
Abstract
Introduction:
The arachnoid tissue is a critical component of cerebrospinal fluid removal. Failure of that function results in hydrocephalus, a serious medical condition. The purpose of this study was to characterize arachnoid cell transport in culture and on three-dimensional collagen scaffold.
Methods:
Arachnoid cells were harvested from rat brainstems and cultured onto bilayered bovine collagen scaffolds. Cell growth and phenotype (protein expression and morphometry) were determined. Permeability and hydraulic conductivity were quantified.
Results:
Cells harvested from the anterior brainstem surface exhibited arachnoid cell phenotype (positive for vimentin, desmoplakin, and cytokeratin), readily penetrated the collagen scaffold, and doubled approximately every 2–3 days. The transepithelial electrical resistance value for a monolayer of cells was 160 Ω cm2 and the permeability of indigo carmine was 6.7×10−6±1.1×10−6 cm/s. Hydraulic conductivity of the collagen construct was 6.39 mL/min/mmHg/cm2.
Conclusion:
Cells isolated from the anterior brain stem exhibited the same phenotype as those found in the native tissue and exhibited aspects of barrier function found in vivo. These studies suggest that an ex vivo model for the arachnoid granulation can be developed.
Introduction
The arachnoid granulation consists primarily of arachnoid cells with an endothelial cell layer. Within the granulation, the arachnoid cells are organized in two distinct layers. 5 The first is an arachnoid cap made up of tightly packed arachnoid cells several cell layers thick. Below is a porous core several millimeters thick that is mainly collagen extracellular matrix with a sparse population of arachnoid cells. These two layers are contiguous with the arachnoidal membrane that covers the central nervous system. Another arachnoidal layer, the pia, is tightly adherent to the brain. 5 Because they are anatomically more accessible and surgically more easily manipulated, cells here serve as a ready source for harvesting. Between the pia and the arachnoid membrane are sparse strands also made up of arachnoid tissue. The strands connecting the more superficial arachnoid membrane to the pia form a three-dimensional (3D) web-like network (hence the name arachnoid). This space between the two arachnoidal layers is the subarachnoid space in which the CSF circulates. The arachnoid granulation inhibits large molecules from entering into the CSF space while allowing small molecules such as water to pass. 6 The complete role of the arachnoid cells in the flow of CSF is unknown despite over 100 years of study, 6 and only recently has the transport of arachnoid granulation in isolation been explored. 7
Previous studies attempting to understand the function/dysfunction of the arachnoid granulation have been limited.8,9 These studies used arachnoid cells obtained from human donors and characterized phenotype and permeability of cells cultured on synthetic matrices (nonwoven poly[ethylene terephthalate] [PET] and cell culture inserts). These studies demonstrated that arachnoid cells from humans can be cultured outside the body and that arachnoid cells cultured ex vivo exhibit a directionality of water flow (i.e., hydraulic permeability differs based on the direction of flow). These studies are important in establishing the ability to culture arachnoid cells in vitro and the basic transport characteristics of cell monolayers. The previous studies do little to recapitulate the native arachnoid granulation. The objective therefore of this investigation is to culture arachnoid cells in a 3D collagen matrix, characterize their behavior, and compare it to that observed in two-dimensional (2D) culture. The development of an appropriate in vitro culture model for the arachnoid granulation can be used to elucidate the function of the granulation and its role in hydrocephalus, to evaluate new therapies to treat the disorder, and ultimately to aid in the development of a fully tissue-engineered arachnoid granulation.
Materials and Methods
Cell isolation
Arachnoid cells were isolated from 19-day-old rats (Rattus norvegicus) with approval from the Minneapolis Veterans Administration Medical Center and University of Minnesota animal care committees. Animals were anesthetized before sacrifice and the pia-arachnoid tissue was harvested from the anterior portion of the brainstem using microsurgical techniques. The tissue was washed three times in phosphate buffered saline (PBS), cleared of extraneous tissue, and chopped into 1×1 mm2 pieces.
Tissue fragments were incubated at 37°C in humidified atmosphere of 95% air and 5% carbon dioxide in a culture media containing Eagle's essential medium with 10% fetal bovine serum (Remel Inc.), nonessential amino acids (Sigma Aldrich), glutamine (BioWhittaker Inc.), streptomycin, and penicillin (Gibco Invitrogen Corp.) at 10μL/cc each; the medium was changed twice per week. Cyclic AMP (cAMP) (2%; Sigma Aldrich) was added to the culture media for a limited number of cultures. Within 2–3 days cells were seen growing out of the dissected tissue, and after ∼7–10 days the cells became confluent. They were passaged following trypsinization with a 1:2 dilution of 0.05% trypsin-0.02% EDTA (Gibco Invitrogen Corp.) mixed with PBS. Cells were grown on Nunclon 6-well plastic plates (Thermo Scientific, Inc., 9.34 cm2 well surface area). Cells could not be passaged beyond passage 9–10, and most experiments used cells from passage 3 or 4.
The phenotype of the cells isolated from the tissue was confirmed using immunohistology. The cells stained positively for cytokeratin and colocalized for desmoplakin and vimentin; these markers are consistent with the arachnoid cell phenotype. 10 The cells also stained negatively for myosin, S100, GFAP, CD31, SMA, and NeuN, indicating that they were not smooth muscle, glial, endothelial, fibroblast, or neuronal cells, respectively. As reported in other studies,11–13 the cells appear similar to those in tissue fragments.
Collagen matrix preparation
Bovine Type I collagen was used to make porous sponges as per the protocol of Doillon et al. 14 A collagen dispersion (0.5% wt/volume) was made by slowly blending lyophilized insoluble collagen into a mixture with H2O+HCl solution (pH 3.0) at 4°C for 1 min. After deaeration, the dispersion was poured into a pan, lyophilized at −30°C, and dehydrothermally crosslinked in a vacuum oven at 110°C and 2.5 torrs for 5 days. Gamma irradiation (17,500 rads) was used to sterilize the sponges. The microstructure of the scaffold has two distinct regions that result from the freeze-drying process. The upper surface of the matrix has low porosity with cells seeded on this surface typically unable to penetrate deeply (see Supplementary Fig. 1S; Supplementary Data are available online at www.liebertonline.com/tea). The bottom surface of the scaffold has high porosity with an average pore size of ∼100 μm in diameter facilitating cell ingrowth. The scaffold was ∼2–3 mm thick and has a 3D architecture similar to the porous central core of an arachnoid granulation. This type of matrix has been used in previous investigations as a dermal replacement or a template for dermal regeneration,15–17 and been used clinically for the treatment of dermal wounds. 18
Construct seeding and culture
Sterile sponges ∼20 mm in diameter were pre-equilibrated with culture media before seeding with rat cells. Approximately 50,000 arachnoid cells at passage 1–2 were used to seed the collagen sponges. Preliminary studies suggested that this seeding density was optimal for ensuring uniform and rapid population of the sponge. Constructs were cultured up to 21 days.
Histology and immunohistochemistry
Cells were permeabilized in 0.25% Triton X-100, blocked in 2% bovine serum albumin, and incubated with primary antibodies in PBS overnight at room temperature. After rinsing with PBS, cells were incubated with appropriate secondary antibody and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were analyzed using a Biorad MRC-1024 single photon confocal microscope 1024 (Biorad Cell Science). For establishing arachnoid cell phenotype, cells were stained with cytokeratin 18 antibody (C-04, Abcam Inc.), vimentin (Prod #080552; Invitrogen), and desmoplakin I+II (Abcam, Inc.). Double labeling of vimentin and desmoplakin was done to help verify cell identification.
To verify purity of the arachnoid cell isolations, cells were also stained for the presence of potential contaminating cells: muscle, fibroblasts, endothelial cells, glial cells, and neuronal cells. Myosin (MA1-35718; Fisher Sci.) was used to identify muscle cells; S100 (13E2E2; BioGenex) and glial fibrillary acidic protein (GFAP, G-A-5; Cell Marque) were used to identify glial cells; CD31 (1A10; Cell Marque) was used to identify endothelial cells; smooth muscle actin (SMA asm-1; Novacastra) was used to identify fibroblasts; neuronal nuclei (NeuN, MAB377; Millipore) was used to identify neuronal cells. Staining levels for the cells were compared to positive controls (uterus, melanoma, brain, bladder cancer, uterus, and brain for myosin, S100, GFAP, CD31, SMA, and NeuN, respectively) or negative controls (brain for myosin, S100, CD31, and SMA, uterus for GFAP, and muscle for NeuN). The presence of gap junctions in cultures was determined by staining with connexin45 antibody (Prod #MAB3100; Millipore).
Scanning electron microscopy
Samples were fixed using 2.5% glutaraldehyde and 2.5% formaldehyde buffered to pH 7.4 with phosphate, followed by a serial dehydration in alcohol. Samples were dried using a critical point dryer (SPI # 13200-AB), sputter coated (SPI# 11429-AB), and imaged using a Hitachi HS 7S electron microscope.
Western immunoblotting
Confirmation of protein content to immunohistochemical staining was performed using Western blot. Positive controls were human A432cell line (Abcam) and melanoma (Abcam) for desmoplakin and vimentin, respectively. Normoxia (a cell line donated by Dr. Roderick Barke, Minneapolis VA Medical Center: PC-12 [TCC # CRL-1721] adrenal gland from rat) was used as the negative control for both desmoplakin and vimentin. Approximately 1.0×107 cells were centrifuged at low speed for 5 min. Pellets were washed with PBS (prod # 17-512F; BioWhittaker) at room temperature and again centrifuged at low speed. PBS was carefully poured out and ice-cold RIPA buffer (product #89900; Pierce) with inhibitors (Sigma Protease Inhibitor Prod # P-8340; Sigma Aldrich and Aprotinin Prod # A6279; Sigma Aldrich) added and incubated on ice for 30 min. Proteins were denatured by boiling in water for 2–3 min and loaded into an 18 well Precast XT gel cassette (prod #345-0136; BioRad Hercules). Chemiluminescent Blue Ranger marker (prod # 26651; Pierce) and low range Rainbow marker (prod # RPN 755 E; Amersham) were used to reference molecular weights. Electrophoresis was performed using a Power Pac Universal (BioRad). Proteins in the gel were transferred to Hybond ECL nitrocellulose (prod # RPN 2020 D; Amersham) by sandwiching between thick Blot paper (prod # 1703967; Criterion BioRad) and run for 45 min at 10 volts using a Trans-blot SD (prod # 170-3940; BioRad). Nitrocellulose membranes were incubated with Startingblock (TBS) Blocking Buffer (prod # 3754; Pierce) for 60 min on a rocker (Red Rocker Hoefer Scientific Instruments) at room temperature. The primary antibodies (Vimentin, Cytokeratin, and Desmoplakin) were diluted in TBST containing Startingblock at 1:1000 and were incubated overnight in the refrigerator on a rocker. On the following morning the nitrocellulose was rinsed twice with ∼30 mL of TBST, incubated with Donkey Serum Block (prod # D9663; Sigma Aldrich) for 60 min, and washed twice with TBST for 5 min each. A 1:10,000 dilution of the secondary antibody (donkey antiRabbit IgG-Horseradish peroxidase conjugate; Amersham # NA934V) was then used to cover nitrocellulose for 60 min at room temperature. The sample was washed with TBST. Proteins were detected with SuperSignal West Pico (Pierce #34080). Autoradiography films (prod # RPN 2103; Amersham) were developed using a CP1000 (AGFA Mortsel).
DNA content assay, cell number, and morphometrics
DNA content was quantified with the DNeasy Blood and Tissue Kit (Qiagen Sciences). Absorbance was measured, as per the manufacturer's instructions, at the 260 nm wavelength. Cell counts were performed using a hemocytometer (Hausser Scientific). cAMP was used to determine if it altered cell growth (1 mg/mL of media).
Cell morphometrics was performed using ImageJ software from the NIH website. High-resolution digital images of the cells were obtained and maximal cell length and width measured. Width was determined perpendicular to the maximal length. Area was determined from image analysis of cell boundary traces. Process counting was done manually. Between 50 and 100 cells were used for process number analysis and morphometric measurements.
Permeability
Transepithelial (arachnoid) electrical resistance
Transepithelial electrical resistance (TEER) was used as a measure of barrier function for cells grown on a monolayer. 19 Chopstick electrodes connected to an epithelial volt-ohmmeter resistance meter (World Precision Instruments, Inc.) were placed on the basal side and apical side of the membrane. Electrical resistance measurements were performed from day 5 to day 21 in culture and the resistance per unit area was determined by dividing values obtained by the surface area of the construct (∼2 cm2). The TEER value of a blank membrane (pore size 3.0 μm; CoStar Transwell) was subtracted from the apparent TEER values of membranes with cells to obtain the effective TEER value. The membrane allows diffusion of small molecules such as ions.
Hydraulic permeability and dye transport
The permeability of indigo carmine (prod #55928, Sigma Aldrich) was quantified. The arachnoid cell construct was placed in a diffusion chamber (PermeGear Side-Bi-Side Cells) and maintained at 37°C. Each chamber was mixed using a magnetic stirring bar to minimize diffusion boundary layers at the interface of the construct. At 10 min intervals, 20 μL samples were taken from the receiver chamber and the concentration of the dye was determined using a Packard SpectraCount photometric microplate reader (measured at 520 nm)(Packard Instrument Co.).
The apparent permeability, Papp, of the cells to the dye can be estimated based on the following equation:
where dC/dt is the slope of the change in concentration in the chamber as a function of time obtained directly from measurements, Co is the initial concentration of dye on the inlet side of the diffusion chamber, and A is the area through which the dye permeates. 20
The actual permeability (P) of the cell monolayer to dye is derived after correcting for the resistance to dye permeation from the membrane
where Pm is the permeability across the membrane without cells.
To measure hydraulic conductivity, the arachnoid construct (1 cm diameter) at day 10 in culture was placed in a horizontal Ussing chamber. A pressure gradient (3.67 mmHg) was applied on the upstream chamber and water flow through the chamber was measured (final volume–initial volume). These measurements were used to estimate the hydraulic conductivity, Lp, of the construct:
where Q is the flow of water, ΔP is the pressure drop across the membrane (3.67 mmHg), A is the area across which the water flows. 21
Statistics
Analysis of variance was used to analyze cell proliferation (DNA content with time) and changes in cell morphology determined using image analysis. Changes in hydraulic or dye permeability were analyzed using a paired t test. Analysis of variances and t tests were performed using SPSS software.
Results
Arachnoid cell isolation
Arachnoid cells isolated from brain tissue (see Supplementary Fig. 2S for cells on brain stem) contained cytokeratin both in cultures grown on 2D culture wells (Fig. 1A) and in 3D collagen sponges (Fig. 1B), as confirmed by western blotting. Expression levels of desmoplakin and vimentin for the cultures were also confirmed through western blots (see Supplementary Fig. 3S), and demonstrated colocalization of desmoplakin with vimentin consistent with the arachnoid phenotype. 22 To differentiate arachnoid cells from other cell types found in the intracranial cavity, the cells were stained for the presence of myosin, S100, GFAP, SMA, and CD31. The cells stained negatively for these markers, indicating that the cells isolated were not endothelia, neurons, glia, myocytes, or fibroblasts.
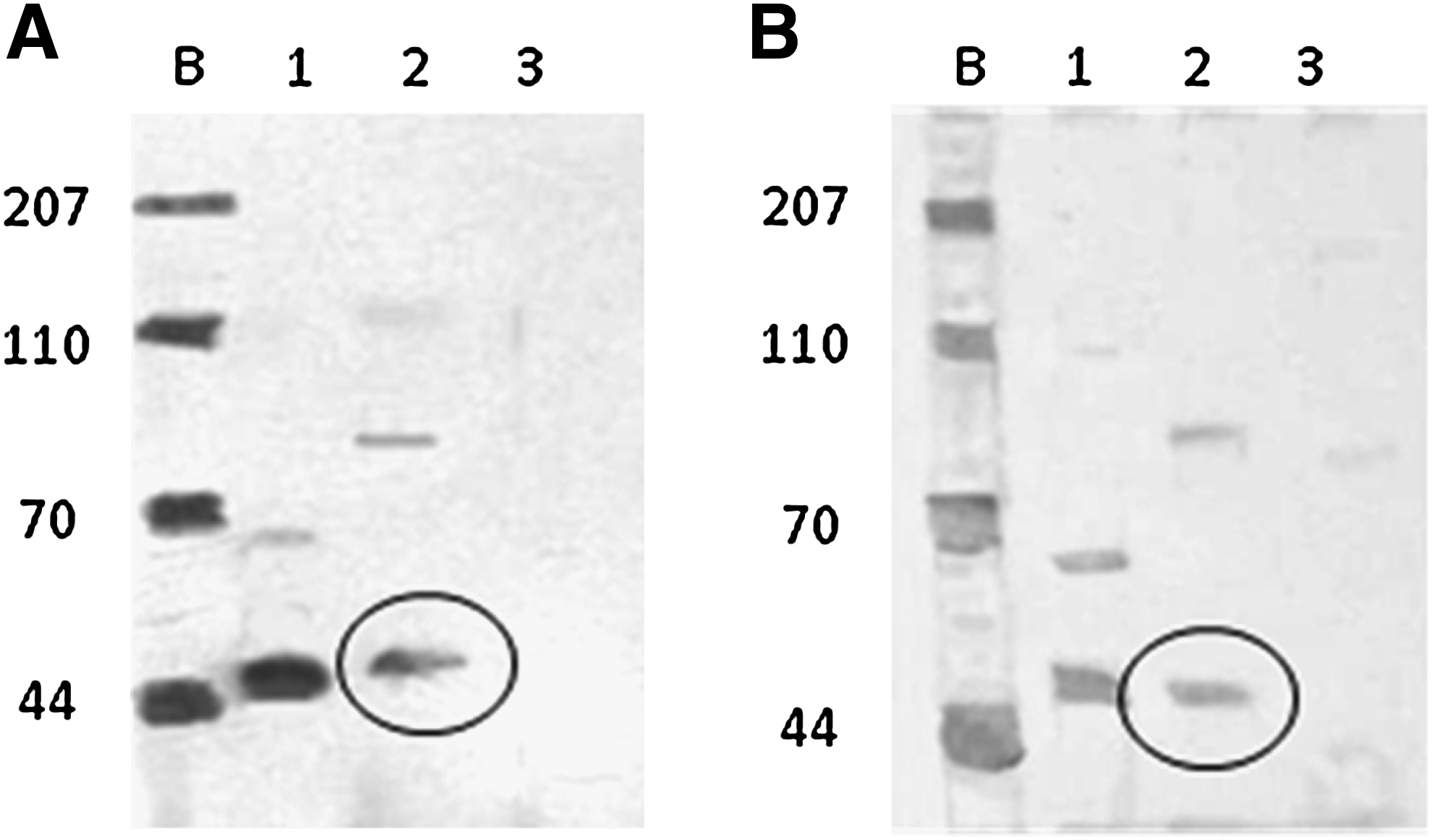
Western blot for cytokeratin isolated from rat arachnoid cells on tissue culture surface
Cell culture on tissue culture plastic
Arachnoid cells on 2D culture plates were predominantly bipolar (Fig. 2A), and stained positively for vimentin (Fig. 2B) and cytokeratin (Fig. 2C). Hematoxylin and eosin staining demonstrated the presence of large nuclei and long spindly processes in greater detail than the immunostaining (Fig. 2D). The cells proliferated with a doubling time of ∼3 days (Fig. 3A) and cell doubling times were consistent (as determined using cell counts and DNA content) (Fig 3B). The addition of cAMP to the culture medium did not alter the growth rate of the cells (p=0.14 by paired t test). Cultures were stained for the presence of gap junction proteins (connexin) at days 5, 10, and 15. Initially, areas of punctate fluorescence indicative of gap junction formation were seen over the cell layer. By ∼3 weeks, connexin staining exhibits two different staining patterns (1) punctate (which was more intense) and (2) diffuse (less intense) (see Supplementary Fig. 4S).
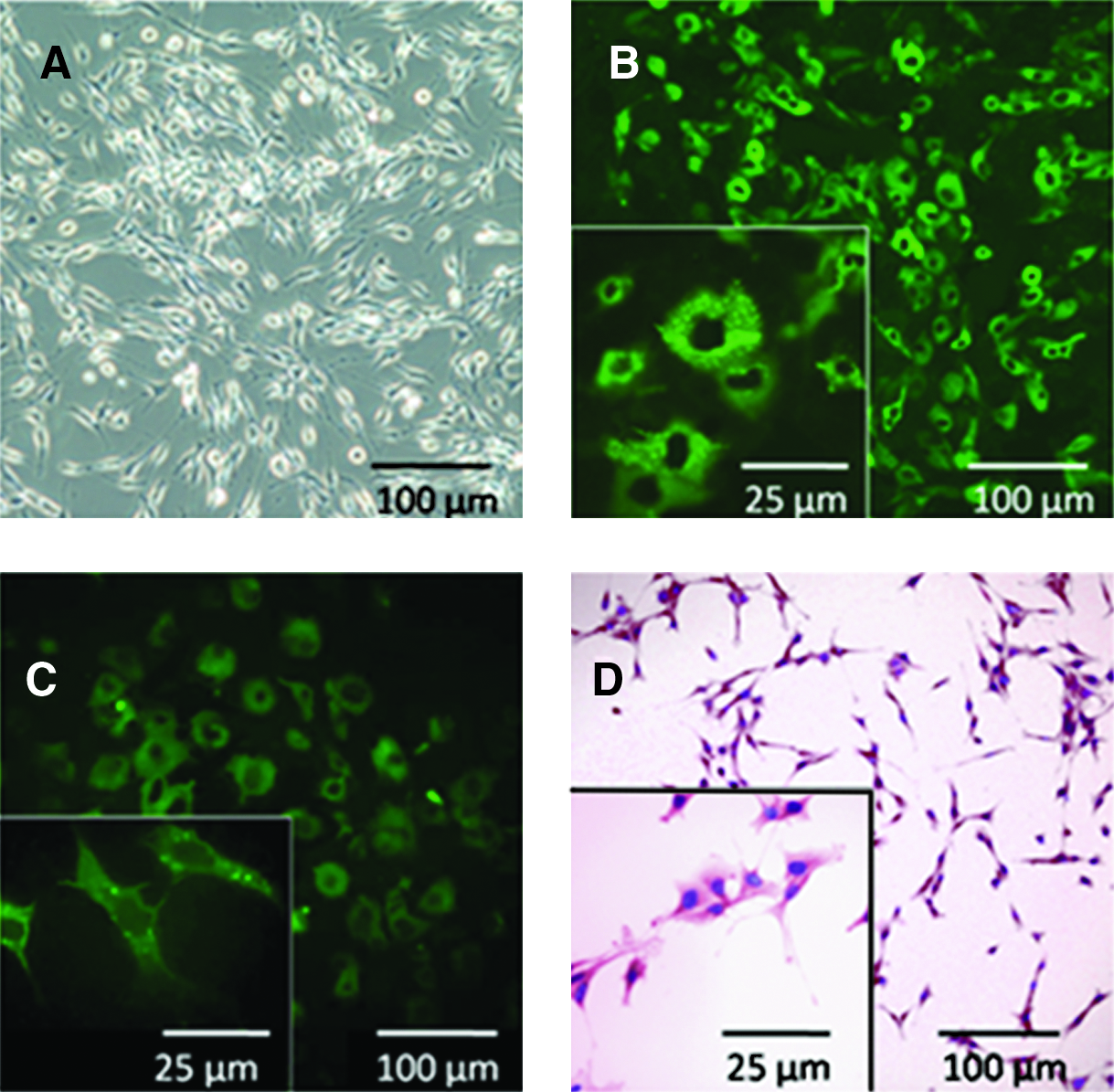
Morphology and staining patterns of rat arachnoid cells on tissue culture plastic:
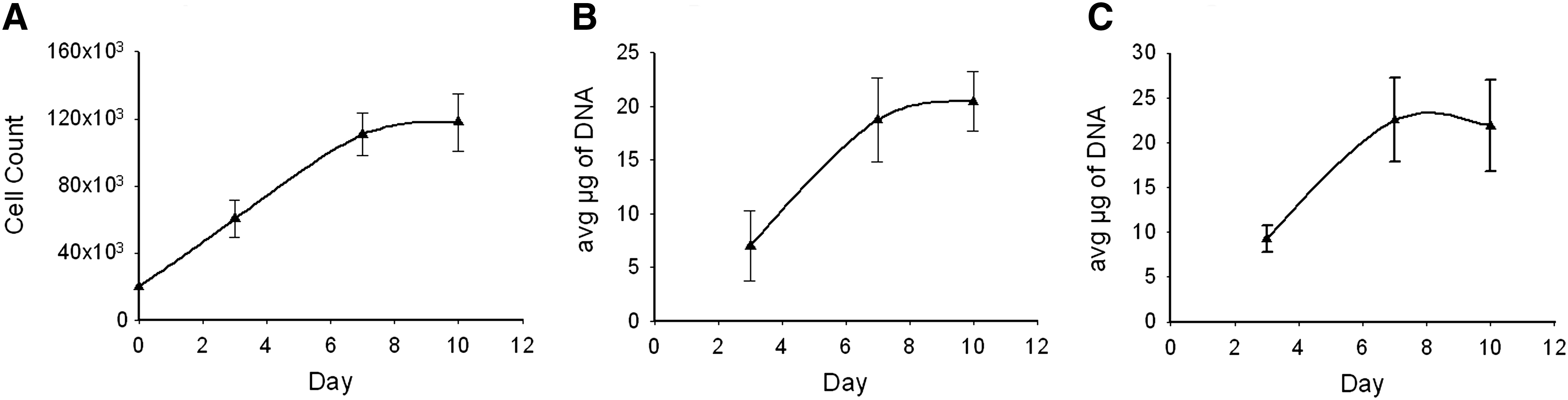
Growth curve for arachnoid cells cultured on tissue culture plastic and collagen scaffold:
Physiological behavior of arachnoid cell monolayers
In the native tissue, the arachnoid granulation forms a functional barrier to the flow of CSF. To quantify the barrier function of arachnoid cell monolayers, we measured (1) TEER 11 and (2) permeability to indigo carmine. 23 TEER values increased with time in culture and achieved the maximum value of 160 Ω cm2 after 15 days in culture and no further increases were observed. Permeability of cell monolayers to indigo carmine was also quantified. The dye was introduced into one side of the diffusion chamber and the concentration of dye in the other chamber was monitored as a function of time (Fig. 4). These data were analyzed using the equation above (Equation 2) to obtain an estimate for the permeability of the cell monolayer. The dye permeability of the arachnoid cells was 6.7×10−6±1.1×10−6 cm/s.
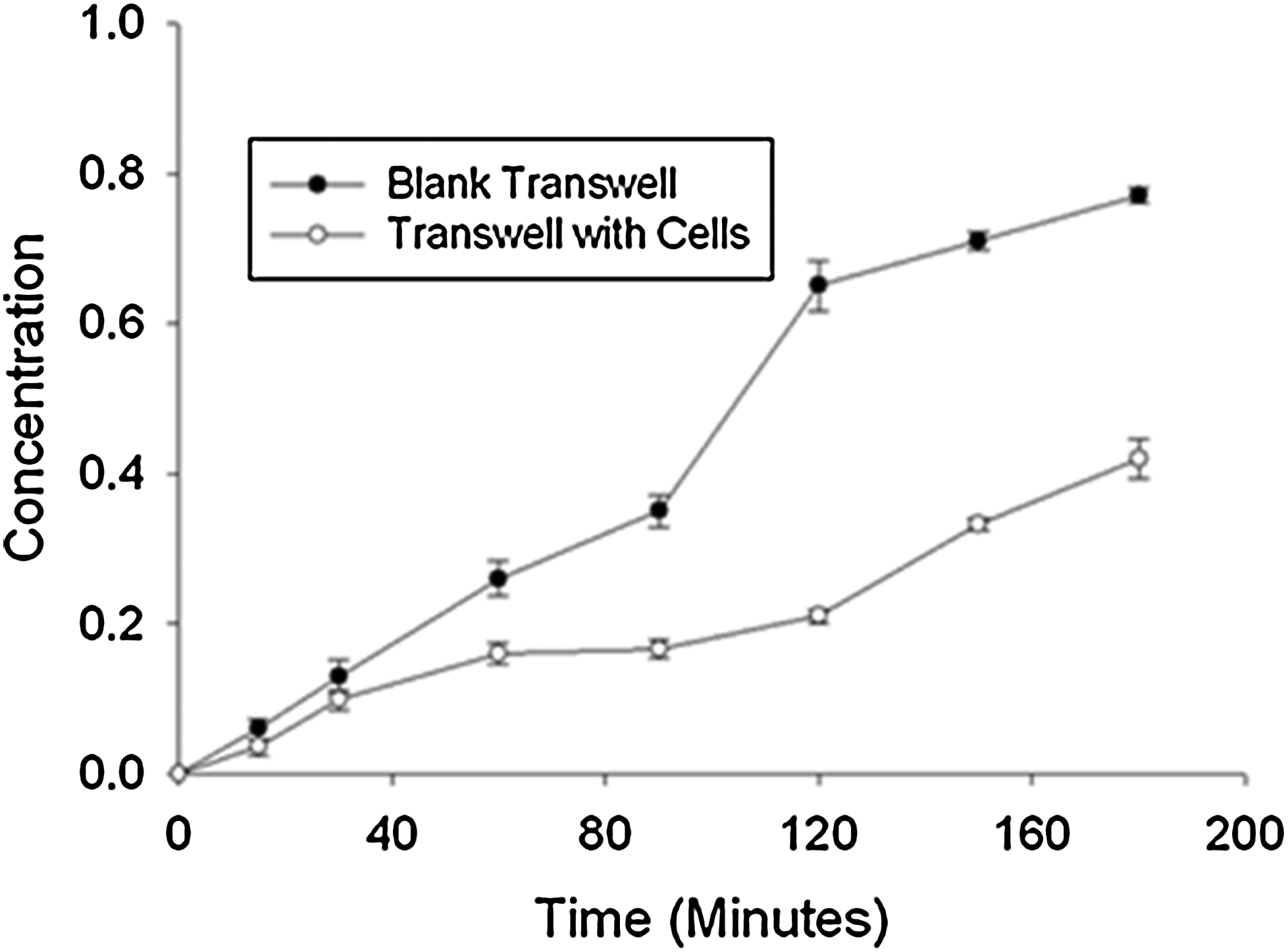
Permeation of indigo carmine through a monolayer of arachnoid cells: Transwell without cells (black circles, n=6) and a Transwell cultured with monolayer of arachnoid cells (open circles, n=6).
Arachnoid culture in a collagen scaffold
The arachnoid cells attached to the collagen scaffold readily (Fig. 5). Preliminary seeding studies were performed to determine the minimum seeding density to achieve population of the matrix. Seeding densities of 10,000, 20,000, and 100,000 cells per well or ∼5,000 to 50,000 cells/cm2 were tried. A seeding density of 50,000 cells/cm2 resulted in the most rapid repopulation of the matrix. Increasing seeding levels to higher values did not increase the rate at which the matrix was repopulated. DNA content in the matrix reached steady state within 7 days (Fig. 3C). Confocal microscopy was used to determine the spatial distribution of cells throughout the matrix (Fig. 5A, B) and the phenotype of the cells (e.g., expressing desmoplakin and vimentin as shown in Western blotting) (Fig. 5C, D). Cells populated the collagen sponge uniformly (see Supplementary 3D video) and electron microscopy revealed that cell morphology was consistent with that observed in native tissue (Fig. 5B). Even though cells penetrated well into the scaffold, cell distribution remained sparse throughout the scaffold, which is consistent with the cell distribution observed in the arachnoid core. 24
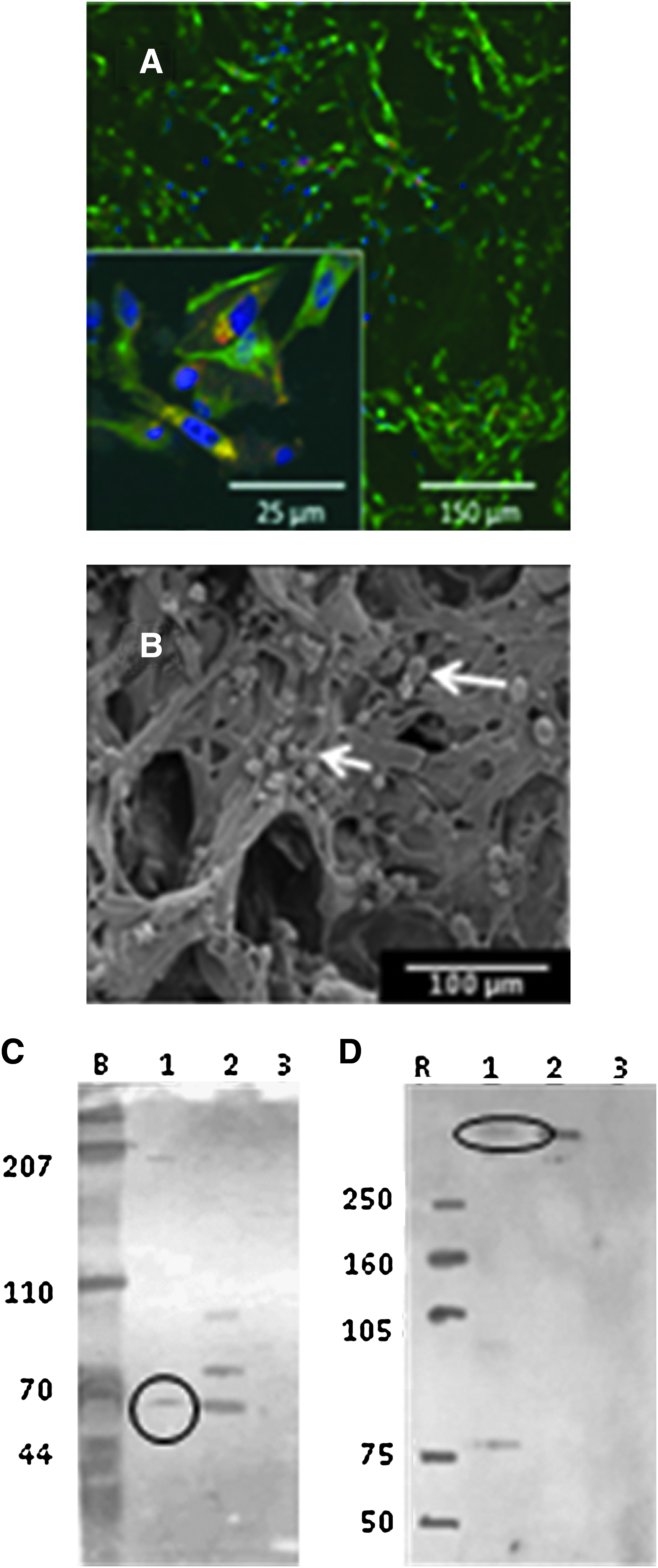
Arachnoid cells grown on porous collagen scaffolds with pore size of ∼100 μm:
Morphometric analysis of cells grown on 2D and 3D surfaces
Arachnoid cell body geometry on 2D cultures was determined quantitatively at 6, 12, and 18 days after seeding (Fig. 6). Specifically, length, width, and area of the cells were quantified. The cell area did not change with time in culture, but cells became significantly shorter over time (day 6–18) (p=0.02). The number of cell processes did not differ over that same period and the variation of length appears to occur both with cells of low and high passages (passage >8). Likewise, the process formation was not different between the 2D cultures compared to the collagen scaffold preparations (Fig. 5S compared to Fig. 5B). The morphology of the cells cultured in the construct was also determined using scanning electron microscopy. Most cells exhibited extensive bipolar process formation, which appeared to penetrate into the scaffold (Fig. 5B). Arachnoid cells of similar shape were also found on the apical (nonseeded microporous) side (Fig. 1S). The number of processes was not significantly different between cells grown on 2D culture plates compared to that of cells grown in a 3D collagen sponge (Table 1).
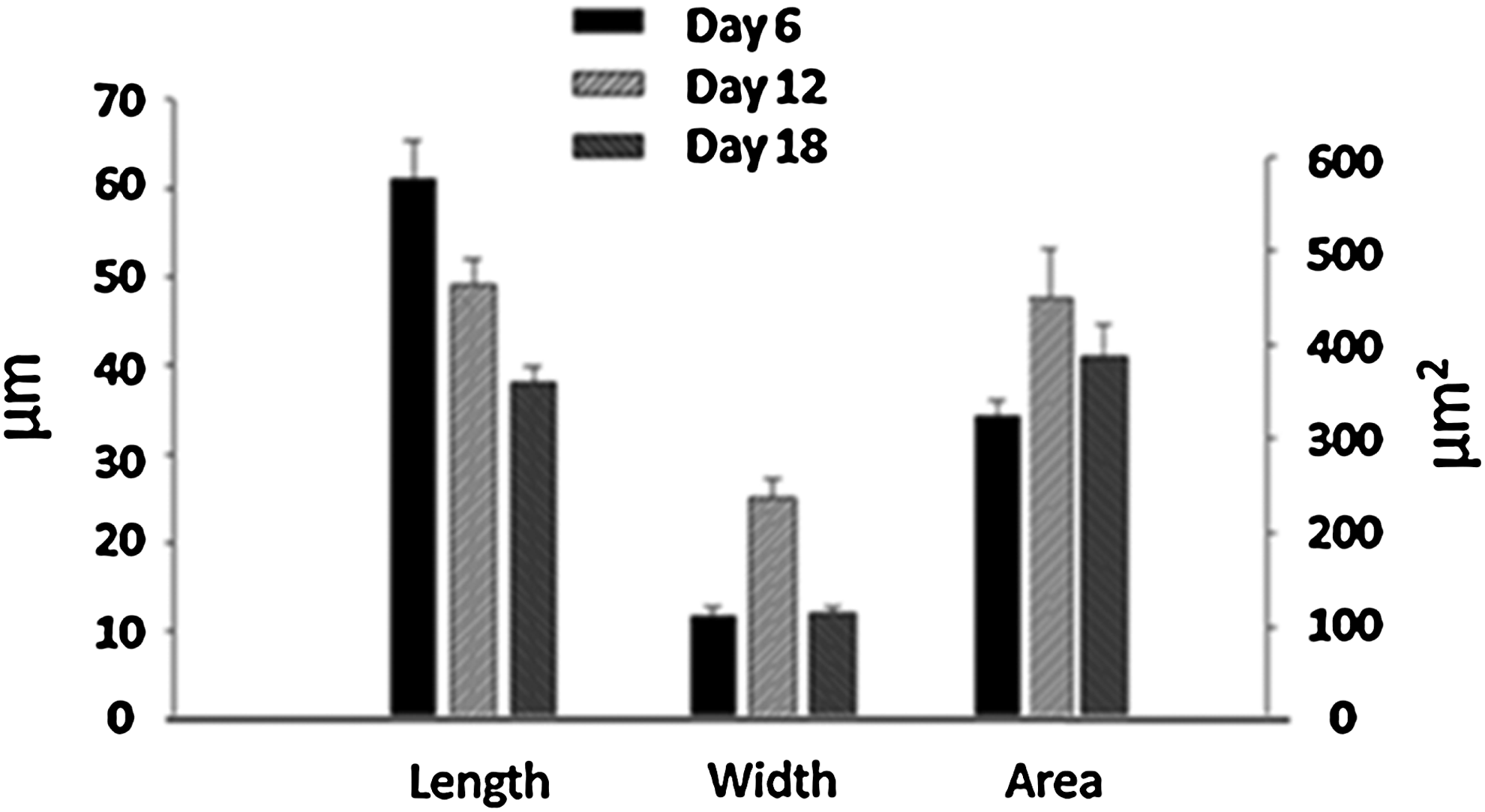
Morphometric analysis of arachnoid cells. Length, width, and area of arachnoid cells grown on tissue culture plates. Error bars indicate standard error.
Standard error of the mean is given in parentheses.
2D, two-dimensional; 3D, three-dimensional.
Physiological behavior of arachnoid construct
An arachnoid construct after 10 days in culture was placed on a Transwell insert and inserted in an Ussing chamber. The flow of water through the construct was measured for a constant pressure difference (3.67 mmHg). The hydraulic permeability for the construct was 6.39±1.53 mL/min/mmHg/cm2. The hydraulic permeability of the collagen matrix alone was 7.07±1.56 mL/min/mmHg/cm2. The difference between the two permeabilities was not statistically significant (two-tailed independent t test, p=0.256).
Discussion
Cell culture on tissue culture plastic
Cells isolated from the tissue exhibited arachnoid cell phenotype: cytokeratin positivity, coexpression of desmoplakin and vimentin, and formation of gap junctions when cultures become confluent. We have also established that the arachnoid cultures that were isolated were not contaminated with other cells present in the native tissue (fibroblast, muscle, endothelial, glial, and neuronal cells). Expression of vimentin and desmoplakin found in the cultures that were isolated in this investigation exhibited similar phenotype to arachnoid cells isolated from human granulation, 25 spinal, 26 fetal 11 tissue, and other locations in the brain. 27
A unique aspect of native arachnoid cells is their ability to form the CSF–blood barrier. Previous investigations have demonstrated that human granulations exhibit tight junction formation, which has also been associated with barrier formation. 12 Zonula Occludens-1 (a component of tight junctions) and E-cadherin (a calcium dependent cell adhesion molecule) have been found in previous studies of arachnoid cell culture. 28 Also, desmosomes, which are made up of multiple proteins, including desmoplakin, have been observed in arachnoid cultures using transmission electron microscopy. 13 The role of calcium is uncertain. In vivo manifestations of calcium in granulations typically are in isolated concretions that are not within the pathways of water movement. In this study, we have shown that gap junctions (as determined by connexin staining) develop in culture and become more numerous with time in culture.
The barrier capability of cell monolayers cultured in vitro has been studied using a variety of methods, including TEER. TEER values for monolayer cultures of Madin–Darby Canine Kidney Cells (MDCK II) and human colonic adenocarcinoma cells (caco-2) range between 100 and 500 Ω cm2, respectively. 29 In this investigation, TEER values of 160 Ω cm2 are well within the range of values measured for other models of blood–brain barrier. It is noteworthy that the ECV304 30 and HCMEC/D3 cell lines have been used as an in vitro model of the blood–brain barrier, 31 and TEER values for these cells have been measured at ∼110 Ω cm2. With TEER of 160 Ω cm2, we demonstrate the first CSF–blood barrier formation capability in an in vitro model.
Transport function of the arachnoid monolayer also has been quantified via the permeation of dyes. By way of comparison, the permeability of Lucifer Yellow for caco-2 cells was 2.5–5.0×10−6 cm/s and MDR-MDCK cells was 6.0–8.0×10−6 cm/s. 20 A recent study demonstrated permeability of Lucifer Yellow for human arachnoid cells in cell culture inserts to be 7–14×10−6 cm/s. 9 The permeability of indigo carmine in arachnoidal cells measured in this investigation was well within the range of these previously measured permeabilities (6.7×10−6 cm/s).
Arachnoid culture in a collagen scaffold
Arachnoid cells that were seeded into the matrix rapidly repopulated the matrix (∼7 days). Cells grown on a collagen sponge (diameter of 16 mm) contained slightly more DNA content on average than cells grown on tissue culture plastic (36 mm in diameter) after the same time period (7 days). This suggests that cell densities from collagen sponges are growing at a rate similar to or slightly higher than cells grown on tissue culture plastic. Overall, the density of cells on the surface of collagen sponges (viewed by scanning electron microscopy) appear to be consistent with the sparse density of arachnoid cells in the native tissue. 32 As with arachnoid cells cultured on tissue culture plastic, arachnoid cells cultured in the collagen matrix exhibited the expected phenotype (expression of desmoplakin and vimentin). Cells cultured in the construct were principally bipolar. This cell shape is consistent with that observed in certain native tissues, 33 and there was little difference in morphometry between cells cultured on tissue culture plastic and in the collagen matrix. The hydraulic permeability measured in this investigation (∼6×10−6 cm/s) was slightly higher than that measured in another study with human arachnoid cells (∼3×10−6 cm/s). 9 It is currently unclear as to the reason for the decrease in hydraulic permeability measured in this investigation although they are within the same range. Because the PET is a nonwoven fibrous mat, a direct comparison is difficult. 8 PET is a synthetic matrix that replicates neither the biological signaling of collagen nor the spongy architecture of the native arachnoid granulation. In contrast to previous studies, we were able to recapitulate the two distinct layers in the arachnoid tissue, the core (collagen sponge studies) and the cap (monolayer studies), and demonstrated function of the cells in these two contexts. The use of a biocompatible and degradable material such as collagen is also important for any potential future uses (or implantation). Minute contaminants on nonbiologic substances in the brain are extremely difficult to treat; therefore, infections of artificial ventriculoperitoneal shunts almost always require removal. This complication is one of the most common complications of shunting. 3 Therefore, the utilization of collagen would be an important step in the creation of a bioartificial construct as a means for diverting CSF.
The density of cells in the construct is much less than that in cell monolayers but mirrors more closely the density of arachnoid cells in the core region of the native granulation. Additional studies are currently being pursued to stratify cell density in the construct to mirror that in the native tissue.
In summary, the results of this study demonstrate that arachnoid cells can be isolated from native rat brain tissue and successfully cultured ex vivo in monolayers and collagen scaffolds. The cells express arachnoid cell phenotype and demonstrate aspects of normal arachnoid granulation function (barrier to the flow of water and larger molecules). This in vitro model of the arachnoid granulation has the potential to help us both understand the causes/development of hydrocephalus and develop tissue engineered alternatives to conventional methods of treatment.
Footnotes
Acknowledgment
This work is supported by a grant from the What-If program of the Biomedical Engineering Institute, Minneapolis, Regents Scholar program of the University of Minnesota, and the Augustine Endowment to the Biomedical Engineering Graduate Studies Program for Medical Professionals.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.