
Abstract
Recent clinical trials have led to the worldwide suspension of aprotinin, the most commonly used antifibrinolytic agent in fibrin-based tissue engineering. For future clinical applications of fibrin-based scaffolds, a suitable, alternative fibrinolysis inhibitor must be identified. The present study aimed to evaluate tranexamic acid (trans-4-aminomethyl-cyclohexane-1-carboxylic acid [t-AMCA]) as an alternative fibrinolysis inhibitor to aprotinin for cardiovascular tissue engineering applications. The effects of various concentrations of t-AMCA (30–160 μg/mL) and aprotinin on fibrin gel-lysis were spectrophotometrically quantified in vitro. Cytotoxic effects of t-AMCA and aprotinin on carotid artery–derived cells, in addition to their influence on fibrin gel mechanical strength, were examined. Further, the influence of t-AMCA versus aprotinin on three-dimensional fibrin-based constructs was analyzed using light microscopy, scanning electron microscopy, and transmission electron microscopy. The results demonstrated that neither t-AMCA (30–160 μg/mL) nor aprotinin elicited cytotoxic effects on cultured cells. Although aprotinin showed reduced fibrinolysis in the presence of plasmin compared to t-AMCA, no significant difference was obtained under standard culture conditions. Additionally, t-AMCA had no negative influence on the mechanical stability of fibrin gels, which also demonstrated excellent cell morphology, tissue development, and ultrastructure. The results from the present study demonstrate that t-AMCA may be a suitable alternative to aprotinin for controlling the in vitro degradation rate of fibrin-based tissue-engineered constructs.
Introduction
The basic approach to cardiovascular tissue engineering is to seed tissue-synthesizing cells into or onto a scaffold, which serves as a temporary extracellular matrix (ECM) configured in the three-dimensional (3D) shape of the structure to be repaired/replaced, to condition this structure in vitro, and finally to implant the structure into the patient. Using autologous cells, the tissue-engineered construct retains complete immunological integrity with high biocompatibility and a low risk of infection for the patient.2,3
The use of fibrin gel as a natural, biodegradable, and autologous scaffold in cardiovascular tissue engineering combines various promising advantages and has been investigated in our research group3–5 and other groups6–10 for various applications. Fibrin gel is a biodegradable polymer that can be produced from the patient's own blood, avoiding the potential risks of foreign body reaction and virus infection.2,4,11 Fibrin is easily synthesized in vitro by mimicking the final stage of the blood coagulation cascade through the combination of thrombin, fibrinogen, and calcium chloride, resulting in a polymerized fibrin gel similar to a physiological clot. Due to rapid gelation of the fibrin–cell suspension, a homogenous distribution of cells can be achieved, and the potential for injection molding 2 makes it possible to form customized 3D cardiovascular structures for the individual. In addition, fibrin has demonstrated excellent properties for cellular growth and tissue development as a 3D matrix structure.4,11
The degradation rate of fibrin-based tissue-engineered constructs in vitro can be delayed by addition of fibrinolytic inhibitors. A commonly used, thoroughly evaluated antifibrinolytic agent is aprotinin—a polypeptide serine protease inhibitor that slows or stops fibrinolysis by inhibiting kallikrein, plasmin, and platelet-activation factors.12,13 Optimal tissue development and collagen formation by aortic myofibroblasts has been demonstrated at supplemental aprotinin concentrations of 20 μg/mL (130 KIU/mL) in cell culture medium. 4 However, the recent association of aprotinin with increased risk of complications, including renal failure, myocardial infarction, heart failure, stroke, encephalopathy, or even death in clinical application, have led to the suspension of worldwide marketing of aprotinin. 14 With regard to future clinical applications of fibrin in tissue engineering, a suitable, alternative fibrinolysis inhibitor needs to be identified. A safe alternative fibrinolysis inhibitor recommended for cardiac surgery is the clinically approved, tranexamic acid (trans-4-aminomethyl-cyclohexane-1-carboxylic acid [t-AMCA]). 14 t-AMCA is an antifibrinolytic agent that competitively inhibits the activation of plasminogen to plasmin via reversible binding to the lysine-binding site on plasminogen. At much higher concentrations, t-AMCA acts as a noncompetitive inhibitor of plasmin. It prevents fibrinolysis by blocking engagement of the activators of fibrinolysis, plasminogen, and plasmin, with fibrinogen and fibrin. 15
The overall goal of the present study is to evaluate the potential of t-AMCA as a safe, alternative antifibrinolytic drug to aprotinin for the development of fibrin-based cardiovascular structures.
In cardiac surgery, aprotinin is usually administered at a loading dose of 2 million KIU followed by continuous infusion of 500,000 KIU/h, and an additional dose of 2 million KIU, whereas the concentration of aprotinin supplemented to cell culture medium is 130 KIU/mL. 4 To determine a suitable t-AMCA concentration for supplementation to cell culture medium, the ratio of the mean aprotinin concentration in blood commonly used in cardiac surgery to the aprotinin concentration used in in vitro cell culture 4 was determined. The same ratio was used to calculate a suitable t-AMCA concentration. The concentration of 130 KIU/mL aprotinin corresponded to 80 μg/mL t-AMCA. The t-AMCA concentrations used for our in vitro studies ranged from a low-dose of 30 μg/mL to a high-dose concentration of 160 μg/mL.
A number of t-AMCA concentrations (30–160 μg/mL; ∼0.2–1 mM) were analyzed to determine their effects on (i) the inhibition of fibrinolytic degradation of fibrin gels, (ii) cytotoxicity (necrosis, proliferation, and apoptosis), (iii) their potential application in cell-seeded 3D fibrin gel structures, and (iv) their influence on the mechanical properties and structure of fibrin gels.
Materials and Methods
Cell expansion and culture
A mixed population of smooth muscle cells/fibroblasts was isolated from juvenile sheep carotid artery under sterile surgical conditions. The arterial tissue was washed with phosphate-buffered saline (Gibco, Karlsruhe, Germany), and endothelial cells were subsequently removed by using 2.4 IU/mL dispase (Roche Diagnostics, Mannheim, Germany). The arteries were then cut into 1-mm rings, bathed in primary cell culture medium (Dulbecco's modified Eagle's medium [DMEM] with 10% fetal bovine serum [FBS], and 1% antibiotic/antimycotic solution; all from Gibco), and maintained in a humidified incubator at 37°C and 5% CO2. Upon confluency, cells were serially passaged by trypsinization (0.25% trypsin/0.02% ethylenediaminetetraacetic acid solution; Gibco).
Fibrinogen solution preparation
Lyophilized, plasminogen-free fibrinogen from sheep plasma (Sigma, Seelze, Germany) was dissolved for 2.5 h in purified water (MilliQ™, ∼20 mg/mL; Millipore, Schwalbach, Germany). After dissolving, the fibrinogen solution was dialyzed against Tris-buffered saline (TBS) in a 6000 to 8000 MW cut-off membrane (Novodirect, Kehl, Germany) overnight. The fibrinogen concentration was determined by measuring the absorbance at 280 nm (extinction coefficient, 1.55) using a spectrophotometer (Tecan Infiniti M200; Tecan Deutschland, Crailsheim, Germany). The final concentration of fibrinogen solution was diluted to 10 mg/mL with purified water (MilliQ).
Unseeded 3D fibrin gel production
Fibrin gels were cast in 24-well plates with a total volume of 1000 μL. After the addition of 350 μL of TBS and 75 μL CaCl2 (50 mM) per well, 75 μL thrombin (40 U/mL; Sigma) and 500 μL fibrinogen (10 mg/mL) were added, gently mixed, and incubated for polymerization at 37°C. The final fibrinogen concentration was 5 mg/mL.
Seeded 3D fibrin gel culture
Fibrin gels (500 μL, n = 4 for each concentration) were cast in 24-well culture plates by mixing 175 μL TBS containing 1 × 106 carotid artery–derived cells with 250 μL fibrinogen solution (10 mg/mL) and 37.5 μL 50 mM CaCl2 in TBS. Polymerization of each gel was initialized by adding 37.5 μL of thrombin solution (40 U/mL), and proceeded for 30 min at room temperature. Fibrin gels were subsequently incubated with DMEM supplemented with 10% FBS, 1% antibiotic solution, and 1 mM L-ascorbic acid 2-phosphate (Sigma). Culture medium was also supplemented either with t-AMCA (Cyklokapron®; Pfizer, Karlsruhe, Germany) at a concentration of 30, 80, 100, 130, or 160 μg/mL, or with aprotinin (Bayer Vital, Leverkusen, Germany) at 130 KIU/mL. Control gels (n = 4) were cultured in the above culture medium without supplementation of fibrinolysis inhibitors. After 60 min of incubation at 37°C and 5% CO2, fibrin gels were gently detached from the culture dish and subsequently cultured for 12 days. Culture medium was replaced every 2 days. After 12 days, one half of each gel was prepared for light microscopy analysis, whereas the other half was prepared for transmission electron microscopy (TEM) analysis.
Fibrinolysis (with plasmin and without plasmin)
Fibrinolysis was evaluated in two ways using a modification of the method described by Smith and Morrissey 16 : in the first procedure (accelerated experiment), 2 μL plasmin (80 nM) was added before thrombin addition to the clotting mixtures to simulate the in vivo situation (n = 3 for each t-AMCA concentration and aprotinin); in the second procedure (simulating the in vitro cultivation), the fibrinolytic activity of standard cell culture medium (DMEM with 10% FBS and 1% antibiotic/antimycotic solution; all from Gibco) and cells was evaluated without additional plasmin supplementation (n = 3 for each t-AMCA concentration and aprotinin).
Fibrin gels (200 μL) were cast in triplicate in 96-well culture plates by mixing 70 μL TBS, 15 μL CaCl2 (50 mM), 15 μL thrombin (40 U/mL; Sigma), and 100 μL fibrinogen solution (10 mg/mL). After polymerization at room temperature (2 h), gels were overlaid with culture medium (phenol red–free DMEM with 10% FBS; Gibco) supplemented either with t-AMCA at a concentration of 30, 80, 100, 130, or 160 μg/mL, or with aprotinin at 130 KIU/mL. Clot formation and lysis, reflected by an initial increase in optical density (OD) immediately after addition of the supplements, was followed by a decrease in turbidity, and was monitored by measuring the turbidity (A405) at room temperature as a function of time until the gels were completely lysed. Absorption was measured every 30 min (Tecan Infiniti M200).
Biomechanical analysis
The mechanical properties of the cell-free fibrin gels were examined using a custom-designed burst pressure device. Fibrin gels (n = 6) were cast in 24-well plates with a total volume of 1000 μL, and allowed to polymerize for 1 h at 37°C. The gels were then incubated with medium containing aprotinin (130 KIU/mL) or t-AMCA (80 or 160 μg/mL) to prevent fibrinolysis. To reduce the high number of samples, only the medium concentration (80 μg/mL) and the highest concentration (160 μg/mL) of t-AMCA were tested. Burst pressure tests were then performed in triplicate after 2 and 20 h incubation at 37°C. Each gel was fixed in a special burst pressure chamber (Applied Medical Engineering, Helmholtz Institute, Aachen, Germany), and water was pumped through the gels by a peristaltic roller pump (IPC4; Ismatec SA Labortechnik, IDEX Corporation, Glattbrugg, Switzerland) connected to the chamber at a constant flow rate (6 mL/min). The induced pressure was measured with a pressure sensor (Juma Midas, Fulda, Germany) connected to a PC using the software LabVIEW (version 7.1; National Instruments, Austin, TX). The highest pressure value measured before gel failure was defined as the burst pressure. The results are presented as values relative to the control (i.e., gels supplemented with 130 KIU/mL aprotinin), which was set at 100%.
Cytotoxicity assays
Lactate dehydrogenase release (necrosis assay)
To examine the cytotoxic effect of t-AMCA on the viability of carotid artery–derived cells, a commercial assay kit was used (CytoTox® 96 Non-Radioactive Cytotoxicity assay; Promega, Mannhein, Germany). This colorimetric test measures the release of lactate dehydrogenase (LDH) upon cell lysis into the culture supernatant. Cells were plated in a 24-well plate at 3.0 × 105 cells/well in medium (DMEM with 10% FBS; Gibco) and were allowed to proliferate for 48 h before treatment. Culture medium was removed, and test medium (DMEM without FBS; Gibco) supplemented with aprotinin (130 KIU/mL) or t-AMCA (30–160 μg/mL) was added to each well. As a negative control, cells were cultured in medium without supplementation of fibrinolysis inhibitors. The amount of LDH released into the culture supernatants was measured after 4 and 24 h of incubation at 37°C. Medium supernatants (150 μL) from each fibrin gel were collected, centrifuged, and transferred (50 μL) to a 96-well plate. Reconstituted substrate mix (50 μL) was added to each well, and the plate was incubated for 30 min at room temperature in the absence of light. After the addition of 50 μL stop solution to each well, the absorbance was measured at 490 nm using a multiwell spectrophotometer (Tecan Infiniti M200). The amount of maximum release of LDH was considered as the amount released by total lysis of untreated cells by freezing the cells for 45 min at −20°C, followed by thawing at 37°C. LDH release was expressed as the percentage of maximal LDH release and calculated according to the following formula: cytotoxicity (%) = OD490 LDH release − blank/OD490 maximum LDH release − blank × 100. The LDH positive control solution (bovine heart LDH) included in the commercial kit was used as a positive control.
MTT assay (proliferation assay)
To determine the effects of t-AMCA on cell proliferation, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltertrazolium bromide (MTT) assay was performed. 17 Briefly, carotid artery–derived cells were plated in a 24-well plate (1.0 × 105 cells per well) in DMEM culture medium and incubated under standard conditions for 48 h. Culture medium was removed, and test media were applied at a concentration of 130 KIU/mL for aprotinin and 30–160 μg/mL for t-AMCA. The viability of cells was determined after 2, 4, and 6 days using the MTT assay. The MTT solution (5 mg/mL in Ca2+- and Mg2+-free phosphate-buffered saline, 40 μL; Sigma) was added to each well and incubated at 37°C for 1 h. The colored formazan crystals were dissolved by adding 400 μL of isopropanol solution (containing 10% formic acid) per well, and the OD was measured at 550 nm using a spectrophotometer (Tecan Infiniti M200). A blank control containing only isopropanol solution was also measured. The assay was performed in triplicate for each treatment, and the mean value with standard error was determined. To calculate the relative viability, the mean absorbance of the sample was divided by the mean absorbance of the control and multiplied by 100.
Caspase-Glo 3/7 assay (apoptosis assay)
The induction of apoptosis was determined by caspase 3/7 activity measurement using the apoptosis Caspase–Glo® 3/7 assay (Promega). This luminescent assay specifically identifies whether caspase-3 and -7, which play key effector roles in apoptosis, are activated. Carotid artery–derived cells were seeded in a white-walled 96-well plate (Nunc, Langenselbold, Germany) at a concentration of 5 × 103 cells per well in medium. Cells were incubated for 24 h at 37°C, medium was removed, and test media were applied for 1 h (data not shown) or 4 h with t-AMCA (30–160 μg/mL) or aprotinin (130 KIU/mL). As a positive control for caspase activation, carotid artery–derived cells were exposed to ultraviolet light, a known inducer of apoptosis, for 2 s as recommended by the product manufacturer. Thereafter, 100 μL of caspase–Glo 3/7 reagent was applied to each sample and incubated for 1 h at room temperature before luminescence was measured (Tecan Infiniti M200). Assays were performed in triplicate for each treatment.
Light microscopy
For light microscopical examination of general tissue morphology and development, Carnoy's fixed, paraffin-embedded, cell-seeded fibrin gels cultivated in the presence of fibrinolysis inhibitors (t-AMCA [30–160 μg/mL], or aprotinin [130 KIU/mL]) were sectioned at 4 μm thickness and were subsequently analyzed by hematoxylin & eosin staining. Sections were viewed using routine bright field light microscopy (AxioImager; Carl Zeiss, Jena, Germany). Images were acquired using a high-resolution CCD color camera (AxioCam MRc; Carl Zeiss).
TEM
Cell-seeded fibrin gels cultivated in the presence of fibrinolysis inhibitors (t-AMCA [30–160 μg/mL], or aprotinin [130 KIU/mL]) were fixed by immersion in 2% glutaraldehyde in 0.1 M Sorenson's buffer (pH 7.4) at room temperature for 24 h, followed by postfixation in 1% osmium tetroxide (Plano, Wetzlar, Germany) in 0.1 M sodium cacodylate buffer for at least 2 h. The specimens were then dehydrated through a graded series of alcohols before being embedded in epoxy resin (Epoxy-Embedding Kit; Fluka Chemie, Deisenhofen, Germany). Ultrathin sections (70–100 nm) with a gold interference color were cut using an ultramicrotome (Reichert Ultracut-E; Reichert-Jung, Heidelberg, Germany), collected on copper mesh grids, and stained with uranyl acetate (Ultrastain-1; Leica, Vienna, Austria) and lead citrate (Ultrastain-2; Leica) using a stainer (LKB Ultrastainer; LKB-Produkter AB, Bromma, Sweden). Ultrastructural examination was then performed using a transmission electron microscope (Philips EM 400T; Philips, Eindhoven, The Netherlands) at an accelerating voltage of 60 kV.
Scanning electron microscopy
Cell-free fibrin gels cultivated in the presence of fibrinolysis inhibitors (t-AMCA [160 μg/mL], or aprotinin [130KIU/mL]) were fixed by immersion in 2% glutaraldehyde in 0.1 M Sorenson's buffer (pH 7.4) for at least 24 h. The gels were dehydrated in acetone and critical-point dried (E-300 Critical Point Dryer; Polaron Equipment, Watford, United Kingdom). Thereafter, the gels were mounted on stubs before being sputtered with gold (Balzers, SCD-030; BAL-TEC AG, Balzers, Lichtenstein) and investigated with a scanning electron microscope 515 (Philips XL-30 ESEM FEG; Philips) at an accelerating voltage of 10 kV.
Statistical analysis
Values are expressed as the mean ± standard error of the mean. The statistical significance was determined using the Student's t-test. Differences were considered to be statistically significant at p < 0.05.
Results
Fibrinolysis
The ability of various t-AMCA concentrations to retard the degradation of fibrin gels was assessed in an in vitro clot lysis assay using two approaches. In the first approach, plasmin was added to the clotting mixture immediately before thrombin addition, whereas in the second approach, fibrin clot polymerization and fibrinolysis was investigated in the absence of plasmin. As shown in Figure 1A, fibrin gels formed in the presence of plasmin were resistant to lysis when aprotinin (130 KIU/mL) was supplemented to medium over the complete test period (10 h). In the presence of t-AMCA (30–160 μg/mL), fibrin gels displayed a lower resistance to lysis. The clots were completely dissolved after approximately 3 h. The mean time to 50% lysis for fibrin clots cultured in medium supplemented with 160 μg/mL t-AMCA was 1.77 h, 1.66 h for 130 μg/mL, 1.31 h for 100 μg/mL, 1.42 h for 80 μg/mL, and 0.83 h for 30 μg/mL. In the second approach, where fibrin clots were formed without plasmin addition (Fig. 1B), the supplementation of t-AMCA to the medium inhibited fibrin degradation at all tested concentrations. The mean time to 50% lysis for fibrin clots formed in the presence of both aprotinin (130 KIU/mL) and t-AMCA (160 μg/mL) was approximately 28.5 h. In the presence of 80 μg/mL, 100 μg/mL, and 130 μg/mL t-AMCA, the mean time to 50% lysis was 25.9 and 24.4 h for fibrin clots formed in the presence of 30 μg/mL t-AMCA, respectively. The fibrinolytic activity was blocked by t-AMCA, with no significant difference to the blockage of activity by aprotinin.
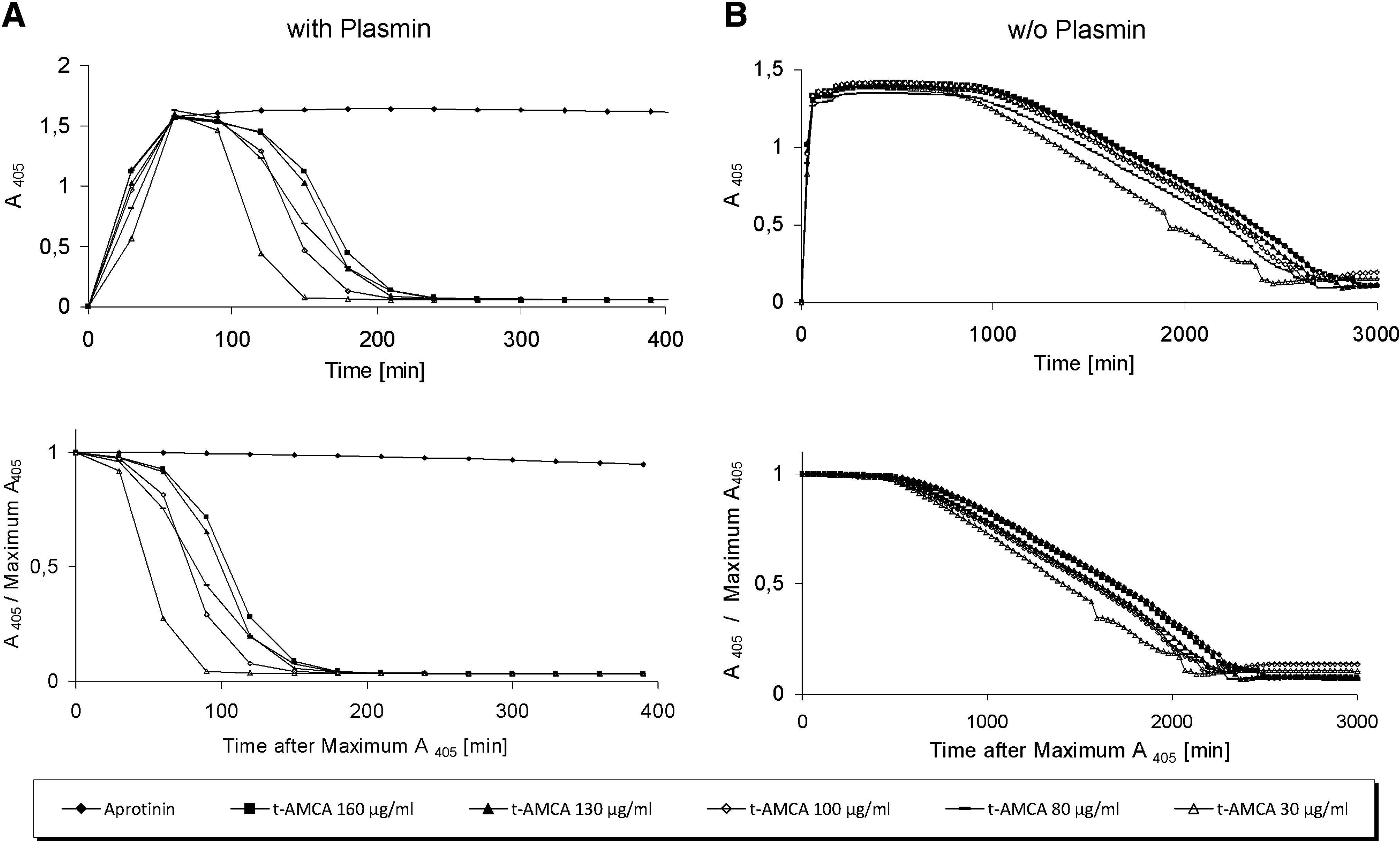
Impact of tranexamic acid (t-AMCA) versus aprotinin on fibrinolysis. (
Mechanical properties
We examined the effect of t-AMCA on the mechanical stability of fibrin gels by incubating fibrin gels for 2 and 20 h with either 80 or 160 μg/mL t-AMCA, followed by burst strength measurements (Fig. 2). After 2 h of incubation at 37°C, fibrin gels cultured in medium containing 160 μg/mL t-AMCA exhibited the highest burst pressure (116 ±5.97 mmHg, n = 6). Fibrin gels cultured in medium supplemented with 20 μg/mL aprotinin (100 ± 8.21 mmHg, n = 6) or 80 μg/mL t-AMCA (102.9 ± 20.51 mmHg, n = 6) showed medium range burst pressure values. After 20 h incubation, the burst pressure of fibrin gels was comparable for all tested conditions (100 ± 4.71 mmHg, n = 6 for aprotinin-supplemented culture; 95.57 ± 5.44 mmHg, n = 6 for 80 μg/mL t-AMCA–supplemented culture; and 99.46 ± 7.33 mmHg, n = 6 for 160 μg/mL t-AMCA–supplemented culture).
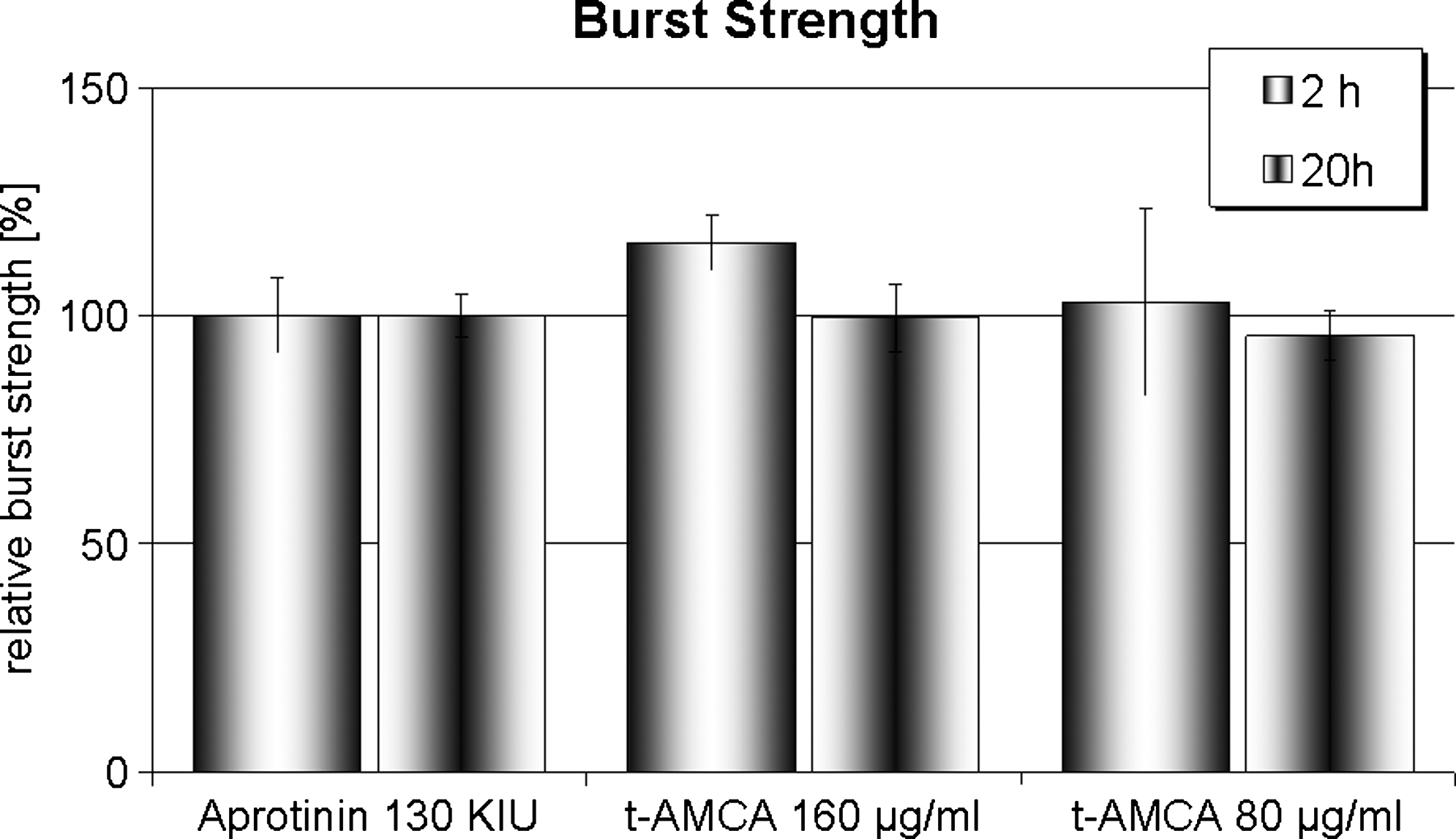
Impact of t-AMCA compared to aprotinin on burst strength of fibrin gels. Burst strength of fibrin gels after 2 or 20 h incubation with medium supplemented with aprotinin (20 μg/mL) or t-AMCA (80 or 160 μg/mL). Data shown represent the mean values ± standard error of at least triplicate experiments.
Cytotoxicity assays
Figure 3A illustrates the respective effects of t-AMCA on LDH release. The exposure to different concentrations of t-AMCA for 4 or 24 h showed no significant difference in LDH release compared to the untreated control cells and the aprotinin group.
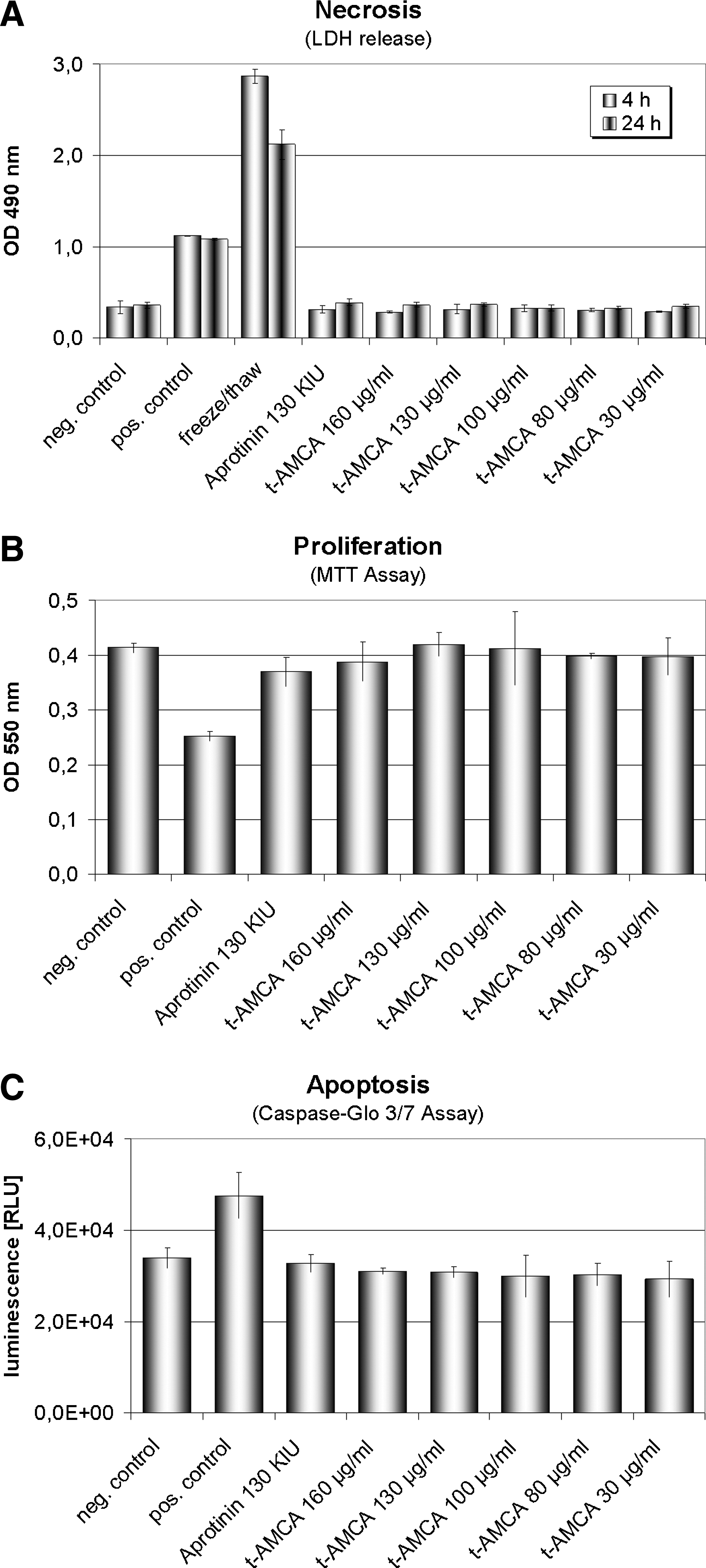
Cytotoxicity assays. (
No significant decrease in cell viability was found in the t-AMCA groups compared with the untreated control (Fig. 3B). None of the tested concentrations of t-AMCA or aprotinin (130 KIU μg/mL) affected growth of the carotid artery–derived cell populations.
In addition, the Caspase–Glo 3/7 assay demonstrated no significant increase in apoptosis between cells treated with t-AMCA compared with untreated controls, whereas the ultraviolet light–treated control showed a significant positive increase in apoptosis (Fig. 3C).
Conventional histology
The microscopic evaluation was used to assess the impact of t-AMCA on cell-seeded 3D fibrin gel structures. Figure 4 shows hematoxylin & eosin staining of carotid artery–derived cells incorporated into a 3D fibrin gel after a culture period of 12 days. The medium was supplemented with 80 μg/mL t-AMCA. The gel demonstrated a homogenous distribution of proliferating cells.

Hematoxylin and eosin stain of carotid artery–derived cells incorporated into a three-dimensional fibrin gel structure. Transverse section through a cell-seeded fibrin gel after a culture period of 12 days in medium supplemented with 80 μg/mL t-AMCA. Scale bar: 50 μm.
TEM
To assess whether the addition of t-AMCA may have an influence on cell morphology/viability, TEM analysis was performed. Figure 5 shows TEM micrographs of fibrin gels after a 12-day culture period in medium supplemented with either aprotinin (130 KIU/mL), or the maximally tested concentration of t-AMCA (160 μg/mL). Cells were well incorporated into the fibrin gel matrix of both samples and exhibited a healthy morphology, and no evidence for cell necrosis was detected at any tested concentration (30 up to 160 μg/mL). In comparison to aprotinin-supplemented gels, t-AMCA–supplemented gels (up to 160 μg/mL) also demonstrated a higher cell density throughout the entire gel structure, with a more extensive ECM.
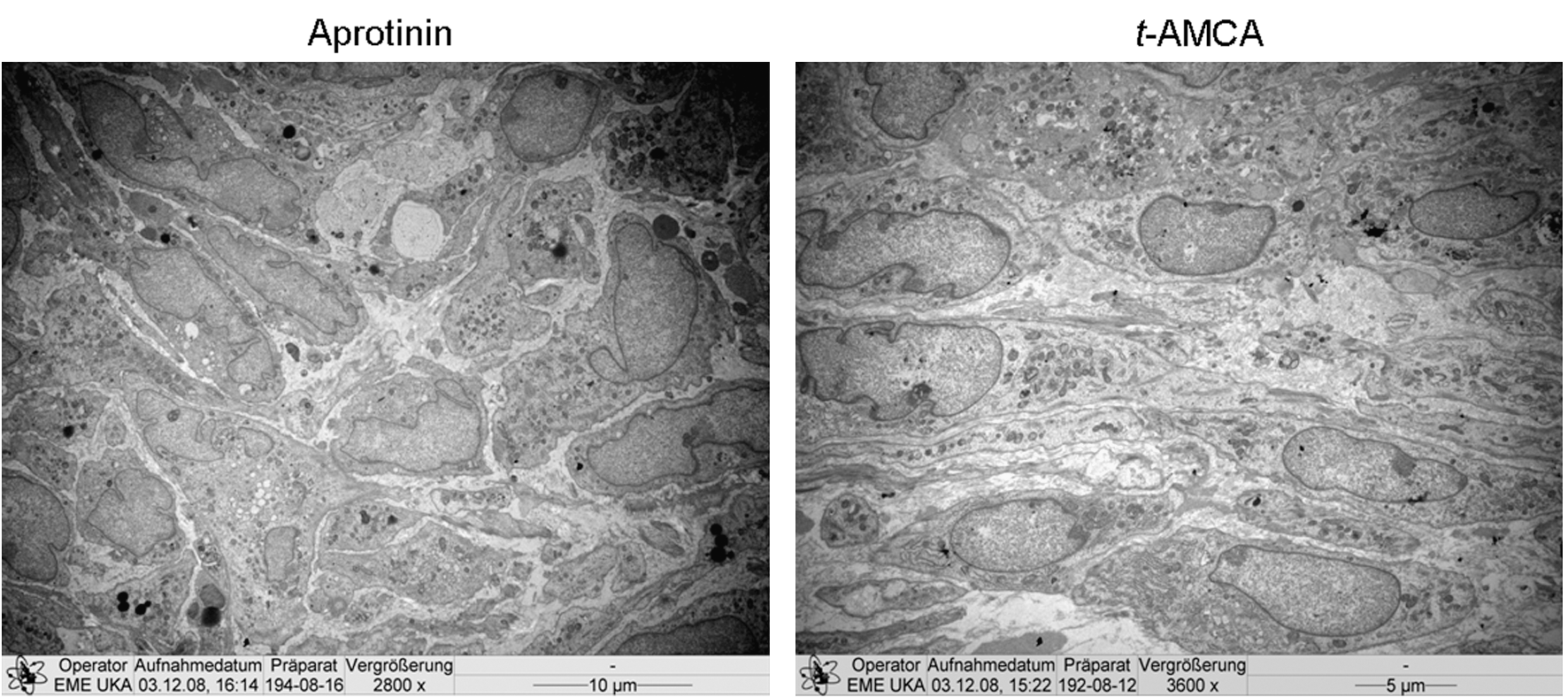
Transmission electron micrographs of three-dimensional fibrin gel structures. Transverse section through a cell-seeded fibrin gel after a culture period of 12 days in medium supplemented with aprotinin (130 KIU/mL) or t-AMCA (160 μg/mL). Scale bar: 10 or 5 μm.
Scanning electron microscopy
Scanning electron microscopy analysis of fibrin gels cultured in the presence of aprotinin (130 KIU/mL) or t-AMCA (160 μg/mL) demonstrated no significant differences in the 3D ultrastructure of the gels (Fig. 6). The fibrin structures were composed of a dense network of long, thin fibers of similar size in both aprotinin- and t-AMCA–supplemented gels.

Scanning electron microscopy pictures of fibrin gels. Fibrin gels were formed in the presence of aprotinin (130 KIU/mL) or t-AMCA (160 μg/mL). Magnification × 15,000. Scale bars: 2 μm.
Discussion
The application of an optimal scaffold material is an important issue in tissue engineering. Fibrin gel has shown many promising results in tissue engineering applications. 18 To preserve the scaffold function of fibrin to maximize the time for cell proliferation and ECM production, fibrinolysis inhibitors have to be used to retard the degradation of fibrin-based scaffolds. Aprotinin, t-AMCA, and ɛ-amino-caproic acid have been analyzed as supplements to the cell culture medium to slow down the degradation, thereby stabilizing the fibrin gel structure.19–22 These antifibrinolytic agents are also currently used in surgery to reduce postoperative bleeding and transfusion requirements.23–27 The serine protease inhibitor aprotinin is a widely used antifibrinolytic drug for tissue engineering applications.4,9 Aprotinin is a naturally occurring, 6000 D molecular weight protein isolated from bovine lung. 28 Aprotinin has been shown to reduce the degradation rate of 3D tissue-engineered fibrin gels and to induce an increase in collagen synthesis and improved tissue development. 4 However, recent clinical trials have led to the suspension of worldwide marketing of aprotinin.14,29 Consequently, alternative antifibrinolytic compounds are desired for fibrin-based tissue engineering applications. The aim of the present study was to evaluate the antifibrinolytic efficacy and potential cytotoxic side effects of t-AMCA in comparison to aprotinin as the gold standard for fibrin-based tissue-engineered cardiovascular structures. Our findings can be summarized as follows.
First, t-AMCA displays similar potencies at inhibiting clot lysis to aprotinin in vitro. If plasmin, an enzyme that degrades fibrin clots, is added simultaneously to the clotting mixture to simulate the in vivo situation, aprotinin is much more potent in inhibiting fibrinolytic activity than t-AMCA. Since plasmin is a serine protease, it is not surprising that aprotinin, an inhibitor of serine proteases, possesses greater antifibrinolytic properties than t-AMCA, which is predominantly a competitive inhibitor of plasminogen activation and, only at much higher concentrations, a noncompetitive inhibitor of plasmin. 30 The results show that the tested concentrations of t-AMCA (30–160 μg/mL) are not high enough to act as a noncompetitive inhibitor to plasmin, but high enough to delay the rate of fibrin degradation for tissue engineering approaches in vitro. The fibrinolytic activity was inhibited in a dose-dependent manner, indicating that the antifibrinolytic potency of t-AMCA lowers with decreasing concentration.
Second, our results demonstrated that fibrin gels cultured in the presence of t-AMCA possess similar mechanical stabilities to those gels supplemented with aprotinin, indicating that the fibrin clot structure was not modified by t-AMCA, and that t-AMCA has no negative influence on the mechanical stability of fibrin gels. Since the most significant drawback of fibrin gels in tissue engineering is structural weakness, 18 the resistance of the gels to mechanical stress and fibrinolytic degradation is quite important. In the closely related field of surgery, t-AMCA has also been considered as a substitute for aprotinin in fibrin sealant products, 31 and beneficial effects have been reported as well as adverse results. Concerning the mechanical properties of the fibrin sealants, Vankemmel et al. 32 evaluated the efficacy of t-AMCA and aprotinin for fibrin sealant use applied to vasal re-anastomosis, and reported comparable mechanical properties of sealants developed using either t-AMCA or aprotinin as an antifibrinolytic additive. In compliance with the results of the present study, the authors suggested that t-AMCA could act as an appropriate substitute for aprotinin.
In addition to analyzing the effects on fibrinolysis and fibrin clot strength, we examined whether or not t-AMCA carries the risk of cytotoxicity when supplementing cultures of carotid artery–derived cells. The data generated from the in vitro assays indicate that the different concentrations of t-AMCA tested neither increased the level of necrosis nor affected cell population growth or induced apoptosis. In contrast, some studies on fibrin sealant products containing t-AMCA indicate that it may be responsible for various adverse reactions. Studies by Furtmüller et al. 33 and Schlag et al. 34 found that the use of fibrin sealants containing t-AMCA carries the risk of neurotoxicity when used in neurological applications. Another study examined the effect of t-AMCA incorporated in fibrin sealant on the cell behavior of neuronal and nonneuronal cells. 35 Different concentrations of t-AMCA were shown to have no effect on initial cell adhesion when t-AMCA was incorporated in the fibrin gels. However, high concentrations (300–450 mM) of solubilized t-AMCA resulted in detachment of all cell types tested (neuronal and nonneuronal) from matrix-coated dishes, indicating that high concentrations should not be used in fibrin sealant products. A more recent study by Furst et al. 36 reported cytotoxic effects of t-AMCA on a fibroblast cell-line (MRC5). Further, the tensile strength of the fibrin sealant and the formation of fibrin fibers were also affected in the study. A possible explanation for these reported adverse effects may be the high concentration of t-AMCA required in commercial fibrin sealants to prevent lysis in vivo (∼95 mg/mL), which is three orders of magnitude higher than concentrations used in our tissue engineering approach. Additionally, the fibrinogen concentration of commercially available fibrin glues tested by Furst et al. is approximately ∼5 to 10 times higher than the concentrations employed in our study, which may have resulted in a significant increase of the diffusion coefficient and decrease of nutritional supply to the cells. Since t-AMCA is a commonly clinically used antifibrinolytic agent with no serious side effects,14,37 the probability for cytotoxic side effects in the development of fibrin-based tissue-engineered structures in vitro should be low.
Cell morphological analysis revealed that supplementation of t-AMCA to culture medium does not affect cell morphology or tissue development within fibrin gel constructs. No toxic degradation or inflammatory reactions were detected, and fibrin gels supplemented with medium containing the highest t-AMCA concentration (160 μg/mL) also displayed excellent tissue development, cell morphology, and ultrastructure. While all of the results presented in this study are limited to the in vitro situation, the application of a fibrinolysis inhibitor in fibrin-based cardiovascular tissue engineering is solely to prevent early degradation of the scaffold in vitro as new tissue is created by the cells before surgical implantation of the graft. The authors have recently demonstrated sufficient neotissue synthesis by cells in a fibrin-based heart valve prosthesis to allow for surgical implantation in the pulmonary trunk after 4 weeks of conditioning in vitro. 38 The valve prostheses, which were conditioned in aprotinin-supplemented medium, demonstrated an excellent histological structure after an implantation period of 3 months, despite almost complete degradation of the fibrin scaffold in vivo. The present study should also be extended to the in vivo situation, however, to demonstrate that conditioning in t-AMCA–supplemented medium can result in constructs with adequate mechanical properties for implantation. Further studies are also necessary to determine whether or not t-AMCA can facilitate the in vitro preparation of scaffolds seeded with other cell types for various tissue engineering applications.
Conclusion
Fibrin-based tissue-engineered constructs have the potential to significantly improve current state-of-the-art treatment modalities for cardiovascular diseases. However, the removal of aprotinin from the worldwide market necessitates the identification of a suitable fibrinolysis inhibitor for in vitro development of such constructs. t-AMCA, currently used in cardiac surgery, neither interferes with the mechanical properties of fibrin-based constructs nor elicits a cytotoxic response in cells seeded within such constructs. In addition, fibrin-based constructs cultured in the presence of t-AMCA display excellent tissue development, cell morphology, and ultrastructure. In summary, the results from the present study demonstrate that t-AMCA may be a suitable alternative to aprotinin for controlling the rate of fibrin degradation of tissue-engineered constructs in vitro.
Footnotes
Acknowledgments
The authors would like to thank Dipl.-Ing. Stefanos Diamantouros for helpful assistance and discussion. We would also like to acknowledge the entire cardiovascular tissue engineering team of the Helmholtz Institute for Biomedical Engineering, Aachen, for excellent collaboration. The authors also extend their thanks to Dipl.-Ing. Manfred Bovi and Dr. Jörg Bornemann, Department of Pathology, University Hospital Aachen, for valuable assistance with SEM and TEM, respectively. The study was financially supported by the Fördergemeinschaft Deutsche Kinderherzzentren e.V.
Disclosure Statement
No competing financial interests exist.