
Abstract
Due to their immunomodulatory functions, mesenchymal stem cells (MSCs) have great potential for clinical applications to prevent rejection in organ transplantation and to prevent graft-versus-host disease in hematopoietic stem cell (HSC) transplantation. Since dendritic cells (DCs) play an important role in modulating diverse T cell responses, including rejection and graft-versus-host disease, the goal of this study was to investigate whether MSCs modulate DC differentiation from HSCs and if this effect could be one of the mechanisms for MSCs' immune-modulating functions. Our results demonstrate that differentiation of HSCs into mature DCs is inhibited in the presence of MSCs. Similar frequency of dendritic precursors in the cultures, either with or without MSCs, suggests that the inhibition of MSCs on the differentiation of mature DCs from HSCs could be due to the arresting of maturation at the dendritic precursor step. Reduced levels of cyclic AMP, adenosine 3′,5′-cyclic monophosphate (cAMP) and β-catenin in DC-like cells from the cocultures are detected, suggesting that induction of apoptosis and inhibition of differentiation could be the basis for the inhibition of mature DCs from HSCs by MSCs. Further, our results demonstrate that DCs derived from HSCs in the presence of MSCs are functionally impaired, especially for those after direct contact with MSCs. To investigate the basis of functional impairment, our data show downregulated tumor necrosis factor-α and transforming growth factor-β1 secretion and upregulated interleukin-6 (IL6) and IL1β secretion in the cultures with MSCs. Together, MSCs can inhibit differentiation of mature DCs from HSCs by arresting them at the precursor stage and induce their apoptosis. Further, HSC-derived DCs in the presence of MSCs are functionally impaired, which could be partly due to the upregulation of IL6 secretion.
Introduction
MSCs can suppress various immune cells, 12 possibly through secretion of soluble immunosuppressive factors such as hepatocyte growth factor, prostaglandin E2, transforming growth factor-β1 (TGF-β1), indoleamine 2,3-dioxygenase, nitric oxide, and interleukin-10 (IL10),4,13–15 by MSCs themselves or immune cells in response to MSCs. Cell–cell contact also plays an important role in the immunosuppressive activities of MSCs. 16
Antigen presenting cells are initiators of immune responses because they direct cellular and humoral immune responses against self and nonself antigens. 17 Immature dendritic cells (DCs) loaded with antigen can silence T cells by inducing apoptosis or increasing the regulatory T cell population.18,19 For a better utility of MSCs as an immune-suppressive agent, we are interested in the mechanism of how MSCs affect the commitment and differentiation of hematopoietic stem cells (HSCs) into different subsets of DCs, the most important antigen presenting cells in mediating T cell responses. Since how MSCs affect the differentiation of HSCs into various subsets of DCs was inadequately studied and seldom reported in the literature, the aim of this study was to define the effects of MSCs on dendritic-lineage commitment of HSCs. Here, we show that MSCs inhibit differentiation of HSCs into various subsets of DCs at the precursor stage, reduce the number of DC subtypes by alteration of cell-cycle state, and induce apoptosis of precursor cells. Further, cyclic AMP adenosine 3′,5′-cyclic monophosphate (cAMP) and β-catenin signaling pathways may be involved in these processes. Taken together, allogeneic MSCs may be applied not only in tissue regeneration, but also in GvHD prevention during transplantation by regulation of DC formation from the recipients.
Materials and Methods
Isolation of CD34+ HSCs and T cells from human umbilical cord blood
Umbilical cord blood (UCB) and peripheral blood was collected after institutional review board approval and patient consent. UCB mononuclear cells were isolated by Ficoll-Paque PLUS (GE Healthcare Bio-Sciences AB) gradient centrifugation. CD34+ and CD3+ cells were purified sequentially by using commercially available MicroBeads (Miltenyi Biotec Inc., Germany) as per the manufacturer's suggestion. Purified CD34+ cells and CD3+ T cells were washed, immunostained, and analyzed by BD FACSCaliburTM flow cytometer (Becton Dickinson Biosciences). For every experiment, CD34+ HSCs from each donor were divided into three groups: HSC alone, HSC-MSC contact coculture, and HSC-MSC transwell coculture. HSCs were cultured in a DC differentiation medium, which is Roswell Park Memorial Institute (RPMI)1640 complete medium based. Purified CD3+ T cells were cultured in the RPMI1640 complete medium for T cell proliferation assay. Details of the RPMI1640 complete medium are indicated in the Coculture section below.
Culture of MSCs
MSCs were isolated and clonally expanded from human bone marrow as previously reported. 20 Their surface immunophenotype and differentiation capabilities into different mesenchymal lineages were confirmed. MSCs were cultured in IMDM (Sigma-Aldrich) supplemented with 10% GIBCO™ embryonic stem (ES) cell-qualified fetal bovine serum (Invitrogen Corporation), 2 mM L-glutamine, 100 U penicillin, 100 μg/mL streptomycin (Sigma-Aldrich), and 10 ng/mL fibroblast growth factor-2 (R&D Systems, Inc.).
Coculture of MSCs and HSCs
Before coculture, MSCs were irradiated (30 Gy), counted, and seeded in 12- or 24-well culture plates (Becton Dickinson) overnight in the RPMI complete medium, consisting of the RPMI1640 medium (Gibco BRL/Invitrogen Life Science), 10% AB+ serum (isolated from a healthy donor), 2 mM L-glutamine, 100 U penicillin, and 100 μg/mL streptomycin (Sigma). UCB HSCs were cultured with irradiated MSCs at a ratio of 1:1 in the DC differentiation medium, which consisted of the RPMI complete medium containing a cytokine cocktail of 100 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF), 2.5 ng/mL tumor necrosis factor-α (TNF-α), and 10 ng/mL TGF-β1 (PeproTech, Inc.)21–23 for 7 days. HSCs alone were cultured in parallel as controls. To evaluate the contribution of soluble factors and cell–cell contact to cell differentiation, transwell chambers (Becton Dickinson) with pore sizes of 0.4 μm were used. The irradiated MSCs were cultured in the lower chamber, while HSCs were seeded in the upper chamber.
Immunostaining and FACS analysis
After 7 days of culture, cells were harvested for flow cytometric analysis. Suspension cells from MSC contact culture were characterized as hematopoietic cells; thus, suspension cells from MSC contact culture were carefully recovered and subjected to analysis. Flow cytometric analysis was performed using a FACSCalibur flow cytometer (Becton Dickinson Biosciences). CellQuest software (Becton Dickinson) was used for data acquisition and analysis. Expression of surface molecules, including CD34, CD4, CD14, CD1a, CD11c, CD116, CD123, E-cadherin, HLAII, and CD86, was assessed according to individual corresponding isotypes. The fluorescence-conjugated antibodies against CD34, CD4, CD123, CD116, and HLAII and their corresponding isotype control antibodies were purchased from BD Biosciences. Phycoerythrin (PE)-conjugated anti-human E-cadherin antibody and its corresponding isotype control antibody were purchased from R&D systems, Inc., while antibodies against CD14, CD1a, CD11c, and CD86 and their corresponding isotype control antibodies were purchased from eBioScience, Inc.
To detect β-catenin expression by flow cytometry, cells were fixed in 3.7% formaldehyde (Sigma) and permeabilized using 0.1% Triton X-100 (Sigma) for 20 min each. Subsequently, cells were incubated with a fluorescein (FITC)-conjugated anti-human β-catenin IgG2a antibody (BD Biosciences). Cells stained with FITC-conjugated IgG2a antibody were used as isotype controls. To analyze flow cytometric data, cell debris and large cells from forward scatter versus side scatter display were gated out and the R2 gate was set at any positive intensity above isotype control staining from each group.
Cell cycle analysis
Suspension cells from different cultures were harvested for cell cycle status analysis. Cell cycle status was examined using the CycleTest PLUS DNA Reagent Kit, according to the manufacturer's protocol (Becton Dickinson Immunocytometry Systems). Briefly, cell suspensions were centrifuged at 1200 rpm for 5 min. After centrifugation, cells were mixed sequentially with trypsin buffer and trypsin inhibitor plus RNase buffer and incubated at room temperature for 10 min in each solution. Propidium iodide (PI) solution was then added after which the cells were incubated at 4°C in the dark before flow cytometric analysis. The distribution of cell cycle phase in the whole cell population was assessed according to PI intensity peaks on FL2-Area DNA histogram. The numbers shown in Table 2 represent the cell numbers in different cell cycle phases.
Cell apoptosis assay
The apoptotic state of DC-like cells was detected using the Annexin V-FITC kit, according to the manufacturer's protocol (Beckman Coulter). Cells were washed with ice-cold phosphate-buffered saline and resuspended in 1× binding buffer (5–50 × 104 cells per 100 μL), and 5 μL of Annexin V-FITC and 2.5 μL of PI were added and incubated at 4°C in dark for 10 min. Then, 400 μL of 1× binding buffer was added, and cells were analyzed by flow cytometry after 30 min.
Intracellular cyclic adenosine monophosphate level determination
Intracellular cAMP concentration was measured using the cAMP HTS Immunoassay Kit (Millipore), according to the manufacturer's protocol. Briefly, 2–4 × 105 DC progenies from different cultures were lysed for 10 min. Cell lysate or cAMP standard was added to the wells precoated with anti-rabbit antibody in triplicate, and then 20 μL of diluted cAMP Alkaline Phosphatase Conjugate Tracer and 50 μL of diluted Rabbit anti-cAMP antibody were added to all wells. After incubation and washing, the plate was developed using a p-NPP Alkaline Phosphatase Substrate. The reaction was stopped and the optical density was read at 405 nm to determine the concentration of cAMP.
Western blot analysis
To analyze β-catenin levels by Western blot analysis, total protein from suspension cells was extracted using lysis buffer (Sigma). The insoluble fraction was removed by centrifugation at 13,000 rpm for 30 min at 4°C. Protein concentration was determined using the Bio-Rad protein assay reagent (Bio-Rad Laboratories). Proteins were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto 0.45 μm nitrocellulose membranes (Bio-Rad Laboratories). Membranes were blocked and incubated with appropriate dilutions of specific antibodies overnight at 4°C. For detection of β-catenin, rabbit anti-human β-catenin monoclonal antibody (Cell Signaling Technology, Inc.) and anti-rabbit secondary antibody conjugated to horseradish peroxidase were used. Membranes were developed with enhanced chemiluminescence reagent (Pierce). Glyceraldehyde 3-phophate dehydrogenase antibody (Sigma) was used as an internal loading control. The relative density of protein bands was quantified by using area densitometry with LabWorks Analysis Software (UVP, Inc.). The β-catenin expression levels in DC-like cells from different groups were normalized against the signal of their glyceraldehyde 3-phophate dehydrogenase expression.
T cell proliferation assay
Allogeneic T cells from UCB were used. A titration of the mitogen, phytohemagglutinin (PHA) (Sigma), was tested at concentrations of 0, 0.625, 1.25, and 5 μg/mL. DCs from different cultures were irradiated at 30 Gy before seeding with T cells. Since 1.25 μg/mL PHA induced the greatest T cell proliferation, this concentration was used for subsequent T cell proliferation assays. Various amounts of DCs (0–4 × 104) and T cells (5 × 104) in a total volume of 100 μL of RPMI 1640 complete medium were seeded per well in a 96-well plate. T cell proliferation assays were done in triplicates. Control groups consisted of DCs or T cells alone with PHA and medium alone. After an incubation period of 72 h, T cell proliferation was detected by CellTiter 96 Aqueous One Solution Cell Proliferation Assay KIT (Promega). Cells were pulsed with 20 μL/per well of AQ solution (Promega) 4 h before the end of the assay, and the optical density at 490 nm wavelength was determined.
Enzyme-linked immunosorbent assay
Concentrations of human IL1β, IL4, IL6, IL10, IL12, TNF-α, and TGF-β in cell culture supernatant were detected using specific capture enzyme-linked immunosorbent assay (ELISA) kits, according to the manufacturer's protocol. ELISA kits detecting IL1β and TGF-β were purchased from eBioScience, Inc., while those for IL4, IL6, IL10, IL12p70, and TNF-α were purchased from BD Biosciences. All samples were performed in triplicates and the data shown represent results from at least three independent experiments. Data were expressed as means ± standard deviation.
LPS stimulation
DC-like cells were collected, washed with phosphate-buffered saline, and stimulated with 100 ng/mL of lipopolysaccharide (LPS; Molecular Probes/Invitrogen) in the RPMI complete medium for 2–3 days. Supernatants were then collected for specific cytokine analysis.
Statistical analysis
Statistical comparison between the control and the experimental groups was carried out with Student's t-test or one-way analysis of variance. p-Values <0.05 were considered to be significantly different.
Results
MSCs inhibit HSC differentiation into DCs
Under DC induction conditions, HSCs develop into different DC subtypes. To investigate whether MSCs affect HSC differentiation into DCs, HSCs purified from human UCB were cultured in either direct or indirect contact with MSCs using transwells in DC differentiation medium. The purity of CD34 + HSC starting population was over 90% (Fig. 1A). After 6–7 days of culture, we observed that HSCs formed cell clusters during DC differentiation, but the number of cell clusters was greatly reduced in the presence of MSCs, especially when grown in direct cell contact (data not shown). As for the surface phenotype, 30%–50% of CD34+ cells were detected in the whole population of cells from MSC cocultures, while only about 10% were observed in the absence of MSCs (Fig. 1B and Table 1). In addition, after MSC coculture, the percentage of cells expressing early markers along DC differentiation, CD117 (c-kit), CD123 (IL3 receptor α), and CD116 (GM-CSF receptor), was 48%–53%, 76%–79%, and 13%, respectively, compared to HSCs grown alone which was 29%, 41%, and 28%, respectively (Fig. 1B and Table 1). Further, the percentage of CD123 expression was downregulated, whereas CD116 expression was slightly upregulated during DC induction (data not shown). Moreover, CD4 expression was reduced in the presence of MSCs (Fig. 1B and Table 1), of which its expression should increase along the course of HSC-DC differentiation (Fig. 1C, D). These data suggest that MSCs inhibited induction of DCs from HSCs.
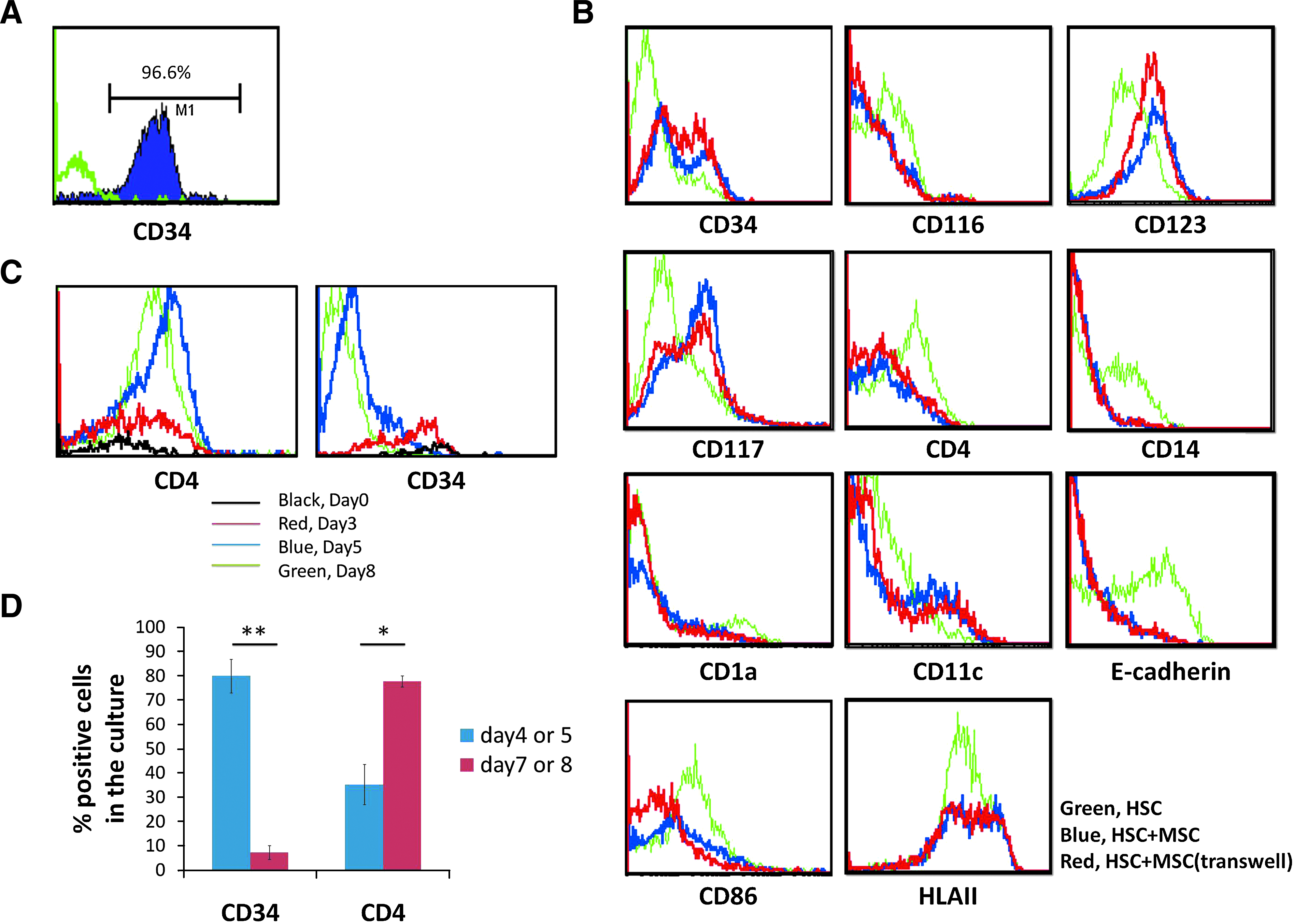
Phenotypic analysis of hematopoietic stem cells (HSCs) and HSC-derived cells cultured alone or with mesenchymal stem cells (MSCs). (
p ≤ 0.008 compared with HSC control.
p ≤ 0.02 compared with HSC control.
HSC, hematopoietic stem cell; MSC, mesenchymal stem cell.
The effects of MSCs on the frequency of other DC progenies were also determined. MSC coculture inhibited expression of CD4, CD14, and E-cadherin, but not CD11c and CD1a (Fig. 1B and Table 1). The percentage of CD1a+Ecad+ cells was also downregulated in MSC cocultures, but not that of CD1a+CD11c+ cells. CD4 expression in CD11c+ cells was markedly reduced by MSC coculture (Fig. 1B). In addition, MSC coculture downregulated CD86 expression upon DC induction (Fig. 1B and Table 1). A decreased mean fluorescence index of HLAII expression was observed without a change in its percentage of expression.
A significant reduction in total cell number of DC progenies derived from both MSC contact and transwell cocultures was observed as compared to the HSC control after 6–7 days in culture (Table 2). Calculation of the absolute number of DC progenies from different cultures demonstrates that MSCs inhibited generation of many myeloid DC subtypes, including CD14+ monocytes, CD4+ DCs (putative pan-DC precursors), CD11c+Ecad+ Langerhans DCs (LCs), and CD11c+ myeloid DCs (mDCs) (Table 3); however, the number of CD11c+CD1a+ LCs precursors was not significantly reduced in the presence of MSCs (Table 3), indicating that MSC coculture did not inhibit the generation of LC precursors, but inhibited them from further differentiation and maturation.
Relative total cell number was determined after normalizing to the HSC control in separate experiments.
p ≤ 0.000004 compared with HSC control.
p ≤ 0.0003 compared with HSC control.
p < 0.05 compared with HSC control.
p < 0.05 compared with HSC control and with HSC + MSC (contact) condition.
The number of different cell types was calculated by total cell number obtained after 7 days of culture multiplied by the percentage of positive cells stained for specific markers. The relative cell number of different cell types in various culture conditions was obtained by normalizing to their corresponding HSC alone control.
p ≤ 0.05 compared with HSC control.
p ≤ 0.006 compared with HSC control.
pan-pre-DC, pan-dendritic cell precursor; mDC, myeloid dendritic cell; pre-LC, Langerhans DC precursor.
Increased apoptosis in MSC cocultures
To determine the possible mechanism contributing to the reduction in total cell number when cocultured with MSCs (Table 2), the cell cycle and apoptotic status of DC-like cells was analyzed. Suspension cells were recovered from different cultures and cell cycle analysis and annexin V staining were performed. MSCs reduced the frequency of the G2/M phase and increased apoptosis in DC-like cells (Table 2). Since activation of the cAMP pathway can prevent CD34+ cell apoptosis, 24 the intracellular cAMP level in DC-like cells from each culture condition was assessed. Reduced intracellular cAMP levels were detected in DC-like cells after coculture with MSCs (Fig. 2A). In addition, β-catenin is a key molecule in the canonical Wnt signaling pathway and is involved in the maintenance of HSC self-renewal. 25 To examine whether the Wnt signaling pathway is associated with MSC-mediated immunomodulatory effects, we analyzed β-catenin expression in DC-like cells isolated from each culture condition. Approximately 13% of cells from the control culture were positive for β-catenin expression; however, all cells were β-catenin-negative after MSC coculture (Fig. 2B, C). Downregulation of β-catenin protein expression in cells cocultured with MSCs was also confirmed by Western blot analysis (Fig. 2C).
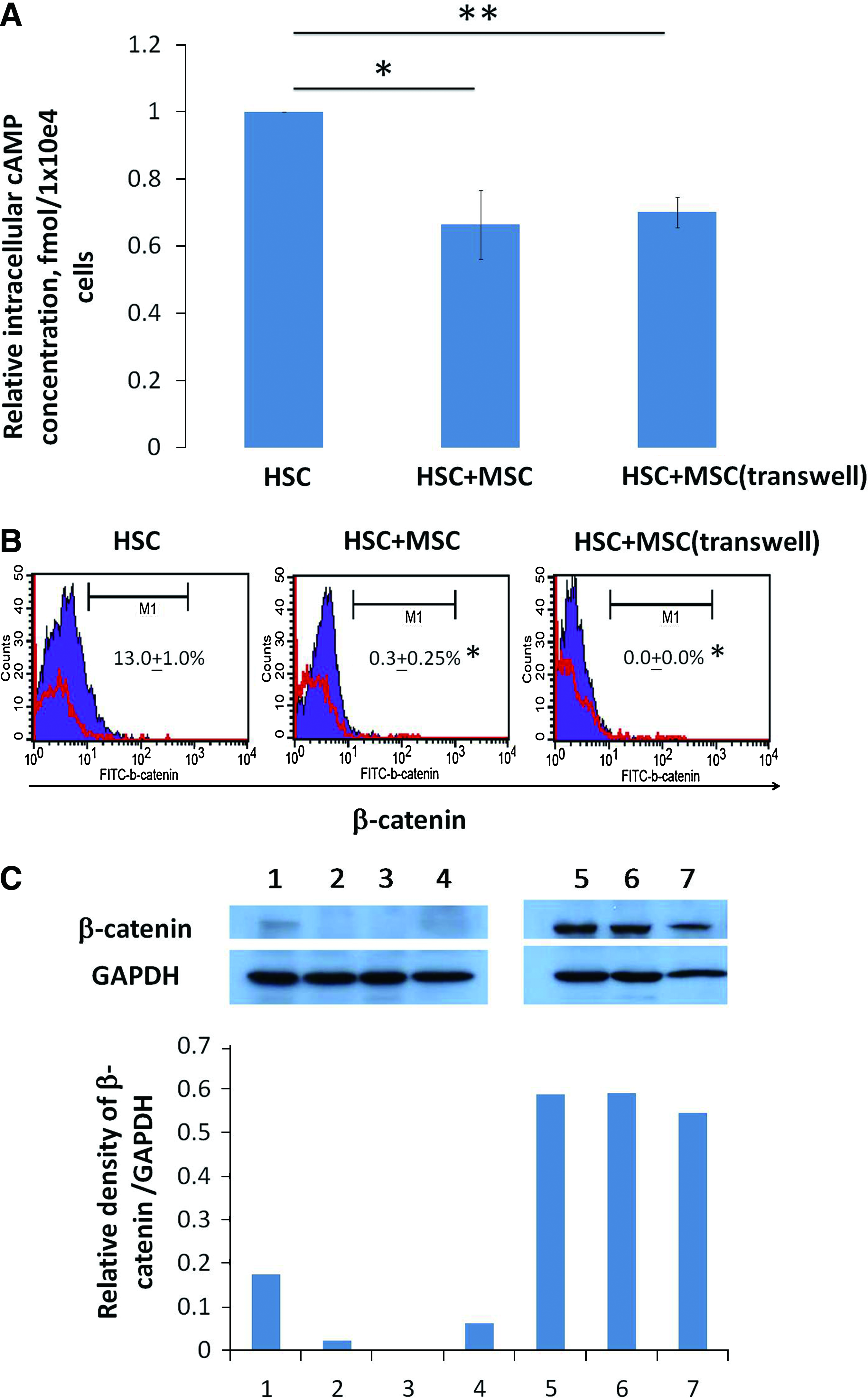
β-Catenin and intracellular cyclic AMP, adenosine 3′,5′-cyclic monophosphate (cAMP) levels in DC-like cells from different cultures. Purified CD34 + cells from UCB were cultured in DC differentiating medium in the absence or presence of MSCs (contact or transwell system) for 7 days and after which the intracellular cAMP level and β-catenin expression of DC-like cells were assessed. (
Overall, cell cycle entry of HSCs during DC differentiation was inhibited and apoptosis was increased by the presence of MSCs. Meanwhile, cAMP level and β-catenin expression were downregulated in DC-like cells cocultured with MSCs, which may account for the increased apoptosis during differentiation.
DC-like cells isolated from MSC cocultures are functionally deficient
To determine whether the function of DC-like cells is altered by MSCs, T cell stimulation assays were performed. After incubating DC-like cells derived from different culture conditions with allogeneic naive T cells, DC-like cells derived from HSCs in direct contact with MSCs displayed a reduced T cell proliferating response after stimulation with PHA (Fig. 3A). A dose-dependent effect of T cell stimulating ability by DC-like cells was observed, but only high doses of DC-like cells from MSC contact coculture showed significant suppressive effect on T cell proliferation. At low cell doses, DC-like cells from transwell coculture showed strong ability to stimulate T cell proliferation, while high doses of DC-like cells showed no difference with that of HSC control culture (Fig. 3A).
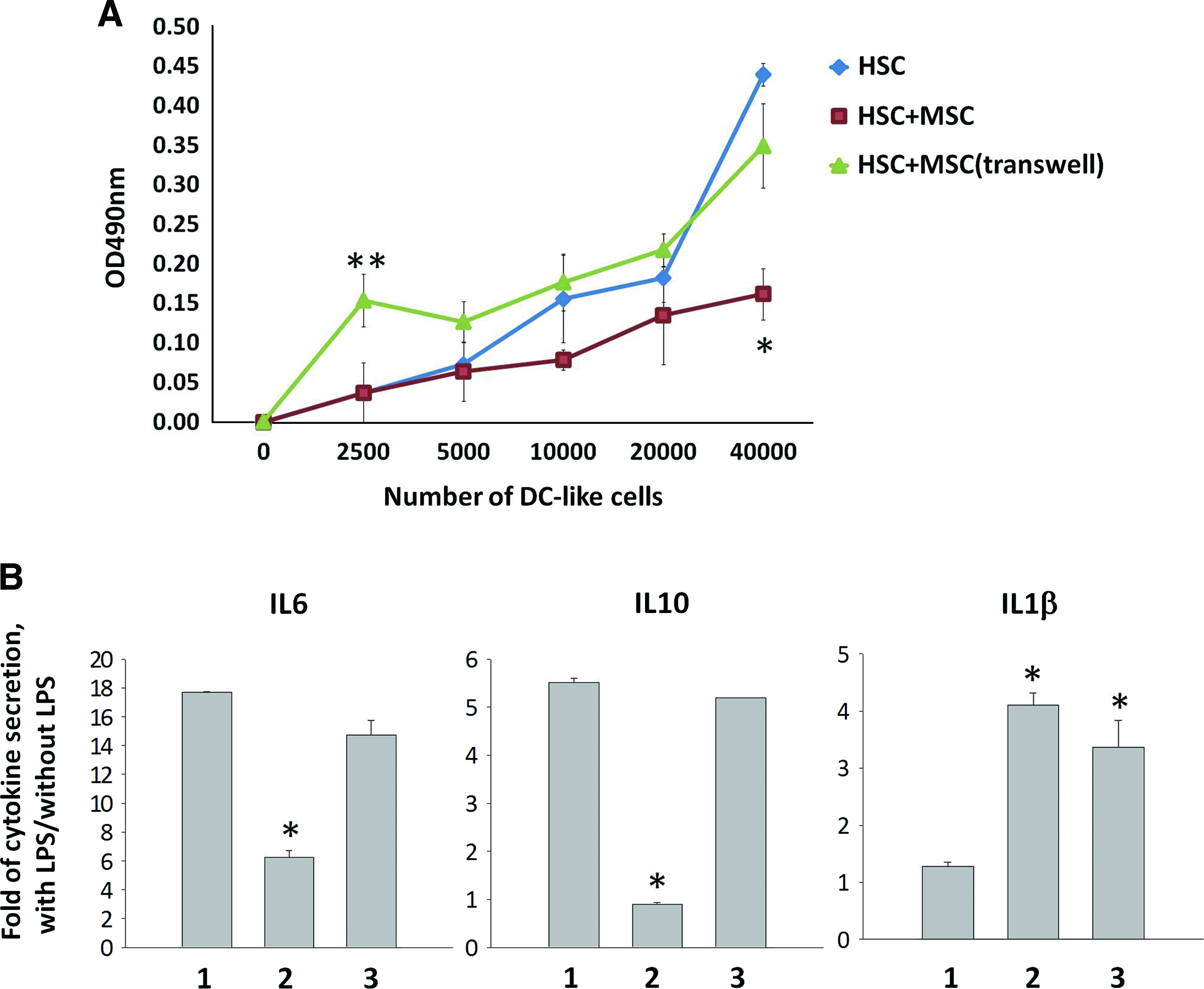
Effect of MSCs on the function of DC-like cells. DC-like cells generated in different cultures were collected, washed, and subjected to allogeneic naïve T cell proliferation assays (
After 7 days of coculture, MSCs were removed, and DC-like cells were further cultured in the RPMI complete medium with and without LPS stimulation for assessment of DC cytokine secretion ability. Whereas reduced IL6 and IL10 secretion by DC-like cells upon LPS induction was observed in DC-like cells from contact coculture, increased IL1β secretion was detected in DC-like cells isolated from both contact and transwell cocultures (Fig. 3B) (suggest indicating groups on graphs, not numbers). TNF-α and IL12p70 was not secreted by DC-like cells from any culture in the presence or absence of LPS (data not shown). These data indicate that the presence of MSCs negatively influenced the ability of DC-like cells to stimulate naïve T cell proliferation and secrete inflammatory cytokines upon LPS stimulation, especially in the contact manner.
MSC coculture reduces pro-inflammatory cytokine and enhances antiinflammatory cytokine expression in HSCs
The involvement of soluble factors were speculated since MSCs cocultured with HSCs in direct contact and in transwells resulted in reduced DC lineage generation and increased cell apoptosis. Thus, capture ELISAs were used to examine inflammatory cytokine levels in these cultures. Among the cytokines analyzed, a significant increase in IL6 and IL1β production were observed in MSC cocultures (Fig. 4A), while TNF-α and TGF-β1 levels were downregulated upon MSC coculture during DC differentiation (Fig. 4A), which may inhibit HSC differentiation into LCs and other DC subtypes. IL10 secretion was similar (Fig. 4A) while IL12p70 was not detectable in any culture (data not shown). Higher IL1β levels were detected in MSC cocultures as compared to HSC control culture, and no IL1β production was detected in MSC-alone culture (data not shown), indicating that IL1β was secreted as a consequence of coculture. Secretion of IL6 by MSCs or HSCs was also examined, and relatively high level of IL6 was detected in the supernatant of MSC culture as compared with that of HSC culture (Fig. 4B). Together, MSCs significantly downregulated TNF-α and TGF-β1 and upregulated IL1β and IL6 levels upon coculture.
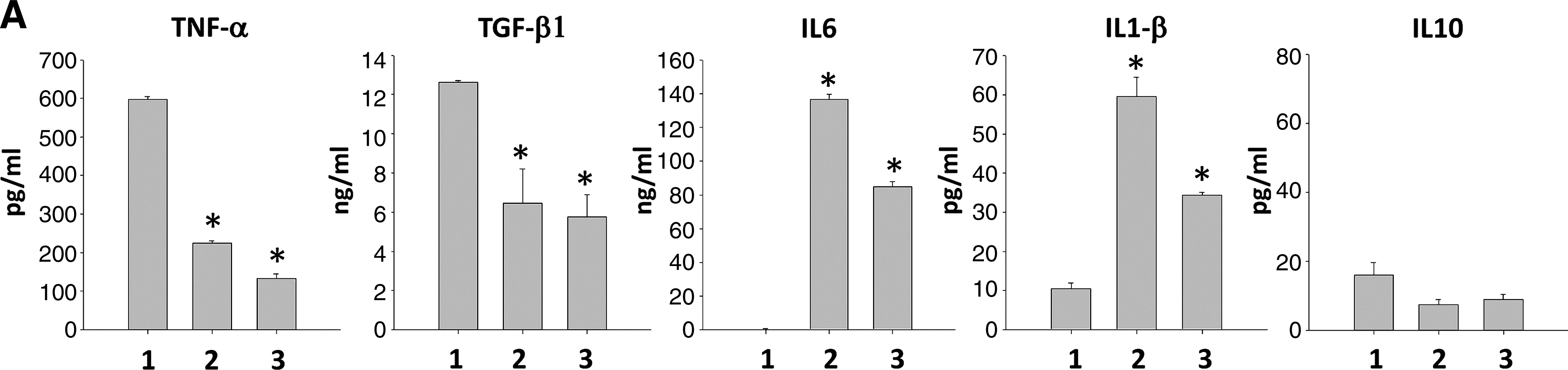
Cytokine profile in different cultures and the effects of interleukin-6 (IL6) on the surface immunophenotype of HSC-derived cells. Purified CD34 + cells from UCB were cultured in the DC differentiating culture medium in the absence or presence of MSCs. (
Discussion
In this study, we showed that coculture of MSCs had an inhibitory effect on HSC differentiation into functional DCs. The DC-like cells derived from HSCs in the presence of MSCs possess the following characteristics: (1) higher expression of CD34, CD117, and CD123 and reduced expression of CD116, CD4, and CD86; (2) lower number of CD14+ monocytes, CD11c+ myeloid DCs, and CD11c+Ecad+ LCs; (3) reduced entry into the G2/M phase of the cell cycle; and (4) lower capacity to induce an allogeneic naïve T cell response and lower secretion of IL6 and IL10 upon LPS stimulation. We also observed that MSC coculture not only inhibited cell proliferation, but also rendered cells apoptotic during HSC-DC differentiation. The inhibitory effect of MSC coculture on HSC-DC differentiation is accompanied with an altered cytokine profile and downregulation of β-catenin levels in DC-like cells. In addition, cell–cell contact between MSCs and HSCs further potentiated these effects.
Several studies have investigated the effect of MSCs on DC maturation and differentiation from monocytes,4,22,26 but very few have assessed the effect of MSCs on HSC differentiation into different DC subtypes. There only are a handful of studies showing the effect of MSCs on CD34+ HSCs toward DC differentiation.21,22 Moreover, the reported culture conditions are different from the present study. For example, apart from GM-CSF and TNF-α, stem-cell-proliferating cytokines such as stem cell factor or flt-3 ligand were included in the DC induction culture medium. Both stem cell factor and flt3 ligand are early acting hematopoietic cytokines that promote cell growth and cell renewal in HSCs. 27 Thus, to minimize the possible effect of cell proliferation, we excluded the stem-cell-proliferating cytokines in this study. To analyze all DC subtypes during differentiation, we added TGF-β1 in our culture system, which is essential for LC differentiation.28,29 Previous studies focused on the production of CD14+ and CD1a+ cells and expression of costimulatory molecules.21,22 In addition to that, we further analyzed different DCs in this study and determined whether MSCs alter early HSC-DC development because MSCs may influence HSC commitment at the early stage of hematopoiesis. Similar to a previously reported study, 21 our results also demonstrated a prevention of CD14+ monocyte generation. While a previous study showed that MSCs prevent differentiation of CD14+ precursors into CD1a+ DCs but not the development of LCs, 22 we observed that MSCs inhibited generation of LCs, but not LC precursors, indicating that MSCs halted LC lineage differentiation at the LC precursor stage.
Expression of CD4 is not only limited to T lymphocytes. CD4 can also be expressed on freshly isolated monocytes and hematopoietic progenitors at different stages of lineage commitment.30,31 In our study, CD4 was not expressed on freshly isolated HSCs, and its expression progressively increased during DC differentiation. No CD3+ T cells were also detectable in the cultures, suggesting that these CD4+ cells generated from HSCs were not T lymphocytes. CD4 expression was markedly inhibited by MSC coculture, indicating that MSCs may inhibit the lineage differentiation of HSCs. In addition, reduced CD4 expression on CD11c+ DCs accompanied with a decreased level of HLAII mean fluorescence intensity after MSC coculture was suggestive of DC suppression.
Reduced cAMP and β-catenin levels accompanied by increased apoptosis were also observed in cells cocultured with MSCs. Elevated cAMP could prevent apoptosis by promoting protein kinase A to phosphorylate glycogen synthase kinase-3β, resulting in decrease of β-catenin degradation and thereby inducing antiapoptotic activity.24,32 Therefore, MSC-mediated reduction in cAMP levels in DC-like cells may result in decreased Wnt signaling, leading to increased apoptosis. Since it has been indicated that β-catenin is essential for DC differentiation, 33 we speculate that MSCs may inhibit DC differentiation and induce apoptosis simultaneously by reducing Wnt signaling. However, the detailed signaling mechanism regulating HSC-DC differentiation and its influence by MSCs needs to be further investigated.
DCs can initiate either tolerance or immunity depending on their maturation or activation state. 34 In spite of a significant reduction of DC number and CD86 and HLAII expression in differentiated DC-like cells, DC-like cells from transwell MSC coculture triggered a stronger T cell proliferation than cells from contact MSC cocultures, suggesting that MSCs reduced the ability of DC-like cells to stimulate T cell proliferation in a contact-dependent manner during hematopoiesis. Surprisingly, T cells stimulated by lower ratio of DC-like cells from transwell coculture promoted the highest cell increase compared to control and contact culture (Fig. 3A). We speculate that it was because the secreting ability of the T cell proliferation stimulant IL6 was not affected while the secreting ability of the other T cell proliferation stimulant, IL1β, was higher in DC-like cells from transwell coculture than in control group (Fig. 3B). Upon LPS stimulation, immature DCs can further maturate through upregulation of MHCII, adhesion molecule, and costimulatory molecule expression, resulting in altered secretion of IL1β, IL6, IL10, IL12, IL18, and IL23.35,36 In this study, MSCs also altered cytokine production by DC-like cells upon LPS stimulation, especially in a cell–cell contact-specific manner, indicating that MSC-primed DC-like cells possess an altered function in cytokine secretion that may affect the following T cell responses after antigen presentation.
In our study, MSC coculture reduced TNF-α and TGF-β1 levels, resulting in downregulation of HSC-DC differentiation. In contrast, MSCs secreted high levels of IL6, which may result from TNF-α- and IL1β-induced NF-κB nuclear translocation. 37 To date, the role of IL6 in DC differentiation is not fully understood; however, IL6 can support generation of myeloid DCs although they are functionally impaired.38–40 In this study, the high level of IL6 secretion by MSCs was detected, suggesting a role involved in the suppressive function of MSCs in HSC-DC differentiation, which is worth to be further explored.
Tissue engineering mainly focuses on the functional and biomechanical stability of the artificial tissues, which contain scaffolds, cells, and biologically active molecules destined for transplantation. Meanwhile, the issue of tissue engineering construct rejection by recipients should also be cautiously considered when allogeneic cells are included. Many studies had focused on tolerance induction during organ transplantation by establishing chimerism in the recipient.41–45 Nevertheless, the arising hematopoietic cells from HSCs injected or from host circulating system may still cause chronic GvHD or the eventual failure of allograft survival in the recipient. Therefore, alternative methods inducing long-term immune tolerance for the grafts based on the hematopoietic stem cell transplantation (HSCT) therapy are worth to be considered. Our findings in this study suggest that MSCs may be able to not only prevent acute immune responses from mature DCs, but also provide a tolerant environment to replace the life-long nonspecific immunosuppressive therapy usually needed in tissue engineering organ transplantation. This study offers new insights into clinical application of MSCs to treat GvHD by preventing rejection and further provide long-term protection after allogeneic organ transplantation to tissue-engineered products using allogeneic cells. Altogether, our results encourage further development of immunotherapy to replace the use of life-long immunosuppressants by means of MSC transplantation.
Footnotes
Acknowledgments
This work was financially supported by HealthBanks Biotech. Co. and grants from the Ministry of Education, Aim for the Top University Plan, and National Science Council (NSC98-2627-B-010-004, NSC98-2314-B-010-001-MY3, and NSC98-3111-B-010-003), as well as Taipei Veterans General Hospital (VGH99E1-014, VGH99S4-001, and VGH99C1-097). We thank Shun-Fen Chen, RN, from the Department of Obstetrics and Gynecology, Taipei Veterans General Hospital, for helping collect cord blood samples. We also thank Mr. Vernon Shih from the Institute of Clinical Medicine, National Yang-Ming University, for proofreading the manuscript.
Disclosure Statement
No competing financial interests exist.