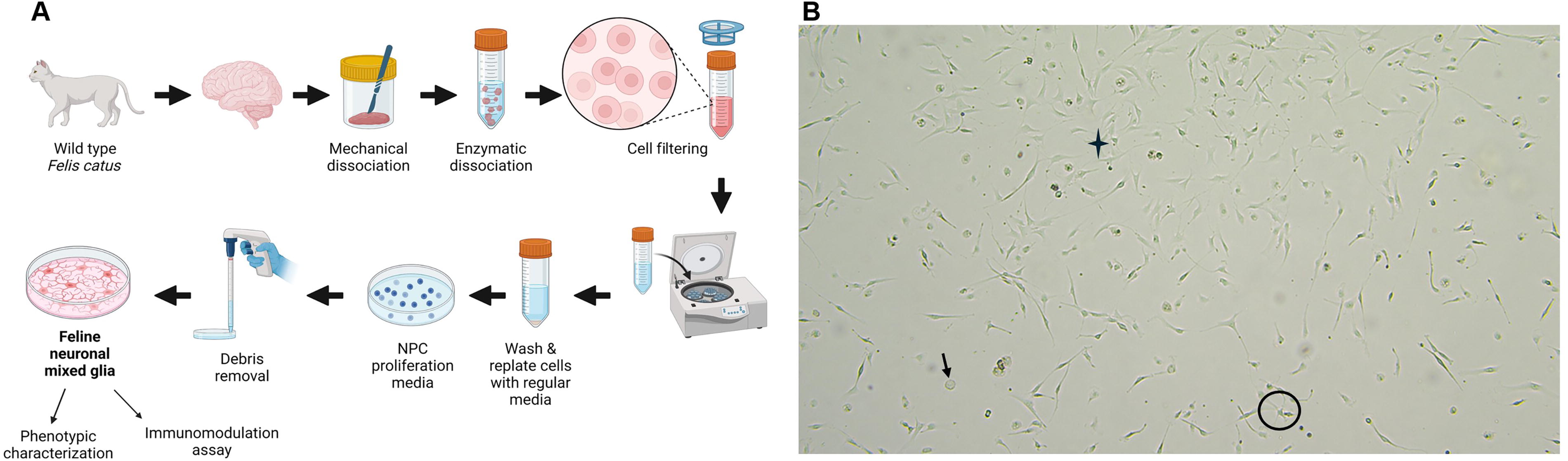
Mixed glia cells were isolated based on modified protocols previously described. 38,41 Three female and two male domestic cats (Felis catus, n = 5), between 3 and 5 months old, were used for this study. All the animals were bred and raised in the Scott Ritchey Research Center at Auburn University, and the protocols were approved by the Auburn University Institutional Animal Care and Use Committee. All animal experiments followed the American Veterinary Medical Association guidelines of euthanasia. After euthanasia, the animals were perfused via the left ventricle of the heart with 1,000 mL cold, heparinized saline solution. Brain (∼20 g of tissue) was removed and immediately was transferred to a sterile beaker with ice-cold Hank’s solution without calcium and magnesium (HBSS −/−, VWR, USA) supplemented with 3% FBS (VWR, USA) and 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco, USA). The tissue processing was performed in aseptic and sterile conditions. Sterile scalpels were used to cut the brain into 2 g portions, and each brain portion was processed separately. Each piece was diced into <1mm3 cubes in Petri dishes with HBSS and antibiotics, using sterile scalpels. Dissociated tissue was transferred in 50 mL centrifuge tubes with 20 mL enzymatic dissociation mix (10 mL enzymatic solution/g of tissue) and was incubated for 30 min at 37°C with agitation. 41 The enzymatic dissociation mix contained 400 U deoxyribonuclease I per gram (Stem Cell Technologies, USA) and 200 U/mL collagenase II (Gibco, USA) in Hibernate A media (Gibco, USA) and was prepared within 1 h of use. 42 The tissue was gently triturated with a 10 mL serological pipette, and an equal amount of regular media, consisting of Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM/F12, Millipore Sigma), 10% FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 1% GlutaMAX (Gibco, USA), was added to stop the enzymatic dissociation. A second trituration step was followed. Cell suspension was passed through a prewet 70 μm nylon cell strainer (VWR, USA), followed by 10 min centrifugation at 275g. The cell pellet was resuspended with neuronal precursor proliferation media (NPC) that contained DMEM/F12, 15% FBS, 1% GlutaMAX, 100 U/mL penicillin, 100 mg/mL streptomycin, 40 ng/mL fibroblast growth factor-2 (FGF-2, R&D biotechne, USA, cat# 3339-FB-025), 40 ng/mL epidermal growth factor (Kingfisher Biotech), and 2 μg/mL heparin (Stem Cell Technologies, USA). 43 Approximately, 1 g of starting material was plated into poly-d-Lysine-coated 100-mm Petri dishes (Corning, USA) with 10 mL of regular media to incubate for 24 h at 37°C in a humidified atmosphere with 5% CO2. The next day, the culture media that contained debris and unattached cells were transferred to a 50 mL centrifuge tube. Cells were washed with 5 mL media to remove debris and were transferred to the same tube for an upcoming 10-min centrifugation at 160 g. Supernatants were discarded, and the cell pellets were resuspended with 5 mL of regular media and plated on top of the 5 mL of media in the coated dishes. Cell cultures were incubated for another 24 h at 37°C. The following day, unattached cells and debris were discarded and washed twice with 5 mL of regular media. Finally, 10 mL of regular media were added to the culture dishes, and the cells were incubated at 37°C for 7–10 days. Minimal media changes were performed.
Cells were harvested for further assays when they demonstrated structural formation and 70% confluency. At that point, they were washed once with HBSS (−/−) and trypsinized with 0.25% trypsin-ethylenediaminetetraacetic acid 1× (Gibco, Canada) for 10 min at 37°C and 5% CO2. Cell scrapers were utilized to aid the detachment of cells, since microglia are known to attach firmly. 44 Trypsin was neutralized with regular media containing FBS, and then the cell suspensions were centrifuged at 300g for 10 min. Finally, cells were stained for viability with acridine orange propidium-iodide and counted via a cellometer (Nexcelom Bioscience, MA, USA) to determine cell density.
Immunocytochemistry on feline mixed glia
A total of 50,000 cells were plated in 4-well glass-chamber slides (Corning, USA) with regular media for immunocytochemistry (ICC) experiments. After 48 h, cells were prefixed for 5 min at room temperature by adding 4% paraformaldehyde (PFA) into the culture media at 1:1 ratio. The mixture was discarded, and 4% PFA was added directly into the wells to fix the cells for 20 min at room temperature. The chamber slides were washed thrice with ice-cold PBS (−/−) before proceeding to a 15-min permeabilization step with 1×PBS and 0.3% Triton X-100 (Sigma-Aldrich, USA) at room temperature. Then, a similar washing step was performed, and 10% goat serum (Thermo Fisher, USA) was added, to prevent nonspecific binding, for 30 min at room temperature. The blocking solution was removed, and 500 μL of the primary antibody (Table 1) solutions in 1% bovine serum albumin (BSA, VWR, USA) in PBS + 0.1% Tween 20 (Millipore Sigma) were added for overnight incubation at 4°C. The next day, slides were washed, and 500 μL of secondary antibody solutions in 1% BSA were added for 1 h at room temperature in the dark. F(ab’)2-Goat anti-Rabbit immunoglobulin gamma (IgG) Secondary Antibody (1:500), Alexa Fluor™ 555, Goat anti-Mouse IgG Secondary Antibody, Alexa Fluor™ 488 (1:500), and Goat anti-Rabbit IgG Recombinant Secondary Antibody, Alexa Fluor™ 488 (1:2,000, Thermo Fisher, USA) were used to target the corresponding primary antibody. The slides were washed, and nuclei were counterstained with mounting medium that contains 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA) before adding the coverslip. Images were acquired using Nikon A1 automated fluorescent microscope and the Nikon instruments-imaging software.
List of Primary Antibodies Used for Immunocytochemistry
Anti-NeuN, anti-NEUronal Nuclei protein; GFAP, glial fibrillary acidic protein; Iba-1, allograft inflammatory factor 1.
FC on feline mixed glia
Phenotypic characterization of mixed glia was additionally performed via FC against feline cell markers using cross-reactive antibodies. 45 One million cells were stained with a viability dye [LIVE/DEAD™ Fixable Near IR (876) Viability Kit, Invitrogen, USA] according to manufacturer’s instructions. Specifically, the CD11b antibody-stained microglial cells, whereas glial fibrillary acidic protein (GFAP) and platelet-derived growth factor receptor beta (PDGFR-β) targeted astrocytes and pericytes/endothelial cells, respectively. Then, cells were blocked with a cocktail of normal cat serum, goat serum, and donkey serum (1:11 dilution, Jackson ImmunoResearch Laboratories, Inc, West Grove, PA) diluted in FC buffer (HBSS −/−, 2% BSA, 3% FBS) for 1 h in ice. 45,46 Subsequently, the cells were washed at 300 g for 5 min and subsequently single stained in FC buffer with the primary antibodies CD11b, PDGFR-β, and GFAP and for 30 min on ice (Table 2). Staining against GFAP required intracellular cell fixation and permeabilization (eBioscience, Invitrogen Set) and an extra blocking step. For staining cells against GFAP, cells were fixed with 100 μL fixation buffer for 30 min in the dark. Then, cells were washed with permeabilization buffer and were blocked with blocking cocktail (FC, animal serums, and permeabilization buffer) for 1 h, protected from light, followed by a similar washing step and GFAP staining. The unbound primary antibodies were washed before adding the diluted secondary antibodies in FC and permeabilization buffer to incubate for 30 min in the dark on ice. Cells were washed, and pellets were resuspended in 200 μL ice-cold HBSS (−/−), before acquiring data using the CytoFLEX LX flow cytometer (Beckman Coulter, Life Sciences). Primary antibodies were secondary stained with Zenon Mouse IgG1-Alexa 647 Labeling Kit as instructed by the company (Thermo Fisher, USA), Alexa Fluor® 647 AffiniPure Donkey Anti-Rabbit IgG (H + L) (1:100), and R-Phycoerythrin AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG, F(ab’)2 fragment (1:100, Jackson ImmunoResearch laboratories, USA). The analysis was performed using CytExpert software. Unstained cells and cells stained only with secondary antibodies served as negative controls. For each sample, 100,000 events and 50,000 singlets were analyzed.
List of Primary Antibodies Used for Flow Cytometry
CD11b, integrin Αlpha M; PDGFR-β, platelet-derived growth factor receptor beta.
Immunomodulation assay
Feline mixed glia were plated at 18,000/cm2 in poly-d-Lysine coated 48-well plates (Corning, USA) with regular media. Cells were treated for 24 h and 48 h with 50 ng/mL LPS from Escherichia coli 0127:B8 (List Biological Labs, Campbell, CA) with or without the addition of 108 EVs/mL isolated under SF, IF, or EV-depleted FBS culture conditions. The dose of the EV treatment was calculated according to the titer obtained from the enzymatic assay (EXOCET). Unstimulated cells in media with vehicle alone (PBS) were used as a negative control. At each time point, the cell culture supernatants were collected and stored at −80°C to quantify the secretion; interleukin 6 (IL-6), IL-1β, and TNF-α were measured in triplicates via commercially available enzyme-linked immunosorbent assays (ELISAs) (Feline DuoSet ELISA assay kits R&D Systems, Inc., Minneapolis, USA), following manufacturer’s protocols. Standard curves and data were acquired via the Agilent BioTek Gen5 microplate reader and software (Agilent Technologies, CA, USA). In addition, cell lysates were stored at −80°C with RNeasy Protect Cell reagent (Qiagen, USA) to stabilize the RNA for RNA extraction.
RNA isolation and first-strand complementary DNA synthesis
RNAs from mixed glia cultured in the presence of MSC-EVs were purified using the RNeasy Plus Mini Kit (Qiagen, USA), according to manufacturer’s instructions. RNA concentration was measured using a NanoDrop Spectrophotometer (Thermo Fisher Scientific) at 260 nm. RNA purity was also evaluated by measuring the 260 nm/280 nm and 260 nm/230 nm ratios, and only high-quality RNA was used for further experiments. A measure of 0.01–0.02 μg of RNA from each sample were utilized for complementary DNA (cDNA) synthesis via the SuperScript III First-Strand Synthesis System (Invitrogen, Thermo Fisher Scientific, Grand Island, NY, USA) according to the manufacturer’s protocol with random hexamers (50 ng/μL).
Real-time polymerase chain reaction of inflammatory factors
Gene expression of the NF-κB subunit p65, Toll-like receptor 4 (TLR4), NOD-like receptor protein 3 inflammasome (NLRP3), inducible nitric oxide synthase (iNOS), and arginase 1 (Arg1) inflammatory factors (Table 3) from mixed glia cultured with the various MSC-EV isolates was evaluated using real-time reverse transcription polymerase chain reaction (RT-PCR). A measure of 100 ng of cDNA was relatively quantified using the SsoAdvanced™ Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) and a QuantStudio 3 Thermal Cycler (Applied Biosystems, Carlsbad, CA). The ribosomal protein housekeeping gene, ribosomal protein L17 (RPL17), is the appropriate endogenous control for feline brain tissue, which was established by our research group. Each well contained 100 ng cDNA, 1× primers, 1× master mix, and RNAase-free water up to 20 mL, and samples were run in triplicates. Forty PCR cycles were conducted based on manufacturers’ guidelines. The primer sequences are listed in Table 3. The data were calculated via the 2-ΔΔCt data analysis method and were normalized to the LPS-stimulated mixed glia with vehicle.
List of Feline Primer Sequences
All primers were purchased from Integrated DNA Technologies (IDT, Iowa, USA).
Arg1, Arginase 1; iNOS, inducible nitric oxide synthase; NF-κB, nuclear factor kappa B; NLRP3, NOD-like receptor protein 3 inflammasome; p65, RelA, NF-κB subunit; RPL17, Ribosomal Protein L17; TLR4, toll-like receptor 4.
Statistical analysis
All the experiments were replicated 3–5 independent times under the same experimental conditions from different animals. The software GraphPad version 10.2.2 (for Windows 64-bit Prism 7.0c, GraphPad Software, Boston, MA, USA) was used for statistical data analysis. Normality, equality of variances, and statistically significant differences among the treatment groups were assessed via repeated measures, multiple comparisons two-way analysis of variance (ANOVA). Repeated measures one-way analysis of variance (ANOVA) was used to identify specific statistical differences between some treatment groups. For nonparametric data, the analysis was performed using the Wilcoxon test. Values lower than P < 0.05 were considered significant. Finally, fold change (2-ΔΔCt) ± standard deviation (SD) represents the mean messenger RNA (mRNA) expression levels.
Results
Cell culture conditions affect the size and yield of isolated EVs from MSCs
The different UC-MSC culture conditions (SF, IF, and FBS) affected the size and yield of the released EVs. Figure 1 graphically depicts the size distribution of the SF-, IF-, and FBS-EV isolates as assessed via NTA. Specifically, EVs derived from IF conditions of MSCs had a median size of 154.2 nm, whereas those exposed to SF and 5% EV-depleted FBS environment had a similar median size of 144.8 nm and 144.6 nm, respectively (Table 4). Overall, the generated particles measure around 200 nm and, thus, are classified as small by the Minimal Information for Studies of Extracellular Vesicles nomenclature (MISEV2023). 47
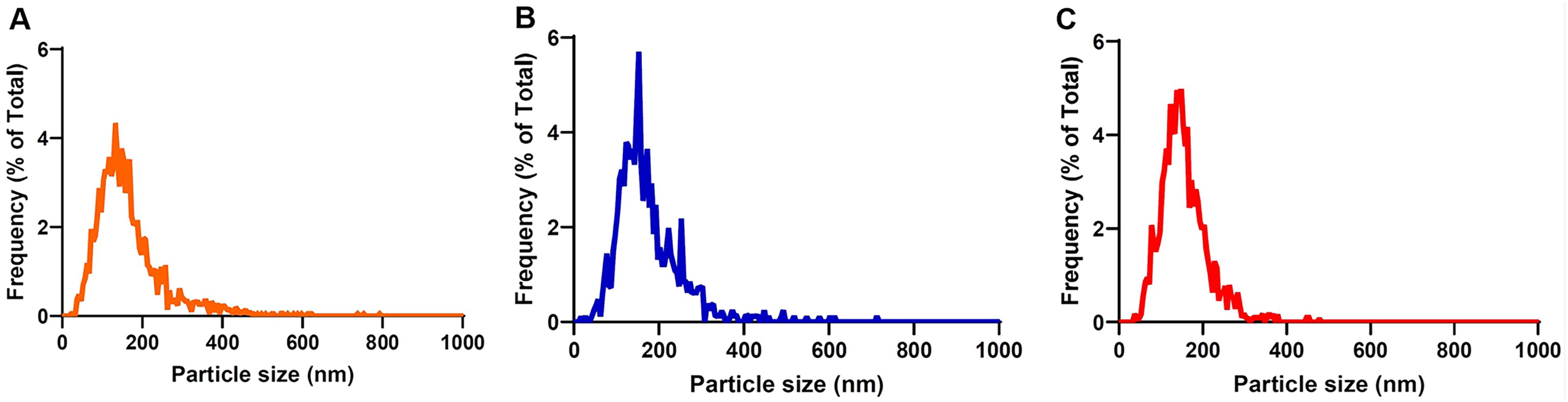
UC-MSC-EVs size distribution isolated from various cell culture conditions (SF, IF, FBS) as determined via ZetaView Nanoparticle Tracking Analysis (NTA) (RoosterBio, Inc., MD, USA). Graphs representing the frequency in % of particle sizes in nm of
Particle Size of the Extracellular Vesicles Secreted by Mesenchymal Stromal Cell Isolates Measured via ZetaView Nanoparticle Tracking Analysis *
RoosterBio, Inc., MD, USA.
FBS, fetal bovine serum; IF, inflammatory; MSC-EV, extracellular vesicles secreted by mesenchymal stromal cells; SF, serum free; UC, umbilical cord.
Nanoparticle (P/mL) and protein concentrations (μg/mL) are highly variable between samples and between quantification methods (NTA or the enzymatic EXOCET assay) (Table 5). However, no statistically significant differences were identified in the particle concentration from the same EV isolate between the quantification methods (Table 5, P = 0.42). Protein concentration was analogous to the NTA particle measurement, where higher protein titer corresponded to higher particle numbers (Table 5). EVs from the FBS group were the most protein concentrated (50,636.8 ± 1,448 μg/mL) compared with the rest of the groups. IF-EVs contained 1,804.8 ± 269.2 μg/mL of protein, whereas the SF product had a protein concentration of 2,039.2 ± 796 μg/mL. However, the EV titer from the enzymatic assay corresponded only to the protein amount of the FBS-EV isolate. The FBS group contained the highest particle concentration in both methods, ranging from 2.83 × 1,010 ± 3.18 × 109 P/mL in NTA (Table 5) and 7.76 × 1,012 ± 1.46 × 108 P/mL in the enzymatic assay. The SF-EV from UC-MSCs had a concentration of 1.40 × 109 ± 1.41 × 108 P/mL, whereas the IF-EVs had a particle concentration of 8.70 × 108 ± 7.07 × 107 P/mL, measured via NTA. However, the quantification via the enzymatic assay revealed the opposite pattern, where the titer of the IF-EVs (4.30 × 1,010 ± 9.5 × 106 P/mL) was higher than the one from the SF group (9.28 × 109 ± 108 P/mL) (Table 5).
Protein (μg/mL) and Particle Quantification (P/mL) of Serum Free-, Inflammatory-, and Fetal Bovine Serum Umbilical Cord Extracellular Vesicles secreted by Mesenchymal Stromal Cells
Protein concentration was measured via MicroBCATM Protein Assay Kit (Thermo Fisher Scientific), whereas the particle concentration was quantified via ZetaView NTA (RoosterBio, Inc., MD, USA) and EXOCET (System Bioscience, USA).
Results sharing a common letter are not statistically significantly different (P = 0.42).
NTA, nanoparticle tracking analysis.
Isolated feline brain cells express mixed glia cell markers
We successfully produced a feline brain in vitro platform to study the immunomodulatory properties of stem cell EV products. The experimental design is summarized in Fig. 2A. Our described cell isolation protocol produced cell cultures with mixed populations, since the cells exhibited round, ramified, and fibroblast-like morphology (Fig. 2B). Cell yield was ∼2 million cells/g of feline brain tissue after a week of culture, with >92% cell viability.
Phenotypic characterization of the isolated feline glia cells was performed via ICC (Fig. 3) and FC (Fig. 4). First, immunofluorescence revealed the successful isolation of neuronal mixed glia from the brains of normal cats (Fig. 3, n = 3). Specifically, our cell isolation method revealed a mixed population of glial cells consisting of microglia as stained with red for allograft inflammatory factor 1(Iba-1), astrocytes as stained with green for glial fibrillary acidic protein (GFAP), and neurons as stained with green for anti-NEUronal Nuclei protein (Fig. 3). Moreover, Iba-1 staining exposed the two main microglial phenotypes, round-amoeboid, and ramified structures, representing the activated and rested microglial cells, respectively. 39 Astrocytes, which are characterized by GFAP expression, appeared to have a star-shaped structure. GFAP is a common astrocytic marker, which is generally dynamically expressed to mechanically support astrocytic cells and the BBB structure. 48 The extensive interaction between microglia and astrocytes is observed in Fig. 3, which is crucial for brain homeostasis in vivo. 49 Oligodendrocytes were absent from the cultures since the oligodendrocyte transcription factor 2 (Olig2) marker was negative in ICC (not reported) and it was further excluded from the analysis.
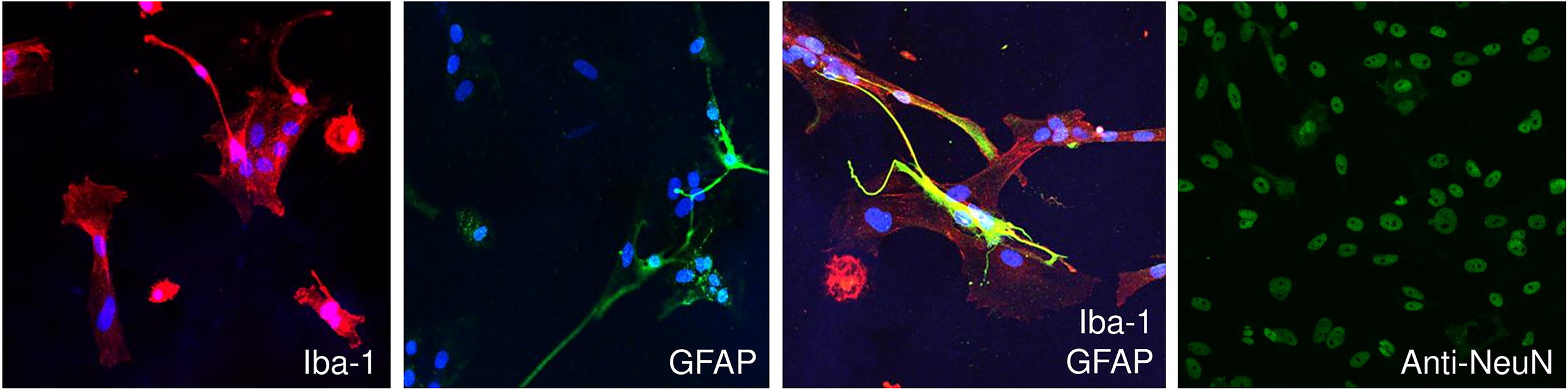
Phenotypic characterization of isolated feline brain cells via immunocytochemistry (ICC). Representative confocal microscopy images of primary mixed glia from one normal cat with microglia (Iba-1, red), astrocyte (GFAP, green), and neuronal (anti-NeuN, green) markers and DAPI staining for the nuclei (blue). Scale bar = 50 μm. Three independent experiments were performed to confirm the method’s reproducibility (n = 3). Iba-1, allograft inflammatory factor 1; GFAP, glial fibrillary acidic protein; anti-NeuN, anti-NEUronal Nuclei protein.
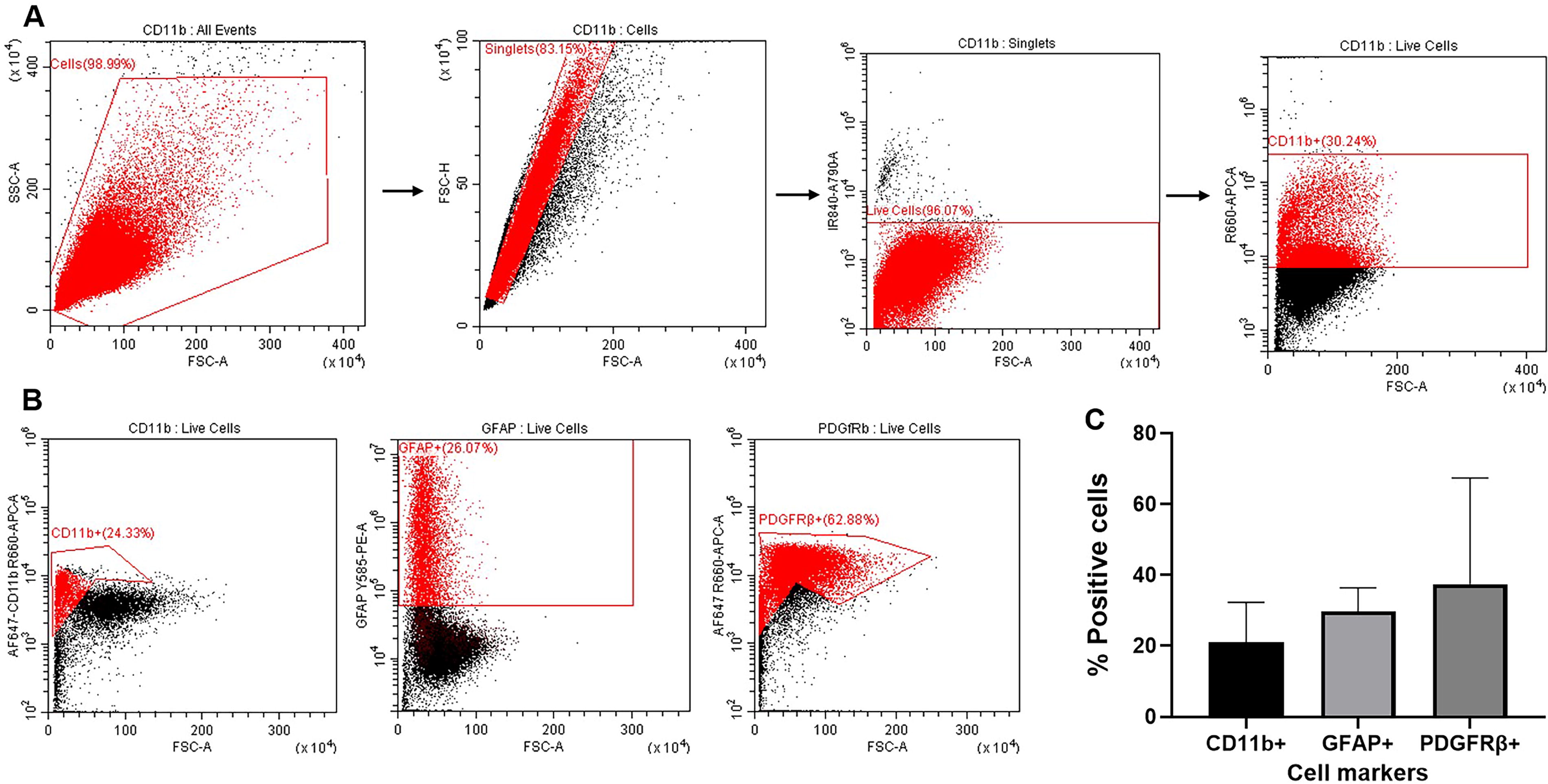
Phenotypic characterization of isolated feline brain cells via Flow Cytometry (FC).
To evaluate the composition of feline mixed glia, quantification via FC was performed (Fig. 4, n = 3). Feline cross-reactive antibodies were used for the quantification of astrocytes, microglia, and other cells of hematopoietic origin cells, such as endothelial cells and pericytes. All events were gated for cell-specific markers, CD11b, GFAP, and PDGFRβ, after excluding doublets and dead cells (Fig. 4A). The isolated feline mixed glia were 20.96% ± 11.35 positive for CD11b (microglia), 29.80% ± 6.63 positive for GFAP (astrocytes), and 37.40% ± 30.05 positive for PDGFRβ (endothelial cells/pericytes) (Fig. 4B, C). The CD11b fraction represents mostly microglia cells—the primary brain immune cells—but also contaminating myeloid-lineage cells, such as monocytes and macrophages. Similar to ICC, astrocytes are the positive cells for GFAP. Endothelial cells and pericytes were quantified by their PDGFRβ expression.
EVs from MSCs decrease the production of main pro-inflammatory cytokines in LPS-stimulated feline mixed glia
The immunomodulatory properties of the MSC-EV isolates derived from SF, IF, and FBS culture conditions were evaluated on LPS-stimulated mixed glia isolated from healthy cats at 24 and 48 h post-addition (Fig. 5, n = 5). A statistically significant difference was not identified for any measured cytokine following the addition of MSC-EVs to LPS-stimulated mixed glia at 24 h (Fig. 5A–C). On the contrary, statistically significant differences were identified for IL-6 and TNF-α at the 48-h time point following the addition of EVs compared with the control group (Fig. 5D, E).
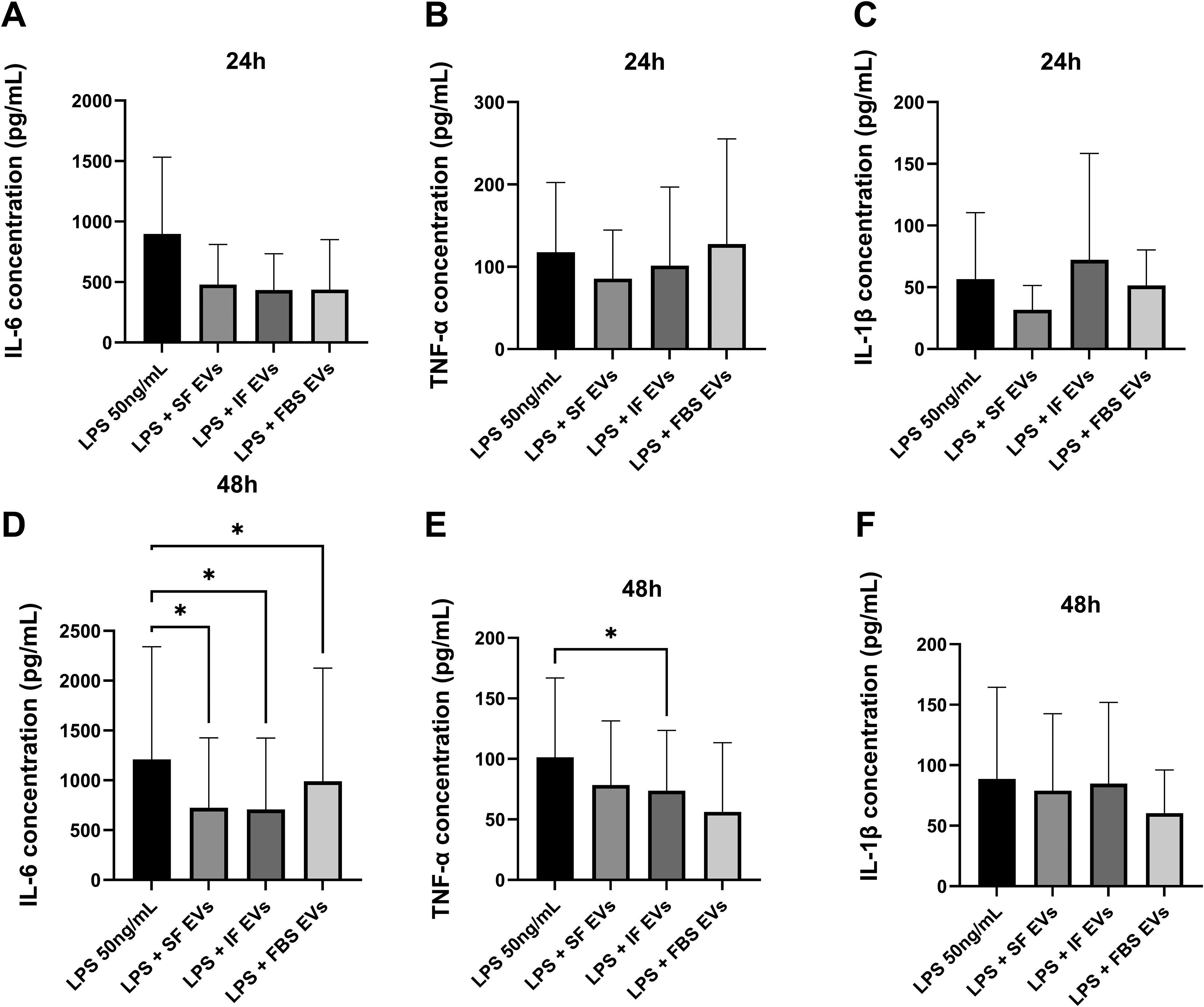
Quantification of
IL-6 concentrations among the groups are depicted in Fig. 5A, D. There were no statistically significant differences between the untreated and EV-treated groups after 24 h incubation (SF, P = 0.15; IF, P = 0.1), but the addition of FBS-EVs appeared to decrease the concentration of IL-6 (437.6 ± 413.4 pg/mL, P = 0.09) compared with the control (897.6 ± 634.3 pg/mL) (Fig. 5A). IL-6 levels were statistically significantly decreased at 48 h following the addition of EVs from SF-EVs (723.9 ± 701.9 pg/mL, P = 0.0383), IF-EVs (706.8 ± 717.8 pg/mL, P = 0.0323), and FBS-EVs (991.1 ± 1,135 pg/mL, P = 0.0364) compared with the LPS control group (1,210 ± 1,130 pg/mL) (Fig. 5D).
mRNA expression of
TNF-α levels among the groups are illustrated in Fig. 5B, E. None of the EV groups after 24 h post-addition to stimulated mixed glia was able to significantly affect the concentration of TNF-α (SF, P = 0.41; IF, P = 0.78; FBS, P = 0.87) compared with the untreated control (Fig. 5B). However, after 48 h of incubation, IF-EVs statistically significantly dampened TNF-α concentration (73.83 ± 49.74 pg/mL, P = 0.0386), compared with the control (101.2 ± 65.70 pg/mL) (Fig. 5E). At the same time point, even though a significance was not found, the addition of FBS-EVs appeared to decrease the production of TNF-α (56.22 ± 57.27 pg/mL, P = 0.05), compared with the control (101.2 ± 65.70 pg/mL) (Fig. 5E).
The concentration of IL-1β among the groups is shown in Fig. 5C, F. No significant differences were noted for the release of IL-1β between the EV-treated and the control groups after incubation for 24 h (SF, P = 0.55; IF, P = 0.82; FBS, P = 0.93) and 48 h. However, it appears that the concentration of IL-1β decreased after 48 h of incubation with all EVs compared with the control group (SF, 78.86 ± 63.67 pg/mL, P = 0.12; IF, 84.73 ± 67.14 pg/mL, P = 0.72; FBS, 60.41 ± 35.71 pg/mL, P = 0.96) (Fig. 5F).
MSC-EVs decrease LPS neuroinflammation in feline mixed glia by modulating common inflammatory signaling pathways
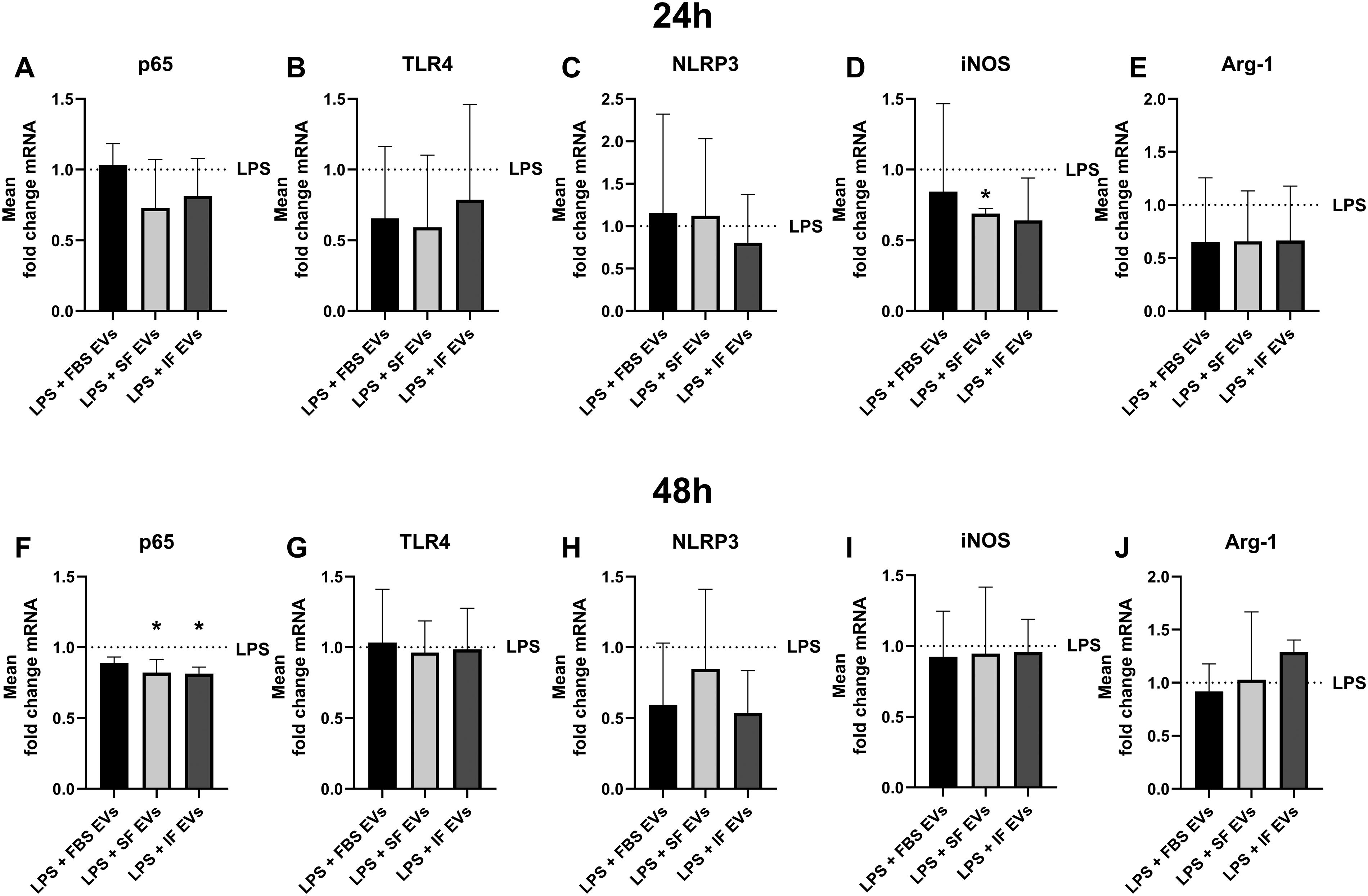
To study the mechanism of action of how the different MSC-EVs decreased the production of pro-inflammatory cytokines in LPS-stimulated feline mixed glia, we relatively quantified the mRNA expression of factors involved in the IF process via reverse transcription quantitative polymerase chain reaction. Specifically, we assessed the gene expression of p65, TLR4, NLRP3, iNOS, and Arg-1 in inflammatory feline brain cells after the addition of SF-, IF-, and FBS-EVs or vehicle treatment for 24 and 48 h (Fig. 6, n = 3).
p65 mRNA expression is illustrated in Fig. 6A, F. After 24 h of treatment, SF-EVs (mean fold change = 0.7299 ± 0.3425, P = 0.2) and IF-EVs (mean fold change = 0.8149 ± 0.2638, 0.43) appeared to decrease the transcription of p65 compared with the control in feline stimulated mixed glia, even though this finding was not statistically significant (Fig. 6A). The addition of SF- and IF-EVs to mixed glia for 48 h statistically significantly downregulated p65 mRNA compared with the untreated group (mean fold change, SF-EVs: 0.8214 ± 0.09143, P = 0.0196; IF-EVs: 0.8141 ± 0.04629, P = 0.0164) (Fig. 6F).
TLR4 mRNA expression levels are shown in Fig. 6B, G. Even though a significance was not identified, the addition of all different isolates of EVs for 24 h appeared to reduce the transcription of TLR4 in LPS mixed glia compared with the LPS group (mean fold change, FBS-EVs: 0.6554 ± 0.5074, P = 0.69; SF-EVs: 0.5915 ± 0.5100, P = 0.61; IF-EVs: 0.7873 ± 0.6739, P = 0.94) (Fig. 6B). Finally, no statistically significant differences were noted from the addition of MSC-EVs in stimulated mixed glia (FBS-EVs, P = 1; SF-EVs, P = 0.99; IF-EVs, P = 1) from cats after 48 h (Fig. 6G).
NLRP3 mRNA expression from all groups is depicted in Fig. 6C, H. The addition of all the EV products did not significantly affect the mRNA levels of NLRP3 after 24-h (FBS-EVs, P = 0.99; SF-EVs, P = 0.99; IF-EVs, P = 0.92) and 48-h treatment (FBS-EVs, P = 0.52; SF-EVs, P = 0.96), but IF-EVs consistently trended to downregulate its levels compared with the untreated groups (mean fold change, 24 h: 0.8034 ± 0.5703, P = 0.92; 48 h: 0.5338 ± 0.3017, P = 0.27) (Fig. 6C, H).
iNOS mRNA expression is graphed in Fig. 6D, I. Only, SF-EVs statistically significantly downregulated the transcription of iNOS after 24 h (mean fold change = 0.6899 ± 0.03626, P = 0.0115), even though all isolates trended to lower its mRNA levels compared with the control group (mean fold change, FBS-EVs: 0.8447 ± 0.6206, P = 0.97; IF-EVs: 0.6408 ± 0.2993, P = 0.39) (Fig. 6D). Finally, no significant mRNA fold changes were observed following the addition of various isolates of EVs for 48 h (FBS-EVs, P = 0.97; SF-EVs, P = 1; IF-EVs, P = 0.99) (Fig. 6I).
The changes in mRNA transcription of Arg-1 following the addition of different isolates of EVs are depicted in Fig. 6E, J. No significant differences were identified for Arg-1 mRNA expression after 24 h (FBS-EVs, P = 0.77; SF-EVs, P = 0.66; IF-EVs, P = 0.71) and 48 h (FBS-EVs, P = 0.94; SF-EVs, P = 1) following the addition of various EV isolates. However, it seems that the addition of IF-EVs tended to increase the gene transcription of Arg-1 after 48 h compared with the EV-untreated mixed glia group (mean fold change = 1.288 ± 0.1142), but this finding was not significant (P = 0.12) (Fig. 6J).
Discussion
The main goal of this study was to determine whether the cell culture conditions of MSCs can affect the characteristics and functionality of the generated EVs and further identify which condition is more efficient in suppressing the inflammatory responses elicited by stimulated mixed glia from cats. First, we proved that EVs derived from different culture conditions of MSCs vary in size and yield, as previously reported. 34 Second, we successfully isolated, characterized, and optimized the stimulation protocols of feline mixed glia to test and compare the immunomodulatory properties of various isolates of EVs when added to stimulated mixed glia. We found that all EV treatments tended to alleviate neuroinflammation in vitro, by suppressing the production of pro-inflammatory cytokines and by modulating the gene transcription of key factors in inflammation after 48 h. We also observed a differential in vitro response against each EV isolate, due to their distinct characteristics. In summary, our results indicate that EVs derived from primed MSCs with inflammatory cytokines (IF-EVs) exhibited an enhanced immunosuppressive function when added to feline stimulated mixed glia compared with EVs from non-primed MSCs.
We previously proved that the characteristics and function of EVs are affected by the culture conditions of the parent cells. 34 This study confirms our previous findings; however, we identified differences in EVs’ median size between our two studies. In this study, we determined the size and the concentration of EVs via NTA, which is considered the golden standard for their characterization. 50 NTA’s ZetaView utilizes a laser in the 488 nm wavelength to assess the Brownian motion of nanoparticles that determines particle concentration and size. Specifically, we found a higher median size of all EV products (<150 nm) compared with the TEM analysis used in our last study (<100 nm). 34 This finding is in accordance with previous results where NTA devices often overestimate the EV size compared with TEM analysis. 50 Specifically, preparation methods used for TEM can cause vesicle shrinkage, whereas NTA methods fail to detect vesicles below 50 nm, resulting in increased mean size distribution value of EVs. 50 We also observed that EVs from inflammatory-primed MSCs were the biggest in size (154.2 nm) compared with the non-primed counterparts (SF; 44.8 nm, FBS; 144.6 nm), which has been reported previously. 51
In addition, nonsignificant discrepancies were observed in particle concentration comparing the two quantification methods, NTA and the enzymatic assay. NTA is based on the physical characteristics of the particle. On the contrary, EXOCET is an enzymatic and colorimetric assay that determines the EV yield, by relying on the acetylcholinesterase (AChE, enzyme) enriched within EVs and it is calibrated via NanoSight. 52 AChE’s presence and the lower particle detection limit of NTA (<50 nm) could explain why the count as determined via the enzymatic assay trended to be higher in the EV isolates compared with the NTA method. Similar to our previous study, primed MSCs with inflammatory cytokines had a higher particle concentration than nonprimed EVs via the enzymatic assay’s measurement. 34 Finally, our studies found that EVs derived from FBS-MSC conditions had the highest protein and particle concentration than among the other EV groups. This is in agreement with our previous study. 34 That could be because the presence of serum promotes vesicle biogenesis, and the depletion of vesicles from the FBS itself might be incomplete. 34 Thus, we can conclude that the EV characterization methods are far from perfect, and researchers need to develop new and reliable methods to manufacture and characterize them, to eliminate the bias in scientific results, and to increase the robustness of EV-based therapeutics.
Our method successfully generated mixed glia cultures from normal cats, which allowed us to test the functionality of different EV isolates. We have decided to test our hypothesis using mixed glia since this population possibly represents more accurately the in vivo CNS environment during neuroinflammatory processes. Specifically, microglial cell protagonist in immune responses in CNS disease, 53 but astrocytes are the main CNS regulatory cells that modulate microglial phenotype and function via factor secretion. 39,48 In addition, the extensive cross talk between glial cells and peripheral immune cells is a consequence of CNS damage and it is vital for the propagation of inflammation in neurological diseases. Thus, should be a target for new therapeutics. 54,55 Therefore, we reasoned to focus our study on the effect of EVs against neuronal-mixed glia cultures, consisting of endothelial cells, pericytes, microglia, astrocytes, and neurons. Those populations were confirmed by our ICC and FC data. Specifically, we found that endothelial and perivascular-like cells were the most abundant cell type present in our feline brain cultures, which is expected in primary cell culture. 43,56 ICC unpublished data confirmed the absence of oligodendrocytes since they require specific culture conditions. Olig2 is a reliable marker since it is an intracellular protein present in all stages of oligodendrocytes’ maturation. 57 Microglia were identified via the expression of ionized calcium binding adaptor molecule 1 and CD11b or Mac-1 surface protein or integrin alpha molecule that regulates cell kinetics. 58 Expression of those markers in the brain corresponds mostly to microglia as commonly used in the literature, 59 which consisted the 20.96% ± 11.35 of feline mixed glia. This agrees with reported results where brain resident, brain infiltrated, parenchymal, or nonparenchymal cells range between 5% and 20% in the entire CNS. 43,46,60,61 Astrocytes represented 29.80% ± 6.63 of the feline mixed glia, similar to the previously reported 20%, based on the expression of GFAP. 61,62 Neurons could not be quantified via FC, because of the absence of feline cross-reactive antibodies. A high SD was noted in the expression of the brain cell markers in FC, which is attributed to the variability in cell culture from the donors. Thus, future studies must optimize mixed glia production and their characterization for translational purposes.
Our protocols for generating feline mixed glia are effective, easy, and affordable. Density gradient and magnetic sorting protocols for cell isolation can be pursued, but they result in low cell yield, and it is challenging to find feline-specific antibodies. Furthermore, the fluorescence activated cell sorting method of cell isolation is possible, but it can activate microglia and decrease cell viability. 43 Due to limitations on the availability of feline-specific reagents, we decided to utilize a widely available protocol of isolation of mixed glia using a dissociation process. We observed that the isolation procedure was still stressful for the primary cells, so they needed at least a week to recover, form structures, and reattach. Cell culture duration was optimized to get sufficient cell numbers and to prevent the domination of contaminating fibroblast-like cells in the culture. 41,63 Initially, we used NPC media to benefit the proliferation of astrocytes and neuronal cells in the primary cultures, without altering the functionality of microglial cells by adding macrophage colony-stimulating factor 43 and permitting extensive fibroblast growth, due to FGF-2 presence.
Our main goal was to assess the anti-inflammatory potential of different MSC-EV isolates against LPS-stimulated feline mixed glia. We treated the cells with LPS, which is a commonly used stimulant, and we screened the appropriate dose of 100 ng/mL (data not shown). 31,64 LPS-induced inflammation and neuroinflammation share mechanistic similarities since both cause microglial stimulation, oxidative stress, and activation of the TLR4 signaling pathway with the subsequent production of pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β. 64 Therefore, we quantified the levels of TNF-α, IL-6, and IL-1β in EV-treated and untreated groups via ELISA, which determines the concentration of protein that contributes to the inflammatory response. Therefore, our data accurately represent the microenvironment in feline LPS-mixed glia. The observed IL-6, TNF-α, and IL-1β concentrations are similar to those previously reported in vitro. 31 Our results proved that all the tested isolates of EVs from MSCs dampened IL-6, TNF-α, and IL-1β levels in feline mixed glia after 24 h and 48 h. However, only IF-EVs significantly decreased in vitro the production of IL-6 and TNF-α after 48 h treatment, which is important in resolving chronic inflammation. Similar findings have been reported in the literature suggesting that primed MSC-EVs have enhanced anti-inflammatory properties. 65 However, statistically significant differences were only detected for some of the tested cytokines. This could be due to the small sample size of the study and the increased variability in responses between biological replicates.
To evaluate the mechanism by which EVs modulated responses, we decided to evaluate the expression of genes mainly involved in neuroinflammation. LPS-induced inflammation stimulates microglia via TLR4 signaling and directly triggers the release of IL-6 and TNF-α. 31 We found that EVs tended to reduce the expression of TLR4 in feline mixed glia; however, a stronger immunosuppressive effect was noted when assayed for cytokine levels via ELISA. Thomi et al. reported that MSC-EVs inhibited the interaction between LPS and TLR4/CD14 in microglial cell lines, 31 but this is not the case in feline glia based on our findings. More studies are needed to thoroughly evaluate the inflammatory pathways in various animal models since there are species differences in gene expression.
We found that SF- and IF-EV treatment significantly repressed the transcription of p65 or rela compared with the untreated control. p65 is the transcriptionally active subunit of NF-κB, which is responsible for the expression of the pro-inflammatory cytokines IL-6, TNF-α, and IL-1β. 66 Therefore, p65 downregulation from EVs might be responsible for suppressing the levels of IL-6 and TNF-α. Zhang et al. similarly reported that the EV treatment decreased the protein levels of NLRP3 and phosphorylated p65 in vivo. 32 We also evaluated the expression of NLRP3 inflammasome in inflammatory glia following treatment with SF-, IF-, and FBS-EVs. This is because the inflammasome cascade positively regulates the production of IL1-β. 31,32 All EV treatments trended to suppress the transcription of nlrp3 in stimulated mixed glia after 48 h. However, this suppression was not significant and could explain why EVs did not significantly decrease the production of IL-1β after 48 h. To conclude, our findings indicate that IF- and SF-EVs alleviate LPS inflammation in feline mixed glia by inhibiting the NF-κB formation and decreasing the production of IL-6 and TNF-α. However, the same culture conditions did not seem to interfere with the inflammasome cascade at least in vitro, as previously shown. 31 Finally, FBS-EV treatment did not have a significant effect in the transcription of p65 and nlrp3.
Finally, we assessed the expression of arg-1 and inos to evaluate the polarization of mixed glia from the M1- inflammatory to the M2 anti-inflammatory state. In microglial cells, iNOS is an enzyme that promotes oxidative stress and competes for the same substrate (arginine) with the Arg1 enzyme, which downregulates the oxide production as an anti-inflammatory mechanism. 67 We found that IF-EVs upregulated—but not significantly—the transcription of arg1 after 48 h, showing an antioxidative effect and reversion of mixed glia from M1 to the M2 phenotype. In agreement with that, Domenis et.al. showed that priming the MSCs with TNF-α/IFN-γ enhances the immunoregulatory properties of the released EVs, by loading them with micro ribonucleic acids (miRNAs) that induce M2 macrophage polarization. 68 A stronger therapeutic effect was not identified because of iNOS competition at 48 h since its mRNA levels were only slightly downregulated. This might explain why IL-1β levels were only slightly decreased after the EV treatment for 48 h since studies suggest that LPS induction of iNOS promotes IL-1β signaling cascade. 69 inos transcription was significantly decreased after 24 h treatment with SF-EVs, meaning a possible microglial resistance to the M1 state. Similar results were observed with adipose stem cell SF-EVs that reduced inOS expression in stimulated macrophages after short incubation. 70 Our results indicate that EVs have an effect on microglial phenotype, but cannot induce total phenotype conversion. In addition, this effect seems to be EV product specific since IF-, FBS-, and SF-EVs regulate microglia’s functionality differently. Another possible reason that EVs did not switch microglia’s phenotype is that we used a mixed population of cells; therefore, it is impossible to evaluate individual cellular responses. However, recent literature rejects the bipolar phenotype of microglia and claims that they gain disease-specific activation profiles. 54 Feline studies that evaluate the expression of those genes are needed since comparison with other species’ gene expression patterns cannot be made. Finally, EVs trended to decrease the expression of the inflammatory biomarkers tested, but in several cases was that reduction significant. The reason could be the differential responses of the biological replicates (feline glia) against the EV treatments.
This study has several limitations. First, the number of animals used for this study was limited (n =3 or 5) in an attempt to follow the principles of the 3Rs (Replacement, Reduction, and Refinement) to ensure ethical and responsible animal use without compromising their welfare while observing meaningful and statistically relevant trends. Moreover, this research represents a pilot investigation to generate preliminary data, establish feasibility, and optimize experimental conditions for future studies regarding stem cell-derived EVs’ anti-inflammatory properties in large animal neuroinflammation models. In addition, the cats were age-matched from both sexes and maintained under strictly standardized conditions to minimize biological variability, which increases the statistical power of the data despite the small cohort. Overall, the sample size was sufficient to test the efficacy of MSC-EVs and achieve significance with moderate power based on published data. 17,71,72 Second, relevant research evaluating the effect of stem cell-derived EVs in feline cells is absent to support our findings further. Furthermore, we used an LPS stimulation protocol to assess the EV-therapeutic effect. However, it should be noted that LPS stimulation of mixed glia evokes a broad and robust immune response after a bacterial infection, which is not the common cause of CNS damage and probably pro-cytokine stimuli would be more relevant. 73 In addition, we chose to subsequently treat the LPS-stimulated mixed glia with the different EV isolates to mimic clinical application and evaluate their effect on the acute phase of injury. We tested only one EV dose (108 P/mL), which was determined using particle concentration instead of protein concentration like most studies. 74 The EV production method has not been standardized yet and so the isolation method of EVs could affect their therapeutic potential and complicate future clinical application. Further research is needed to standardize and optimize the production and characterization methods of EVs. Moreover, future studies should evaluate the exact mechanism by which MSC-derived EVs alleviate disease-related neuroinflammation and identify the responsible miRNAs.
This is the first study to evaluate and compare the effectiveness of different isolates of EVs from MSCs against mixed glia from normal cats. The feline model strengthens the translational ability of our study since it provides a human-relevant platform to thoroughly investigate the effects of stem cell EVs. The next steps consist of evaluating whether various EV isolates could modulate the function of other immune cells, such as peripheral blood mononuclear cells and specifically T regulatory cells. In addition, future studies should explore the effect of EVs on mixed glia functionality isolated from cats with a naturally occurring neurodegenerative disease. Ultimately, the therapeutic effect of EVs on attenuation of chronic neuroinflammation after administration to a small animal model of a naturally occurring neurodegenerative disease is needed to be able to draw clinically appropriate conclusions.
Conclusion
In summary, EVs derived from primed (IF) and nonprimed (SF) human MSCs strongly alleviate neuroinflammation in the feline in vitro model, by decreasing the production of main inflammatory mediators. A similar but less profound effect was observed from FBS-EVs. Most importantly, we found that IF-EVs exhibit an enhanced immunosuppression, unlike the FBS-EVs. Thus, we can conclude that the production method can be crucial and influence their characteristics and biological action; therefore, the EV environment should be taken into consideration when designing research and clinical studies. Finally, this study shows that stem cell EVs can alleviate inflammatory responses elicited by LPS-stimulated mixed glia cells isolated from feline and, thus, might serve as the next-generation therapeutic to treat neuroinflammatory processes that occur in various neurodegenerative diseases.
Footnotes
Acknowledgments
The authors would like to thank Dr. Rie Watanabe and Dr. James Gillespie for contributing to the flow cytometry data acquisition and Dr. Laura Huber for her statistics advice. Finally, this work would not have been possible without the help of the personnel in the feline research colony of the Scott Ritchey Center.
Author Disclosure Statement
The authors declare no conflict of financial or personal interest.
Funding Information
This work was supported by the Animal Health and Disease Research Program, College of Veterinary Medicine of Auburn University.