
Abstract
The osteogenic differentiation potential of umbilical cord blood-derived mesenchymal stem cells (UCB-MSCs) has been documented previously, and partially demineralized bone matrix (pDBM) represents a promising candidate for bone tissue engineering scaffolds. In this study, pDBM scaffolds derived from porcine cancellous bone were evaluated for their ability to support human UCB-MSCs osteogenic differentiation in vitro and bone-forming capacity in vivo to assess the potential use of UCB-MSCs in bone tissue engineering applications. MSCs were isolated from full-term human UCB and expanded, and their cell surface antigen markers and multilineage capability to differentiate into osteoblasts, chondrocytes, and adipocytes were analyzed. The in vitro proliferation and osteogenic differentiation of UCB-MSCs loaded onto the three-dimensional pDBM scaffolds were determined. Critical-sized full-thickness circular defects (5 mm in diameter) created bilaterally in the parietal bones of athymic rats were treated with one of the following: osteogenically induced UCB-MSC/pDBM composites (Group A, n = 8), noninduced UCB-MSC/pDBM composites (Group B, n = 8), pDBM alone (Group C, n = 8), or left untreated (Group D, n = 8). Microcomputed tomography analysis showed that new bone was formed in Group A at 6 weeks postimplantation, and greater bone volume and density were found after 12 weeks. In other groups, new bone formation was not evident after 6 weeks, and no bone union was found at 12 weeks. Histological examination revealed that the defect was repaired by tissue-engineered bone in Group A at 12 weeks, and fibrous union was observed in Groups B, C, and D. These results demonstrate that pDBM can support osteogenic differentiation of human UCB-MSCs in vitro and in vivo, and UCB-MSCs may serve as an alternative cell source for bone tissue engineering and regeneration.
Introduction
One alternative source is umbilical cord blood (UCB), which can be attained by a less invasive method without harm for the mother or infant. 13 There is now growing evidence to suggest that stem cells with the potential to generate mesenchymal cell types exist in UCB. The extensive characterization of these cells has revealed that UCB-derived mesenchymal stem cells (UCB-MSCs) are similar to BMSCs with respect to cellular properties and multilineage differentiation potential.13–18
It was reported that under appropriate inductive conditions, human UCB-MSCs can be directed toward the osteogenic lineage. Exposed to optimal concentrations of dexamethasone, ascorbic acid, and β-glycerophosphate in vitro, these cells can upregulate alkaline phosphatase (ALP) activity, produce osteoblast-associated proteins, and deposit mineralized extracellular matrix (ECM) characteristic of osteoblasts.19,20 Chang et al. reported that human UCB-MSCs have a significantly stronger capacity for osteogenic differentiation than BMSCs in vitro, showing that UCB-MSCs may be a potential cell source for bone tissue engineering. 21 Thus, the delivery of human UCB-MSCs to an orthotopic site for bone regeneration becomes an attractive issue.
Derived from native osseous tissue, partially demineralized bone matrix (pDBM) represents a promising candidate material for bone tissue engineering scaffolds because of its close relation in structure and function with autologous bone. 22 We and others have demonstrated that pDBM supports the in vitro proliferation and osteogenic differentiation of human BMSCs and promotes in vivo osteogenesis of these matrix-incorporated osteoprogenitors.22–26 However, no study has been reported up to now regarding the propensity of pDBM to support osteogenesis of human UCB-MSCs in vitro and in vivo.
We hypothesized that pDBM scaffolds could support osteogenic differentiation of human UCB-MSCs in vitro, and the UCB-MSC/pDBM composites would increase bone formation in vivo. To test this hypothesis, pDBM scaffolds seeded with human UCB-MSCs were kept in an in vitro culture for up to 2 weeks, and cell proliferation and osteogenic differentiation were quantified. Then we implanted the cell/scaffold composites into a rat cranial critical-sized defect and evaluated new bone regeneration at 6 and 12 weeks by microcomputed tomography (micro-CT) and histological analysis.
Materials and Methods
Isolation and culture of human UCB-MSCs
All protocols for human tissue handling were approved by the Research Ethical Committee of Shanghai 9th People's Hospital. Harvested from term deliveries (38–41 weeks) at the time of birth with the mothers' consent, human UCB samples (30–50 mL, n = 21) were added to heparinized tubes (5000 units/mL) and processed within 12 h of collection by a minor modification of methods previously reported.13–15,17–19 Briefly, each 10 mL blood sample was diluted with an equal volume of phosphate-buffered saline, and mononuclear cells (MNCs) were isolated with Ficoll-Pague™ PLUS (1.077 g/mL; Amersham Biosciences, Uppsala, Sweden) density gradient centrifugation (400 g for 25 min). The MNC fraction was seeded in 100-mm dishes (Falcon–B&D, San Jose, CA) at a density of 1 × 106 cells/cm2 and cultured in basic culture media (low-glucose Dulbecco's modified Eagle's medium [DMEM]; Gibco, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS; Gibco), 2 mM
Cell proliferation and flow cytometric analysis
Cell population doubling was analyzed by DNA assay as previously reported. 27 Briefly, UCB-MSCs were expanded for 10 passages in 100-mm dishes as described earlier. Three dishes at each passage were used for DNA assay (Hoechst 33258 dye; Sigma). The cumulative population doublings were calculated using the formula: x = [log10(NH) − log10(NI)]/log10(2), 28 where NI is the inoculum cell number and NH is the cell harvest number. As the cell number was determined from passage 1, the cumulative doubling number was first calculated for passage 2.
Flow cytometric analysis of cell surface antigen markers was performed as previously reported.29,30 Briefly, trypsin-released UCB-MSCs of passages 1 and 3 were collected and washed with flow cytometry buffer (1 × phosphate-buffered saline, 2% FBS, and 0.05% sodium azide). Cell aliquots (1 × 106 cells) were incubated at 4°C for 30 min in flow cytometry buffer containing 20 μL of fluorescein isothiocyanate (FITC)-conjugated or phycoerythrin (PE)-conjugated monoclonal antibodies: CD29-PE, CD34-FITC (BD Pharmingen, San Diego, CA), CD31-PE, CD45-PE, CD105-PE, CD166-FITC (AbD Serotec, Raleigh, NC), and CD106-PE (Santa Cruz Biotechnology, Santa Cruz, CA). For Stro-1 analysis, cells were immunolabeled with mouse anti-human Stro-1 antibody (R&D Systems, Minneapolis, MN), and the secondary antibody is anti-mouse IgM-FITC (BioSource, Camarillo, CA). Labeled cells were analyzed by flow cytometry (Coulter Epics Altra; Becton Dickson, San Jose, CA), and cells stained with isotype controls, FITC- or PE-conjugated nonspecific mouse IgG (Becton Dickson), were used to assess background fluorescence.
In vitro differentiation of UCB-MSCs
To induce osteogenic differentiation, human UCB-MSCs of passage 3 were replated at 2.5 × 105 cells/cm2 in 35-mm dishes (Falcon-B&D) and cultured for 2 weeks in 5 mL of osteogenic medium (consisting of basic culture medium plus 10−8 M dexamethasone, 10 mM β-phospherglycerol, and 0.2 mM ascorbic acid; all from Sigma). The medium was changed twice a week and the in vitro mineralization was revealed by alizarin red staining after 14 days as previously reported. 29
To induce adipogenic differentiation, cells of passage 3 were replated at 2.5 × 105 cells/cm2 in 35-mm dishes and cultured for 3 weeks in 5 mL of adipogenic medium (consisting of basic culture medium plus 0.5 mM isobutyl-methylxanthine, 10 μM insulin, and 200 μM indomethacin; all from Sigma). The medium was changed twice a week and the intracellular lipid accumulation was assessed by oil red O staining after 21 days as previously described.29,31
To induce chondrogenic differentiation, 2.5 × 106 cells of passage 3 were centrifuged in a 15-mL polypropylene tube (Falcon-B&D) at 150 g to form a pellet and then cultured for 4 weeks in 5 mL of chondrogenic medium (1% FBS, 6.25 μg/mL insulin, 10 ng/mL transforming growth factor-β1, and 50 μg/mL ascorbic acid in DMEM) (FBS and DMEM from Gibco; insulin and transforming growth factor-β1 from Sigma). The medium was changed twice a week and the micromass nodules were examined by safranin O staining after 28 days as previously reported. 14
Scaffold preparation and cell seeding
The pDBM scaffolds were prepared from porcine trabecular bone by a modification of methods previously described.23–25 Briefly, cancellous bone samples were sequentially extracted in absolute ethanol for 4–8 h (depending on the clarity of the extract), decellularized by 1% Triton X-100 for 48 h, and degreased in anhydrous ethyl ether for 12 h. Then, the samples were partially demineralized in 0.6 N HCl for 15 min (50 mL/g bone), rinsed in distilled water for 12 h to neutralize the pH and to remove liberated salts, sectioned into disks (5 mm in diameter and 1 mm in thickness), and air-dried for 24 h. To increase group homogeneity, disk scaffolds from all samples were evaluated with a micro-CT system (μCT-80; Scanco Medical AG, Bassersdorf, Switzerland) as previously described. 24 Relatively homogeneous scaffolds, with volume porosity of 75% ± 8%, pore size of 310 ± 54 μm, and tissue volume density of 297.80 ± 34.19 mg HA/cm3, were selected for use in this study. The scaffolds were then sterilized by 60Co irradiation and stored at 4°C until use (Fig. 1).
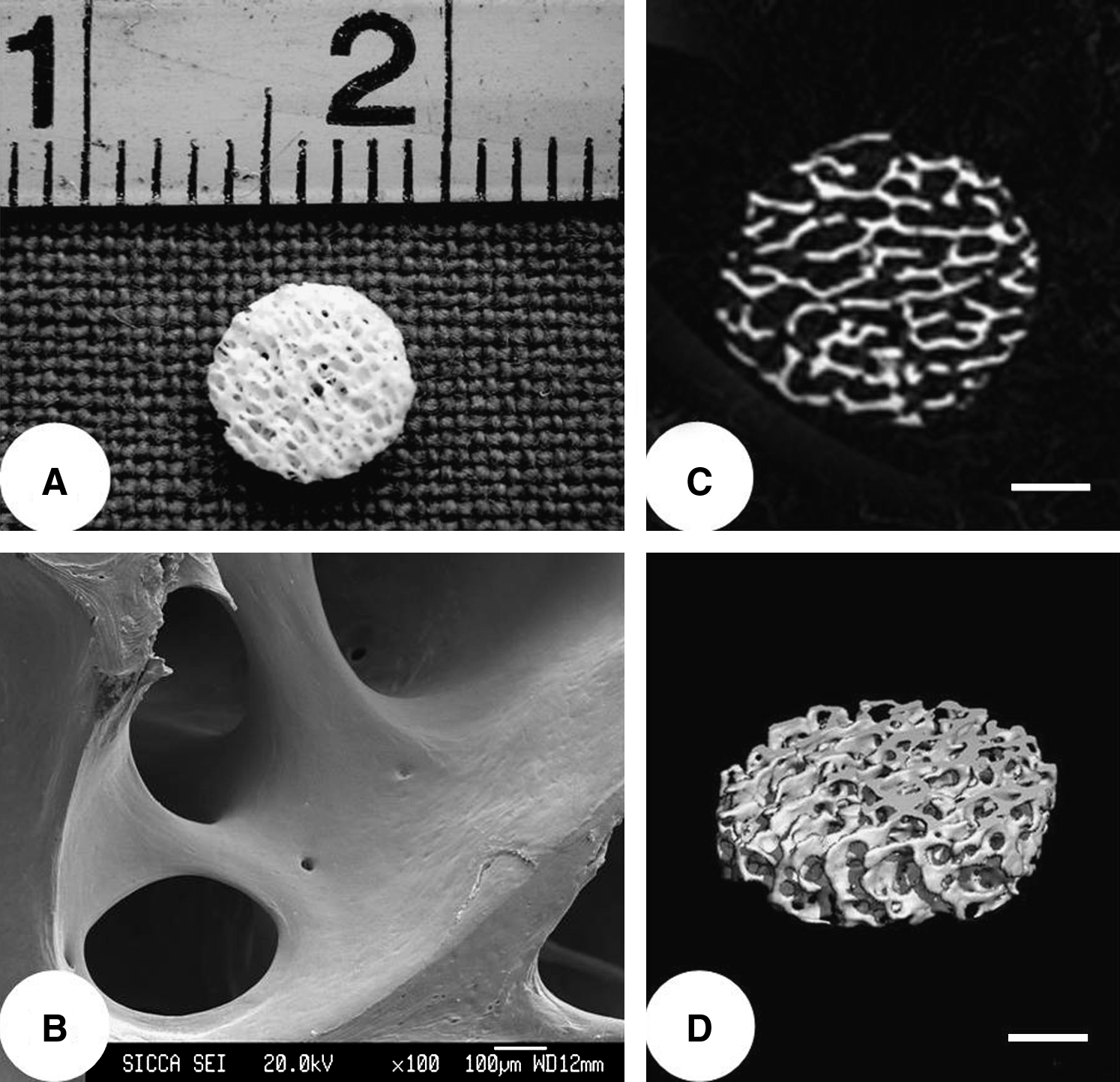
Structural properties of the pDBM scaffold. (
Human UCB-MSCs of passage 3 from four independent cord blood donors were resuspended in basic culture medium at a density of 1 × 107 cells/mL. Aliquots (30 μL) of cells were added into the pDBM scaffolds and incubated at 37°C for 2 h. Then the cell/scaffold composites were transferred into six-well plates (Falcon–B&D). One cell/scaffold composite was subcultured in one well with 5 mL of osteogenic medium, which was changed twice a week. Cell/scaffold composites cultured in basic culture media and scaffolds without seeded cells but soaked under the same osteogenic condition served as controls.
In vitro growth of UCB-MSCs on scaffolds
To visualize the growth and spatial distribution of UCB-MSCs on the pDBM scaffolds, at day 4 after cell seeding, the cell/scaffold composites were fixed with 2.5% glutaraldehyde, postfixed in 0.1% osmium tetroxide, dehydrated through an ethanol series, and dried in a CO2 critical point dryer. The samples were sputter coated with gold and then subjected to scanning electron microscopy (SEM, JXA 8100; Jeol, Tokyo, Japan) examination.
Cell proliferation on the scaffolds was determined by DNA assay at days 1, 4, 7, 10, and 14, as reported previously.24,26,31–33 At each time point, samples were homogenized with a pellet grinder (Fisher Scientific, Ottawa, Canada) and stored at −20°C until just before analysis, when they were digested in 500 μL proteinase K (Sigma) at 56°C for 16 h. The homogenates were then subjected to three freeze–thaw–sonicate cycles (30 min at −80°C, 30 min at room temperature, and 30 min of sonication) for complete extraction of DNA from the cell cytoplasm. DNA content in the lysate was quantified spectrofluorometrically (523 nm) using Hoechst 33258 dye (Sigma), by correlating fluorescence with DNA content using standards containing known amounts of DNA.
In vitro osteogenesis of UCB-MSCs on scaffolds
To assess the osteogenesis of human UCB-MSCs on pDBM scaffolds, ALP activity and osteocalcin (OCN) content were evaluated as follows at days 1, 4, 7, 10, and 14 after cell seeding. At each time point checked, the cell/scaffold composites were homogenized with 1 mL Tris buffer (pH 7.4; Sigma) and sonicated. The cell lysate (0.1 mL) was mixed with 0.5 mL p-nitrophenol phosphate substrate solution (Sigma) and 0.5 mL ALP buffer solution (Sigma). After incubation at 37°C for 15 min, 10 mL of 0.05 N NaOH were added to stop the reaction. The absorbance of the reaction product was measured with a Varioskan multimode detection reader (Thermo Electron, Waltham, MA) at a wavelength of 405 nm and correlated with ALP activity using standards containing known amounts of p-nitrophenol.
Twenty-four hours before measuring OCN levels at each time point, 5.0 mL of serum-free osteogenic medium were replaced in each well, to prevent cross-reactivity of the anti-OCN monoclonal antibodies with bovine serum proteins. The OCN concentration in the medium was measured using a human OCN ELISA kit (Invitrogen, Carlsbad, CA) following the manufacturer's protocol.
The amounts of ALP and OCN produced by each sample of cell/scaffold composite was divided by the total cell number of that sample, which was derived from the DNA measurement, thereby allowing statistical comparisons to be made between the different groups.
Also at 14 days of culture, reverse transcriptase–polymerase chain reaction (RT-PCR) analysis of bone-specific mRNA expression was performed as previously reported. 22 Briefly, total RNA was isolated from the cell-seeded samples using an RNAprep Micro kit (TianGen Biotech, Beijing, China), and complementary DNA was synthesized using PrimeScript 1st Strand cDNA Synthesis kit (TaKaRa Biotech, Dalian, China). Four osteoblast-related genes, ALP, OCN, osteopontin (OPN), and collagen type I (Col I), were amplified, with β-actin as a control. The sequences of primers, individual annealing temperature, and amplicons lengths are shown in Table 1.
ALP, alkaline phosphatase; Col I, collagen type I; OCN, osteocalcin; OPN, osteopontin; F, forward; R, reverse.
Surgical procedure
After 14 days of culture in vitro, the cell/scaffold compos-ites were used for in vivo implantation. Sixteen skeletally mature (7–8 weeks old) Sprague–Dawley, athymic male rats (250–300 g) were used for orthotopic implantation in this study, and all procedures were performed at the facility accredited by the Animal Care and Experiment Committee of Shanghai JiaoTong University School of Medicine. Under anesthesia (intraperitoneal injection of ketamine [60 mg/kg] and medetomidine [3 mg/kg]), the nude rat was placed in ventral recumbency with front and hind legs extended. The surgical area was shaved, cleaned, and scrubbed using standard aseptic surgical technique. Bilateral critical-sized calvarial bone defects (5 mm in diameter) were created as described previously.34–36 Briefly, a midline calvarial incision was made, the subcutaneous fascia was divided, and periosteum was resected by blunt dissection (leaving at least 5 mm from the defect edge). Full-thickness circular defects of 5 mm were made bilaterally in the parietal bones using a dental trephine. Constant saline irrigation was used during this process to cool and clear any remaining debris, and extreme care was taken not to damage the dura mater or the midsagittal sinus. In each animal, one defect was grafted randomly with either an osteogenically induced UCB-MSC/pDBM composite (Group A) or a noninduced composite (Group B), and the opposing defect was grafted with a pDBM scaffold (Group C) or left empty (Group D) (n = 8 for each treatment). The wounds were then closed with resorbable sutures. The animals were returned to their individual cages and observed until fully recovered from anesthesia.
Micro-CT evaluation
Rats were sacrificed using CO2 asphyxiation at 6 or 12 weeks after implantation. The calvarial bone was excised, trimmed, and fixed in 4% paraformaldehyde for 24 h at 4°C. The samples were scanned using a micro-CT system (μCT-80; Scanco Medical AG) in a high-resolution scanning mode (a voxel size and slice thickness of 20 μm). Three-dimensional images were reconstructed, and the local density and volume of the regenerated bone within the defects were calculated using its auxiliary software (Scanco Medical AG).
Histological examination
After micro-CT measurement, the specimens were decalcified in 10% ethylenediaminetetraacetic acid for 2 weeks at room temperature, dehydrated through ascending alcohol gradients, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin and Masson's trichrome according to standard protocols.
Statistical analysis
All the in vitro experiments were conducted in triplicate, and data collected were expressed as means ± standard deviation with n = 3–5. For the in vivo study, four samples in each group were harvested for the micro-CT analysis at each interval (6 and 12 weeks postimplantation). Statistical analysis was performed by one-way analysis of variance, assuming equal variance using SPSS 11.0 software (SPSS Inc., Chicago, IL). A p-value of less than 0.05 was considered statistically significant.
Results
Cell proliferation and flow cytometric analysis
Only 11 different UCB-MSC populations (six males and five females) were established from 21 cord blood samples. About 2 weeks after MNC seeding, colonies of adherent cells with elongated fibroblast-like morphology were observed in culture (Fig. 2A). After primary culture, human UCB-MSCs exhibited a morphologically homogeneous spindle-like population as shown in Figure 2B. The mean population doubling time of UCB-MSCs was 31.3–35.5 h during the culture time tested, with cumulated population doublings of 14.6 times achieved through passages 2–10 (n = 3; Fig. 2C).
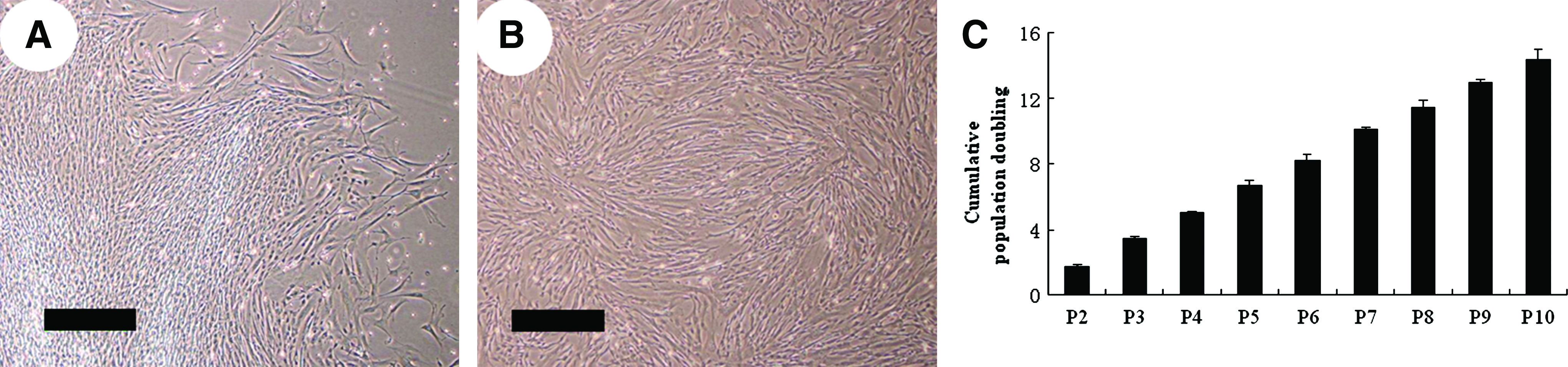
Proliferation and flow cytometric analysis of human UCB-MSCs. (
For further characterization of UCB-MSCs, a panel of cell surface antigen markers was tested using flow cytometric analysis. In consistence with previous reports,14,15,21 UCB-MSCs were positive for CD29 (the adhesion marker), CD105, CD106, CD166, and Stro-1 (the mesenchymal progenitor-specific markers). Additionally, the expression of CD31 (the endothelial cell marker), CD34 (a hematopoietic stem cell marker), and CD45 (leukocyte common antigen) decreased with culture time. Compared with passage 1, cells of passage 3 were negative for these markers, indicating that after continuous expansion and passage, UCB-MSC population was purified and the contamination by hematopoietic or endothelial cells was deleted (Table 2).
Data presented as mean value ± standard deviation with n = 5.
In vitro differentiation of UCB-MSCs
To assess the differentiation potential of UCB-MSCs along the mesenchymal lineage, cells of passage 3 were exposed to the osteogenic, adipogenic, and chondrogenic induction media for 2–4 weeks, respectively. As shown in Figure 3, osteogenic differentiation was revealed by the positive alizarin red staining of cell matrix mineralization. Adipogenic induction resulted in the intracellular accumulation of lipid droplets as visualized by oil red O staining. Chondrogenic differentiation was confirmed by the formation of a sphere in the micromass culture and the secretion of cartilage-specific proteoglycans visualized through safranin O staining. In contrast, no spontaneous differentiation occurred in the control UCB-MSCs cultured in the noninduced media.
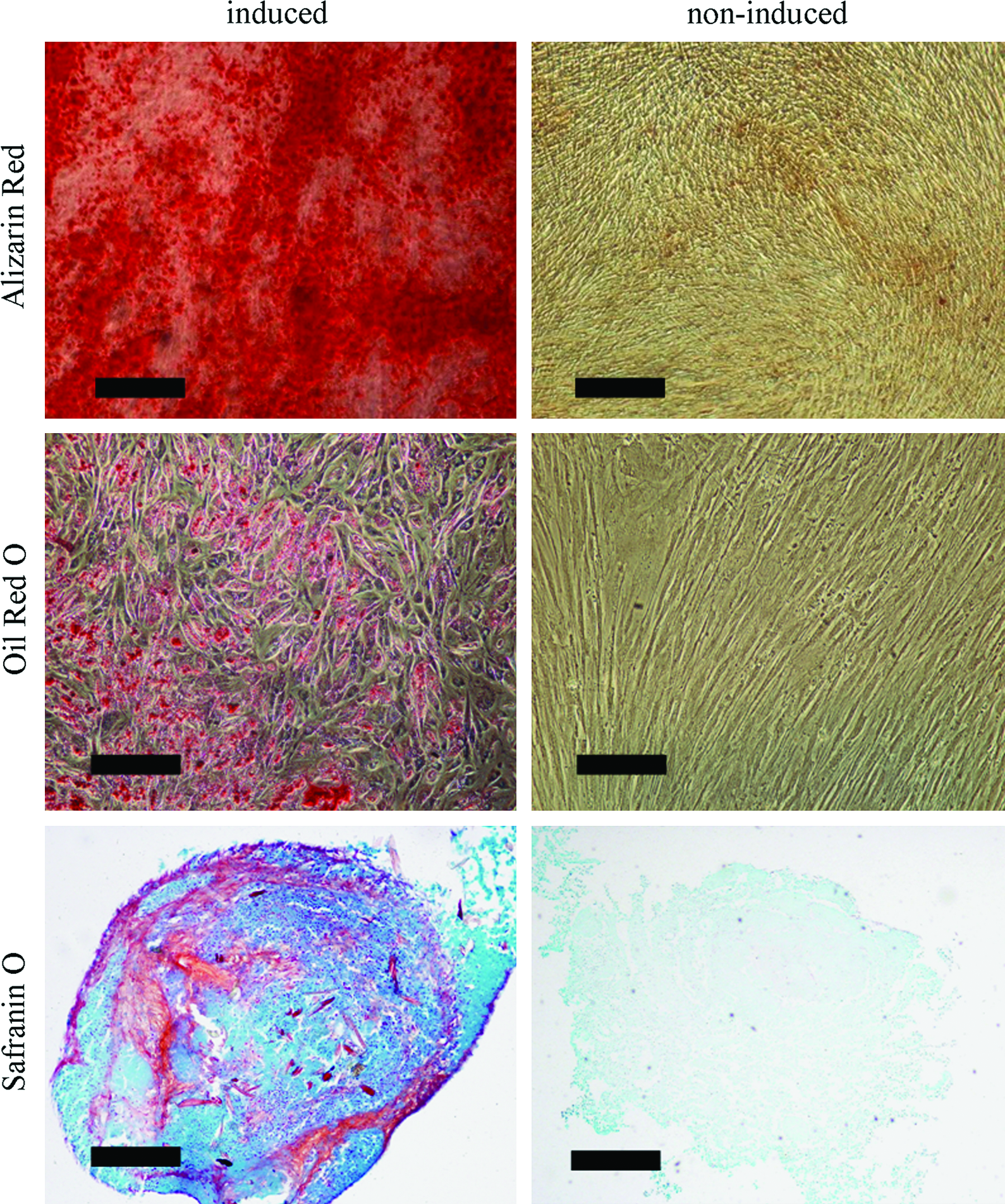
Multilineage differentiation of human UCB-MSCs of passage 3. The osteogenic differentiation was revealed by the positive alizarin red staining of cell matrix mineralization (scale bars: 500 μm). The adipogenic differentiation was confirmed by the positive oil red O staining (scale bars: 200 μm). And the chondrogenic differentiation was identified by the positive safranin O staining (scale bars: 200 μm). No spontaneous differentiation occurred in the noninduced UCB-MSCs. Color images available online at www.liebertonline.com/ten.
In vitro growth of UCB-MSCs on scaffolds
At day 4 after cell seeding, SEM observation showed that both the osteogenically induced and noninduced human UCB-MSCs spread and attached well on the pore surface of pDBM scaffolds, depositing dense ECMs (Fig. 4A, B).
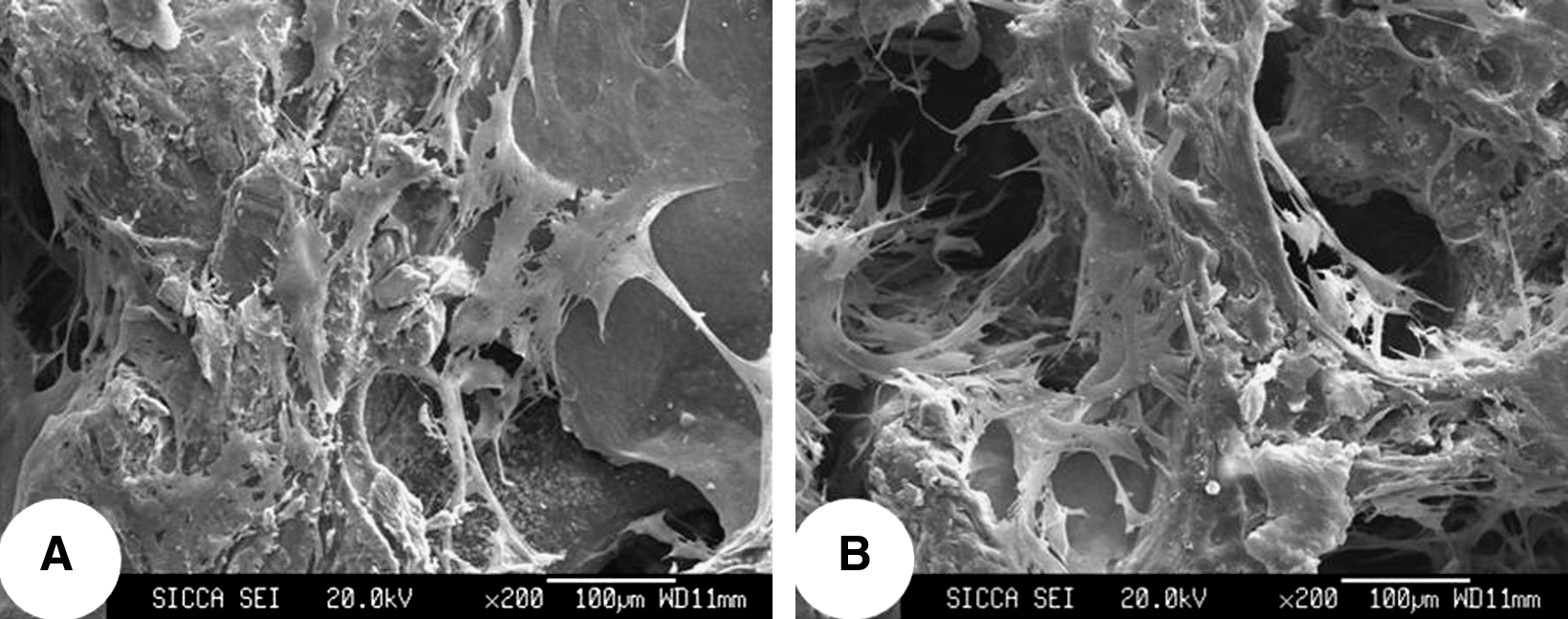
In vitro growth of UCB-MSCs on scaffolds. Scanning electron microscopy images demonstrated that both the osteogenically induced (
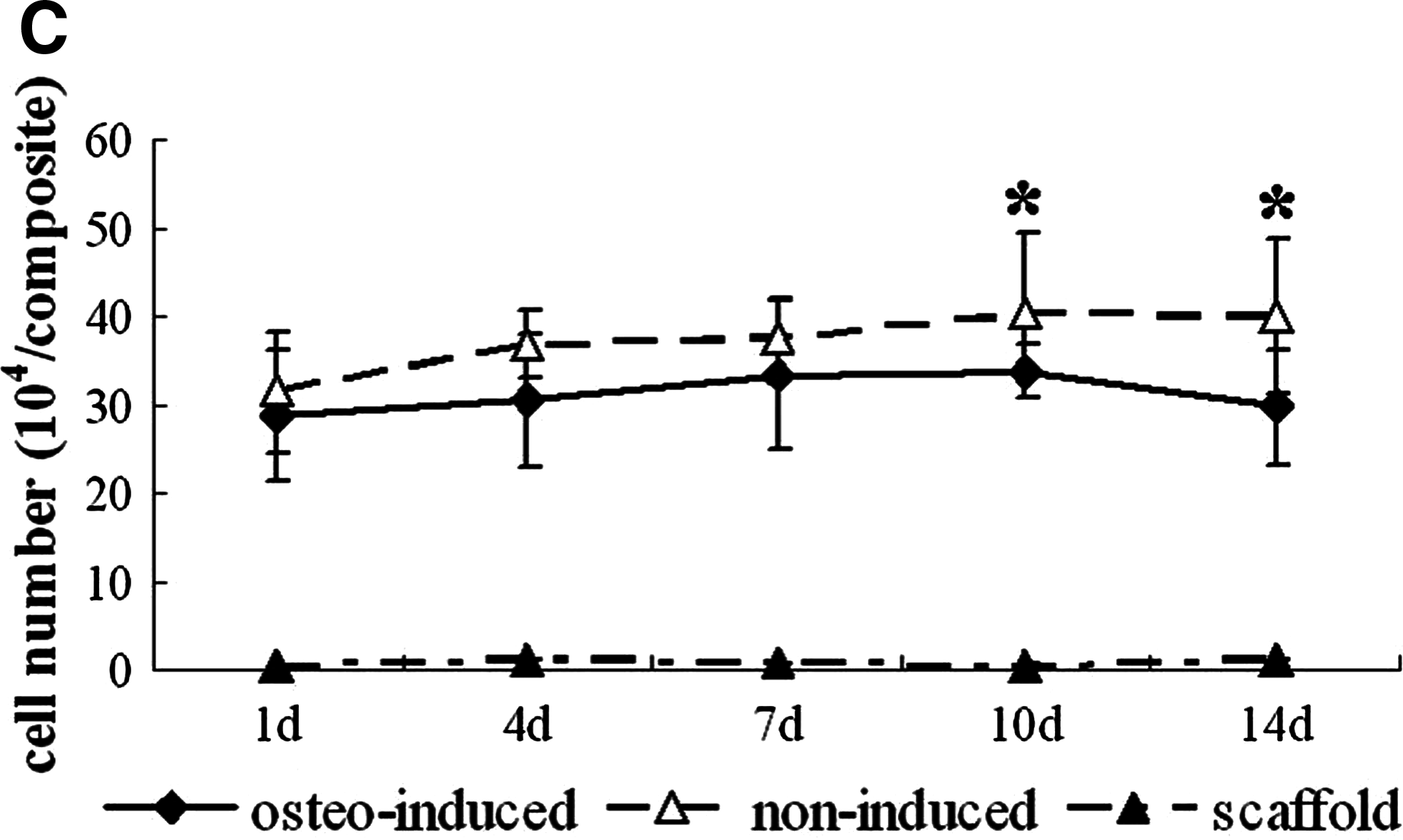
According to the DNA assay, after cell seeding, the osteogenically induced cell number increased gradually from day 1 to 10, with the highest cell number detected at day 10 of cultivation, which was significantly higher than that of day 1 (p < 0.05), followed by a decrease at day 14. The cell number of noninduced composites increased from day 1 to 7, reached the plateau thereafter, and was significantly more than that in the osetogenically induced group at days 10 and 14 (p < 0.05; Fig. 4C).
In vitro osteogenesis of UCB-MSCs on scaffolds
The osteogenic differentiation of UCB-MSCs grown on the scaffolds was evaluated by measuring the expression of ALP and the release of OCN at days 1, 4, 7, 10, and 14 postseeding, respectively. As shown in Figure 5A, compared with the noninduced group, significantly increased ALP activity was found in the osteogenically induced group at days 7, 10, and 14 (p < 0.05), exhibiting a steadily increasing trend within 14 days. The amount of OCN content in the osteogenically induced cells increased from days 1 to 10, reached the peak at day 10, and then began to decrease. In contrast, UCB-MSCs in the control group only produced a minimal amount of OCN for 14 days, significantly lower than that of the induced cells at each time point checked except at day 1 (p < 0.05; Fig. 5B).
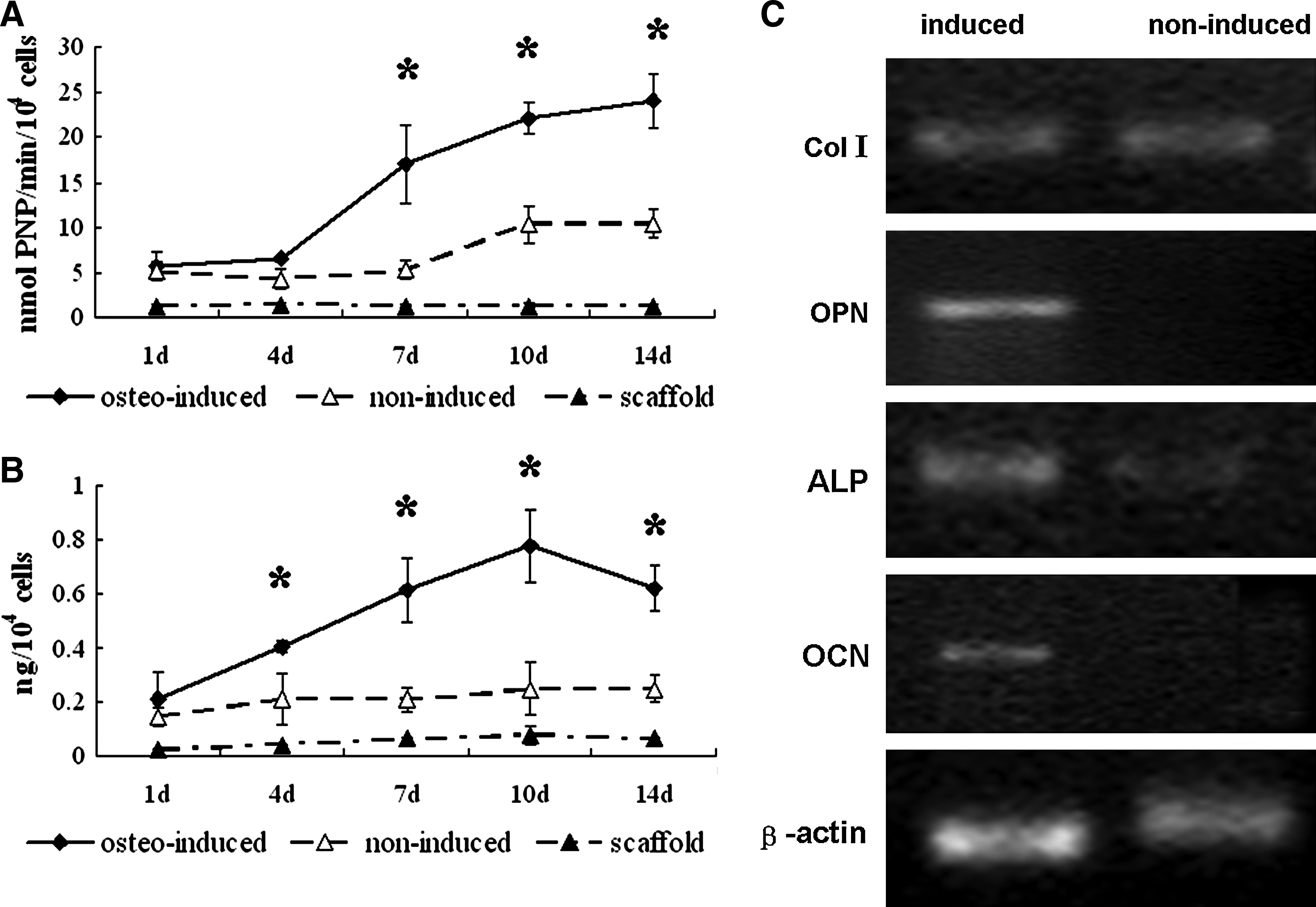
The osteogenic differentiation of osteo-induced and noninduced UCB-MSCs on pDBM scaffolds was evaluated by ALP activity (
RT-PCR analysis was performed at day 14 to evaluate the osteoblastic differentiation parameters of UCB-MSCs on scaffolds. The osteogenic induction conditions resulted in the expression of all four genes analyzed in this study (ALP, OCN, OPN, and Col I). For the control cells, the OCN and OPN transcripts were not detectable. The expression of Col I gene was similar to that of the induced group, but the ALP gene was amplified at a significantly reduced level (Fig. 5C).
Micro-CT evaluation
All of the wounds healed uneventfully without infection, and all animals survived until the day of sacrifice. Micro-CT measurements were taken at 6 and 12 weeks postoperatively, and representative images from each group are shown in Figure 6. After 6 weeks, scattered new bone was found in the defects filled with osteogenically induced cell/scaffold composites (Group A). For the empty defects (Group D) and defects grafted with noninduced cell/scaffold composites (Group B) and pDBM (Group C), no obvious evidence of new bone growth was noticed (Fig. 6A). At 12 weeks, the negative control sites displayed minor evidence of new bone formation in the periphery of the defect edges. No bony union occurred in the defects of Groups B and C, and the residual scaffold was still visible. In contrast, the majority of the defects in Group A were filled with newly formed bone tissue and the implanted pDBM scaffold was completely resorbed (Fig. 6B).
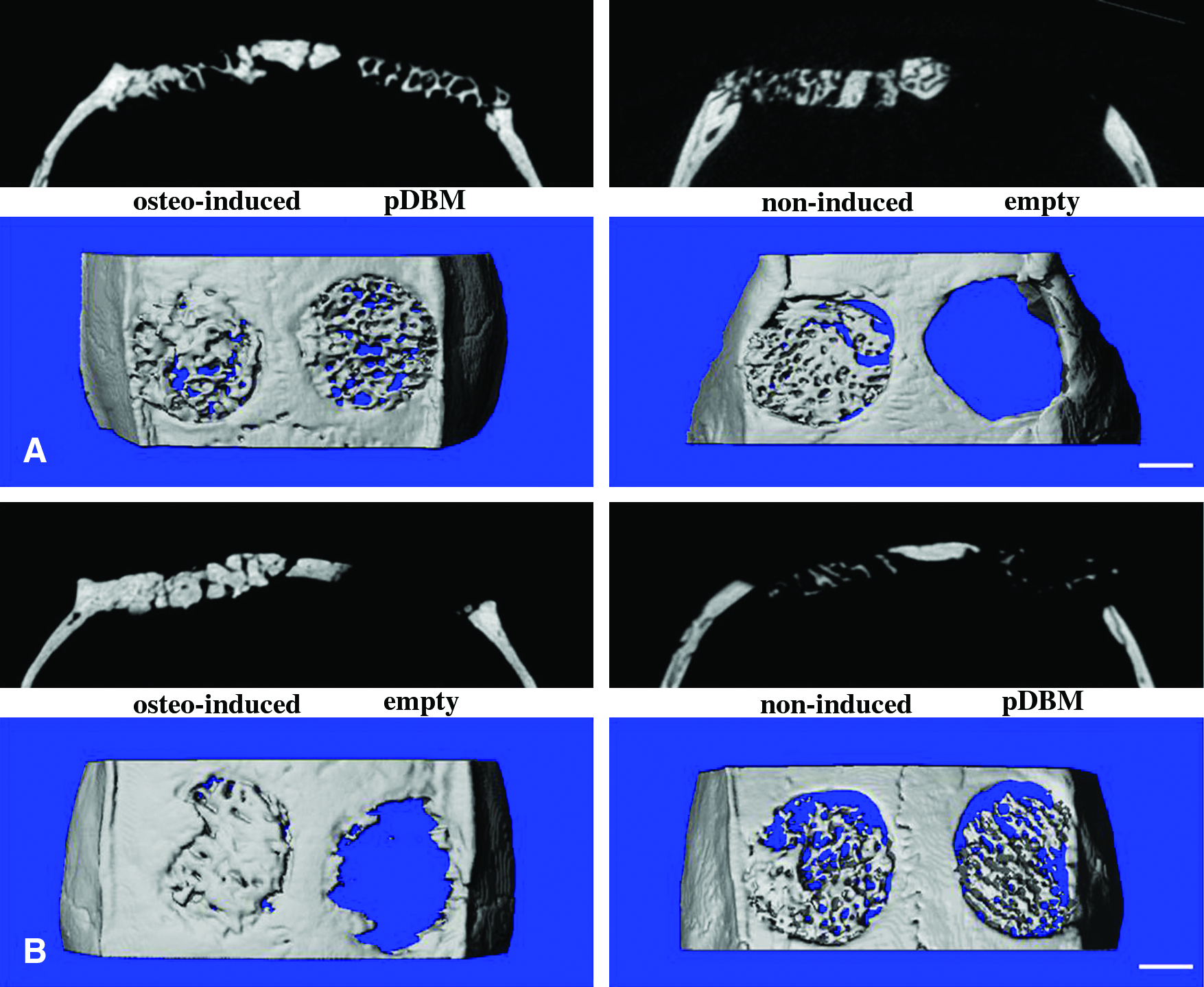
Two-dimensional (upper) and three-dimensional (lower) micro-CT images of the repaired calvarial defects at 6 (
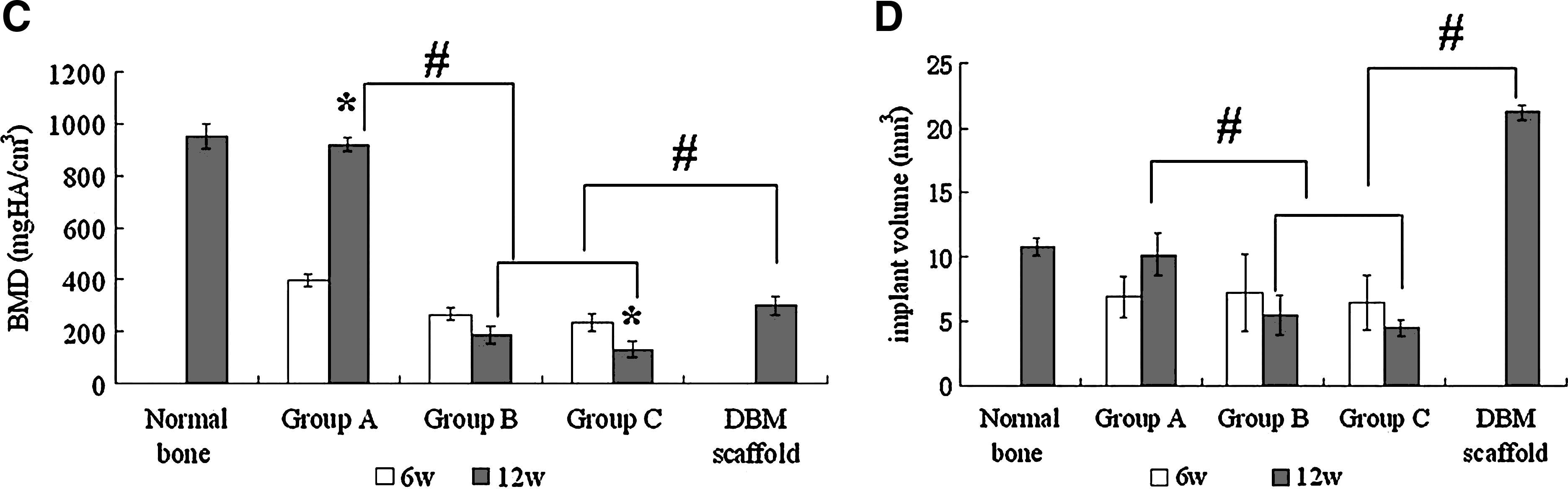
To quantify the new bone regeneration within the calvarial defects, local bone mineral density (BMD) and volume of regenerated bone in Groups A, B, and C were measured and compared with normal calvarial bone (removed from the same parts of three 20-week-old rats without surgery) and pDBM scaffolds. As shown in Figure 6C, quantitative analysis revealed a significant 2.31-fold increase in BMD from 6 to 12 weeks in Group A. The tissue-engineered bone showed a relatively high BMD of 920.41 ± 24.05 mg HA/cm3 at 12 weeks, which was similar to that of the normal bone (952.29 ± 47.15 mg HA/cm3) (p > 0.05), whereas in Group B, the local density decreased from 266.47 ± 23.66 to 186.72 ± 41.66 mg HA/cm3, and decreased from 235.09 ± 35.64 to 131.95 ± 31.26 mg HA/cm3 in Group C. At 12 weeks, the densities of Groups B and C were significantly lower than that of Group A (p < 0.001) and also significantly lower than the density of dry pDBM scaffolds (p < 0.05). For the implant volume analysis, the new bone volume of Group A increased from 6.89 ± 1.56 mm3 at 6 weeks to 10.19 ± 1.68 mm3 at 12 weeks, up to 94.6% of that of the normal calvaria. The volumes of the remaining pDBM scaffolds in Groups B and C decreased from 7.23 ± 2.99 to 5.49 ± 1.59 mm3, and from 6.51 ±2.11 to 4.5 ± 0.62 mm3, which were 25.88% and 21.22% of the original volume of pDBM scaffolds, respectively (Fig. 6D).
Histological examination
Histological examination confirmed the findings of micro-CT observation. At 6 weeks, newly formed osteoid-like tissue (numerous osteocytes with round cell bodies inside a homogenous matrix without lamellation) was found between the fragmented pDBM and thin fibrous sheath in Group A. No new bone formed in Groups B and C, and the residual pDBM scaffolds were surrounded by thick fibrous tissue. In Group D, only a thin layer of fibrous tissue spanned the defect. No evident inflammatory response was observed in all groups (Fig. 7A).

Histological observation of bone regeneration at 6 (
At 12 weeks, the defects were almost bridged by remodeled trabecular bone in Group A, and no residual scaffold was found within the defects. Fibrous union occurred in Groups B, C, and D, with only minimal new bone formation at the periphery of the defects (Fig. 7B). Masson's trichrome staining revealed a combination of osteoid (blue) and mature bone (red) within the defects of Group A. In Groups B and C, the defects were connected with fibrous tissue (green) and the remnants of pDBM particles (red, decellularized lamellar matrix) (Fig. 7C).
Discussion
Repair of large bone defects caused by trauma, infection, cancer, or congenital malformation represents a continuous challenge in orthopedic surgery because autogenous grafts are not available in large amounts and their removal causes morbidity at the donor site.37–39 Tissue engineering approaches have proven effective in bone regeneration recently, and tissue-engineered bone substitutes are considered a potential alternative for autologous bone transplantation.1–6
Identifying appropriate cell sources for tissue engineering is a primary research goal in regenerative medicine. Based on their large ex vivo expansion capacity, differentiation potential, and the ease of accessibility, UCB-MSCs seem to hold promise for cell-based therapy strategies and tissue engineering. The results of this study demonstrate that human UCB-MSCs are capable of proliferating and osteogenically differentiating on three-dimensional pDBM scaffolds in vitro, and forming tissue-engineered bone in an orthotopic site in vivo, suggesting that human UCB-MSCs could be alternative cell sources for bone tissue engineering.
We first examined the proliferation capability and osteogenic differentiation of human UCB-MSCs on the pDBM scaffolds in vitro. SEM findings showed that UCB-MSCs attached and proliferated well on the pore surface, excreting abundant collagenous-like ECM on the scaffolds. The DNA assay results confirmed that pDBM could support the growth of human UCB-MSCs in vitro. It was found that the osteogenically induced cells began to decrease on the scaffolds from day 10, compared with the noninduced cells. The decline of cell proliferation may be explained by the growth arrest and downregulation of proliferation genes accompanied with the osteogenic differentiation of osteoblasts. 31
The ability of pDMB scaffolds to support osteogenic differentiation of human UCB-MSCs in vitro was demonstrated by several osteogenic markers in this study. Higher levels of synthesis of ALP and OCN by UCB-MSCs cultured in osteogenic media were found when compared with the noninduced cells. And the osteogenic differentiation of UCB-MSCs was confirmed by the increased transcript expression of ALP, OCN, and OPN. Interestingly, the noninduced controls also displayed increases in ALP activity during the observation time, but at a significantly decreased level, in consistence with the RT-PCR result of ALP. This induction effect in noninduced cells could be due to matrix-incorporated osteogenic factors (bone morphogenetic proteins and/or other noncollagenous proteins) retained within the pDMB scaffolds.22,24,40,41 It was reported that UCB-MSCs contain different subpopulations with heterogeneous differentiation capacities,15,21 and the flow cytometric analysis of our study confirmed that the seeded UCB-MSCs of passage 3 were not completely homogenous. These matrix-incorporated factors were possibly responsible for the low level of osteogenic differentiation in certain subpopulations within noninduced human UCB-MSCs.
Previous studies demonstrated that in vivo osteogenesis of pDBM in conjunction with an osteogenic cell source could be accelerated and enhanced by promoting in vitro osteogenic differentiation of these cells before implantation.22,24–26 In this study, bony union of the critical-sized calvarial defect was only achieved in the osteogenically induced group. We speculated that in vitro osteogenic differentiation of UCB-MSCs on pDBM scaffolds before implantation may deliver a mature osteoblastic population to the defect sites, capable of more rapid bone formation in vivo. For the noninduced UCB-MSCs group, however, only a relatively small subpopulation of the MSCs was osteogenically stimulated by the matrix-incorporated osteogenic factors retained in pDBM, as shown in the in vitro study, and fibrous union was achieved in the critical-sized defect.
In this study, it was found that osteogenically induced cells seeded onto scaffolds could promote bone formation in vivo, demonstrating that the degradation rate of pDBM scaffolds might well match the new bone formation rate in Group A. However, because of similar density, pDBM was not able to be fully distinguished from the newly regenerated bone by micro-CT scans, and analysis of the degradation rate of the scaffolds was not possible. Thus the comparison of the degradation rate of pDBM between different groups was limited in this study.
Another limitation of this study is the relatively low frequency of MSCs in full-term UCB. Because of the process of hematopoiesis during fetal development, which initiates within the yolk sac before moving through the circulation to other anatomical sites within the developing embryo, there is an inverse correlation between gestation time and the frequency of both hematopoietic stem cells and MSCs within UCB.42–44 Based on our results, MSCs were isolated and expanded from only 53.8% of the full-term UCB samples (11/21), which is consistent with the low isolation efficacy of full-term cord blood reported previously.13,14,45,46 Therefore, further studies of investigating rapid and efficient methods of isolating and expanding MSCs from full-term UCB are needed. On the other hand, human UCB has been found to be less immunogenic than adult marrow and blood cells, 16 suggesting that allogenic UCB-MSCs obtained from more than one single cord might be used to tissue-engineer bone in patients.
In conclusion, this study demonstrates that pDBM supports the proliferation and osteogenic differentiation of human UCB-MSCs in vitro, and tissue-engineered bone composed of pDBM scaffolds and osteogenically induced UCB-MSCs can repair critical-sized calvarial defects in athymic rat models, indicating that UCB-MSCs might possibly serve as an alternative cell source for bone regeneration in clinics.
Footnotes
Acknowledgments
The authors acknowledge Dr. James D. Kretlow (Department of Bioengineering, Rice University) for his insightful comments and polish for the manuscript. This work was financially supported by the National High Technology Research and Development Program of China (2006AA02A123), Natural Science Foundation of China (30800232), and Shanghai Science and Technology Committee projects (07QA14053 and 075407072).
Disclosure Statement
No competing financial interests exist.