
Abstract
Although abdominal aortic aneurysms (AAA) can be potentially stabilized by inhibiting inflammatory cell recruitment and their release of proteolytic enzymes, active AAA regression is not possible without regeneration of new elastic matrix structures. Unfortunately, postneonatal vascular smooth muscle cells (SMCs), healthy, and likely more so, diseased cells, poorly synthesize or remodel elastic fibers, impeding any effort directed at regenerative AAA treatment. Previously, we determined the eleastogenic benefits of oligomers (HA-o; 4–6 mers) of the glycosaminoglycan, hyaluronan (HA) and transforming growth factor-β1 (TGF-β1) to healthy SMCs. Since AAAs are often diagnosed only late in development when matrix disruption is severe, we now determine if elastogenic upregulation of SMCs from late-stage AAAs (>100% diameter increase) is possible. AAAs were induced by perfusion of rat infrarenal aortae with porcine pancreatic elastase. Elastic matrix degradation, vessel expansion (∼120%), inflammatory cell infiltration, and enhanced activity of matrix-metalloproteases (MMPs) 2 and 9 resulted, paralleling human AAAs. Aneurysmal SMCs (EaRASMCs) maintained a diseased phenotype in 2D cell culture and exhibited patterns of gene expression different from healthy rat aortic SMCs (RASMCs). Relative to passage-matched healthy RASMCs, unstimulated EaRASMCs produced far less tropoelastin and matrix elastin. Exogenous TGF-β and HA-o (termed “factors”) significantly decreased EaRASMC proliferation and enhanced tropoelastin synthesis, though only at the highest provided dose combination (20 mg/mL of HA-o, 10 ng/mL of TGF-β); despite such enhancement, tropoelastin amounts were only ∼40% of amounts synthesized by healthy RASMC cultures. Differently, elastic matrix synthesis was enhanced beyond amounts synthesized by healthy RASMCs (112%), even at lower doses of factors (2 mg/mL of HA-o and 5 ng/mL of TGF-β). The factors also enhanced elastic fiber deposition over untreated EaRASMC cultures and restored several genes whose expression was altered in EaRASMC cultures back to levels expressed by healthy RASMCs. However, the activity of MMPs 2 and 9 generated by EaRASMC cultures was unaffected by the factors/factor dose. The study confirms that SMCs from advanced AAAs can be elastogenically induced, although much higher doses of elastogenic factors are required for induction relative to healthy SMCs. Also, the factors do not appear to inhibit MMP activity, vital to preserve existing elastic matrix structures that serve as nucleation sites for new elastic fiber deposition. Thus, to enhance net accumulation of newly regenerated elastic matrix, toward possibly regressing AAAs, codelivery of MMP inhibitors may be necessitated.
Introduction
Due to surgical and graft-associated complications associated with surgical AAA treatment, 6 including aortic neck expansions, nonsurgical AAA treatment modalities are being explored. These include approaches aimed at pharmacologically inhibiting inflammatory cell recruitment and MMP release/activity within AAAs by delivering tissue inhibitors of matrix metalloproteases (TIMPs) 7 or modified tetracyclines 8 (e.g., doxycycline), and chemically crosslinking pre-existing elastic matrix structures.9,10 However, these strategies can at best serve to arrest growth of the AAA but not actively regress the condition. In fact, doxycycline has shown promise in inhibiting inflammatory cell infiltration into the AAA wall and attenuating production and activity of MMPs slowing growth of human AAAs. However, in parallel, it also appears to impede accumulation of new elastic matrix by already highly elastogenically deficient adult vascular SMCs, reducing prospects for regenerative elastic matrix repair leading to AAA regression. 11 In such cases, an additional impetus in terms of elastogenic factors can potentially enhance net accumulation of new elastic matrix.
In prior studies we identified a unique cocktail of elastogenic factors, namely, hyaluronan (HA) oligomers (HA-o; 4–6 mers; 756 Da)12,13 and growth factors (TGF-β1),14,15 that synergistically attenuated healthy rat aortic SMC (RASMC) proliferation, significantly enhance elastin precursor (tropoelastin) synthesis, tropoelastin recruitment and elastic matrix assembly, fiber maturation, and stability. We subsequently investigated the dose-specific effects of the same factors (0–20 μg/mL HA-o and 0–10 ng/mL TGF-β) on elastogenesis by SMCs derived from CaCl2 induced rat aortal expansions (aRASMCs), 16 which simulated very early stage inflammatory human AAAs; we showed the aRASMCs isolated from these expansions to exhibit an activated phenotype in culture and to be amenable to elastogenic stimulation. However, AAAs are frequently diagnosed at advanced stages of development when matrix deterioration is severe. From the standpoint of a clinically applicable paradigm for AAA regression, elastogenic upregulation of SMCs derived from late-stage AAAs (>100% diameter expansion) must be demonstrated. We investigate this aspect in the present study.
Materials and Methods
Induction of aortal injury via elastase perfusion
All animal studies were approved by the IACUC at the Medical University of SC and at Clemson University, where this work was performed before recent relocation of the authors. Animal use conformed to AVM stipulations for humane care of animals. Adult male Sprague-Dawley rats (250–300 g in weight) were acclimatized for 1 week before surgery. The rats were placed under general anesthesia (2%–4% v/v isoflurane) and the infrarenal abdominal aortae surgically exposed and injured via elastase perfusion, using a published method modified for our purposes (Fig. 1).17,18 Briefly, the posterior lumbar aortic branches and the inferior mesenteric arteries were ligated with 9–0 suture (Surgical Specialties Corp., Reading, PA) if they were within the area of interest. The connective tissue surrounding the aortae was circumferentially dissected at the proximal and distal ends to allow placement of vascular clamps (Fine Science Tools, Foster City, CA) and distal silk ties. The aortic diameters were measured using an eyepiece micrometer fitted on a stereoscope (Olympus SZ61; Olympus, Center Valley, PA). Proximal and distal microclips were placed at the renal junctions and iliac junctions, and small aortotomies made using the tip of a 271/2-guage needle. A PE-10 catheter (Scientific Commodities, Inc., Lake Havasu City, AZ) was inserted into the aortal lumen and the distal silk ties tightened to hold the intraluminal catheter in place. The aortae were slowly perfused with porcine pancreatic elastase (20 U/mL; 25 min). The enzyme concentration was chosen based on that adopted by others for inducing AAAs in mice, 19 and our own preliminary optimization studies wherein we found the above conditions of perfusion to represent the minimal enzyme concentration and time necessary to generate a >100% diameter increase in rat aortae. The aortal sections were filled with the elastase solution until the aortae segment between the clamps ballooned to 200% of the original diameter as measured by the eyepiece micrometer. After perfusion, the distal ties were released, the catheter tubing removed, the aortotomies closed with 9–0 sutures, and the proximal and distal clamps removed. After confirmation of reperfusion to the lower limbs, and collection of postperfusion measurements, the intestines were replaced and the incision sites closed in two layers with 4–0 Vicryl suture and 4–0 Ethilon suture (Ethicon, New Brunswick, NJ). The rats were then placed on a heat pad until they became alert, indicating recovery from anesthesia, at which time, 80 μL of Buprenex® analgesic (0.03 mg/mL; Buprenorphine) was injected subcutaneously. The animals were monitored for a few days with administration of Buprenex every 12 h if necessary. Aneurysmal aortae were harvested 14 days postoperative.
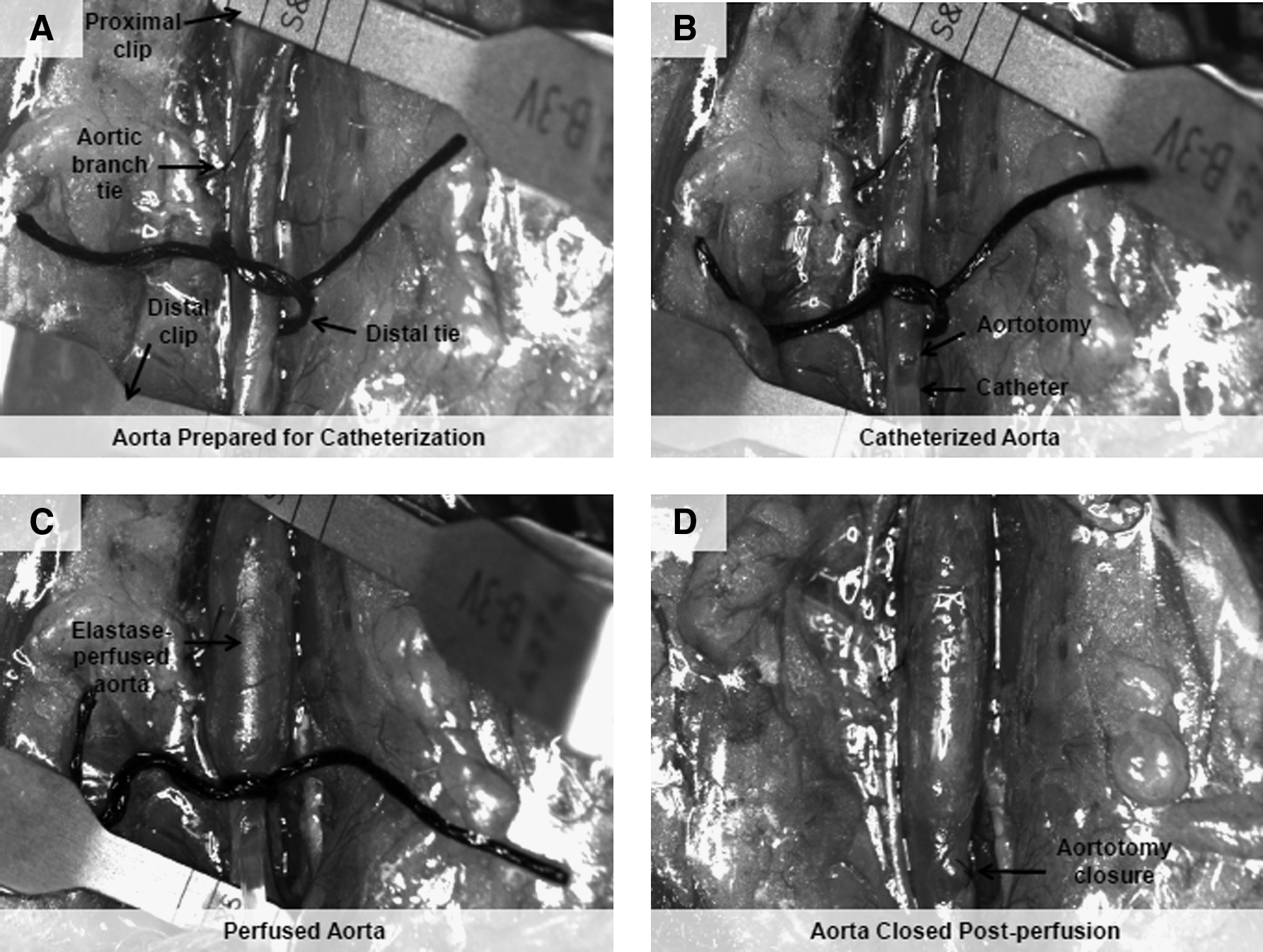
Procedure for abdominal aortic aneurysms induction via elastase perfusion (20 U/mL; 25 min).
Histological and biochemical characterization of aortal expansions
Diameter expansion measurements were conducted on aortae pre- and postperfusion and 14 days postoperative (n=12) from photographs taken at the respective time points and analyzed via ImageJ software (NIH, Bethesda, MD). Histological and biochemical analysis of these expansions was performed. Standard hemotoxylin and eosin staining (H&E; for nuclear and intracellular and extracellular protein observation), modified Verhoff Van Gieson staining (VVG; for elastin observation; ScyTek Laboratories, Logan, UT), and scanning electron microscopy (SEM; for ultrastructural observation of aortic tissue cross-sections) were conducted on untreated control aortae, sham-treated (saline-perfused) aortae, and elastase-perfused aortae (n=3 animals/group). In addition, immunofluorescence labeling was performed to detect presence of inflammatory macrophages via their expression of the CD68 marker (Mouse×rat CD68 Ab from Abcam, Inc., Cambridge, MA). Aortae from the respective animals were harvested 14 days postoperative, rinsed in sterile saline, embedded in optimum cutting temperature (OCT) medium, and stored at −20°C until cryosectioning. Sections (5 μm) were processed for histology or SEM. Standard histological protocols were followed. For SEM, samples were fixed in 2% v/v gluteraldehyde in phosphate buffer, rinsed in phosphate buffer, DI water, and then dehydrated in graded ethanol and finally in hexamethyldisilazane (Sigma Aldrich, St. Louis, MO) (3 min). The histological and SEM samples were imaged on an Olympus BX41 (Olympus, Center Valley, PA) and a Hitachi SEM TM1000 (Hitachi Technologies, Pleasanton, CA), respectively.
MMP activity within elastase-perfused aortae was assessed by gelatin zymography methods 11 and compared to healthy rat aortae (n=3/group). Briefly, aortic tissue was homogenized in RIPA buffer and then assayed for protein content using the BCA assay (Thermo Scientific, Waltham, MA). All lanes were loaded in multiplicate with 25μg of protein in each lane. A prestained molecular weight standard (Invitrogen, Grand Island, NY) was loaded alongside. After development, staining, and de-staining, densities of the bands corresponding to MMP-2 (Zymogen: 72 kDa, Active: 66 kDa) and -9 (Zymogen: 92 kDa, Active: 82 kDa) that appeared on a dark background of stained gelatin, were quantified using ImageJ software, and reported as relative density units.
Isolation and culture of EaRASMC
Expanded abdominal aorta segments were harvested from elastase-perfused adult male Sprague Dawely rats (n=3) at 14 days postoperative, and opened lengthwise, and the intima scraped gently with a scalpel blade. The medial layer was dissected from the underlying adventia, chopped into ∼0.5-mm-long sections, and washed twice with warm phosphate-buffered saline (PBS). The tissue slices were then pooled, enzymatically degraded in DMEM-F12 (Invitrogen) containing 125 U/mg collagenase (Worthington, Biochemicals Lakewood, NJ), and 3 U/mg elastase (Worthington Biochemicals) for 30 min at 37°C, centrifuged (400 g; 5 min), and cultured in T-75 flasks with DMEM-F12 containing 10% v/v fetal bovine serum (PAA Laboratories, Etobicoke, Ontario) over 2 weeks. Primary aneurysmal rat aortic SMCs (EaRASMCs) derived by outgrowth from these tissue explants were cultured over 2 weeks, and the cells passaged when confluence was attained. For experiments, passage 3 SMCs were seeded onto 6-well tissue culture plates (area=10 cm2) at a seeding density of 2×104 cells/well and cultured in DMEM-F12 medium containing 10% v/v fetal bovine serum (FBS; PAA Laboratories, Dartmouth, MA), and 1% v/v Penstrep (South Logan, UT). The total volume of medium added per well was 5 mL.
Experimental design and time points
Passage 3 EaRASMCs were cultured in medium containing supplemented factors (0.2, 2, and 20 μg/mL HA-o and 1, 5, and 10 ng/mL TGF-β; n=6/condition) within 6-well plates (2×104 cells/9.6 cm2) for 21 days, over which time the cells deposited extracellular matrix. Control EaRASMC cultures received no factors. Matrix production by the EaRASMCs was compared with that of identically seeded, healthy, passage 3 RASMCs, which we investigated in a recent study. 20 Spent medium aliquots were removed from each well, at the time of each, twice-weekly medium change, pooled with previously removed aliquots, frozen at −20°C, then biochemically assayed together with their corresponding cell layers when harvested at 21 days of culture.
DNA assay for cell proliferation
The DNA contents within the respective groups of cell cultures were compared to determine the impact of the factors and factor doses on SMC proliferation. For analysis, the cell layers were harvested at 21 days of culture, resuspended in NaCl-Pi buffer, and sonicated on ice, and their respective DNA contents quantified using a fluorometric assay described by Labarca and Paigen. 21 Cell counts were then calculated assuming 6 pg of DNA/cell. 21
Immunofluorescence detection of SMC phenotypic markers
Immunofluorescence techniques were used to observe SMC expression of phenotypic markers of contractile and synthetic activity. RASMCs and EaRASMCs (n=3/marker/cell type) were cultured in sterile, 2-well Permanox® chamber slides (Nalge Nunc International, Rochester, NY) under identical experimental conditions as described for cultures intended for biochemical analysis, though the cell seeding density and treatment doses were adjusted, to account for the reduced substrate surface area and cell number, respectively. The cell layers were fixed with acetone for 10 min at −20°C and blocked with 5% v/v goat serum (30 min). α smooth muscle actin, SM22α, calponin, and caldesmon (contractile phenotype markers), and thrombospondin and osteopontin (synthetic phenotype markers) were then detected with polyclonal antibodies against rat antigens (1:100 v/v; Abcam, Inc.) and observed with FITC-conjugated IgG secondary antibodies (1:1000 v/v; Chemicon, Temecula, CA). The cell layers were cover-slipped with Vectashield mounting medium containing the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA), which labeled the cell nuclei.
Flow cytometry
In preparation for flow cytometry analysis, SMC cultures (n=6 per culture condition; n=1 per marker) were trypsinized, centrifuged (1500 RPM; 10 min), resuspended at 1×106 cells per sample, fixed with 4% w/v paraformaldehyde in PBS (37°C; 10 min), washed, and then permeabilized with 0.1% TritonX-100 in PBS for 1 min. Immunoflourescence detection of SMC phenotypic markers was conducted with the antibodies outlined above. The samples were incubated with primary antibodies (30 min; 4°C), followed by an FITC-conjugated secondary antibody (20 min; 4°C; dark). After labeling, cells were resuspended in 0.5 mL of PBS and kept at 4°C protected from light until analysis. Cytometric analysis was done using a Becton Dickinson FACSCalibur Analytical Flow Cytometer and data processed using Cell Quest Pro 5.2 (BD Biosciences, Franklin Lakes, NJ). Data were collected to corroborate the results we observed with immunofluorescence staining of cell markers. Statistical analysis of flow cytometry data was not performed since the results pertain to only n=1 cultures/marker.
Fastin assay for elastin
A Fastin assay (Accurate Scientific and Chemical Corporation, Westbury, NY) was used to quantify the total amount of elastin deposited within cell layers (matrix elastin), and that released into the culture medium as a soluble precursor (tropoelastin). For each treatment group, tropoelastin in the spent medium was collected and pooled over the culture period and frozen at −20°C. To isolate matrix elastin after 21 days of culture, the cell layers were trypsinized, resuspended in NaCl/Pi buffer, and centrifuged (2500 RPM, 10 min). The cell pellet was digested with 0.1 N NaOH (98°C, 1 h), and centrifuged to yield a less crosslinked, alkali-soluble supernatant fraction (soluble elastin), and a mature, highly crosslinked, insoluble pellet (insoluble elastin). Since the Fastin assay quantifies only soluble α-elastin, the insoluble elastin was first converted to a soluble form before quantification. To do this, the insoluble elastin pellet was dried, solubilized with 0.25 M oxalic acid (95°C, 1 h), and the pooled digests then centrifuge-filtered (3000 RPM, 10 min) in microcentrifuge tubes fitted with low-molecular-weight (10 kDa) cut-off membranes (Millipore, Bedford, MA). All three elastin fractions (tropoelastin, and soluble and insoluble matrix elastin) were quantified using the Fastin assay.
Von Kossa staining
Van Kossa staining was used to observe calcific deposition by EaRASMCs. SMCs (n=2/condition) were cultured in sterile, 2-well Permanox® chamber slides (Nalge Nunc International, Rochester, NY) under identical experimental conditions as described for cultures meant for biochemical analysis, though the cell seeding density and treatment doses were adjusted to account for reduced the substrate surface area and cell number, respectively. After 21 days of culture, the SMCs were incubated with 1% w/v silver nitrate solution and placed under UV light (λ=254 nm; 20 min). After several changes of distilled water, the unreacted silver was removed by rinsing cells with 5% w/v sodium thiosulfate for 5 min, and then PBS. The slides were counterstained with hematoxylin. The appearance of black-stained masses in the cell layers confirmed the presence of calcium phosphate deposits.
Gel zymography
Activity of MMP-2 and -9 were detected in the culture medium by gelatin zymography methods described in section Histological and biochemical characterization of aortal expansions. Briefly, aliquots of culture medium were assayed for protein content using the BCA assay. All lanes were loaded in multiplicate with equal amounts of protein in each lane. A prestained molecular weight standard (BioRad, Hercules, CA) and MMP-2 and 9 standards (Anaspec, San Jose, CA) were loaded alongside. After development, staining, and destaining, densities of MMP-2 and -9 bands on a dark background of stained gelatin were quantified using ImageJ software and reported as relative density units.
Transmission electron microscopy of elastic matrix
Transmission electron microscopy was used to characterize the ultrastructure of the elastic matrix. At 21 days postseeding, all control and test groups outlined earlier were fixed with 2% w/v cacodylate glutaraldehyde (12 h), postfixed in 1% w/v osmium tetroxide (1 h), dehydrated in a graded ethanol series (50%–100% v/v), embedded in Epon 812 resin, sectioned, placed on copper grids, stained with uranyl acetate and lead citrate, and observed on a Hitachi TEM H7600T (High Technologies, Pleasanton, CA).
Statistical analysis
Experimental data (n=6/condition unless otherwise mentioned) was analyzed using Student's t-test. Asterisks in figures denote statistical significance (p<0.05) for each group compared with treatment controls (EaRASMCs cultured with no factors).
DNA microarray analysis
Gene expression was measured in healthy, untreated RASMCs, elastase perfusion-derived EaRASMCs, and EaRASMCs cultured with elastogenic factor supplements (2 μg/mL HA-o and 5 ng/mL TGF-β), using Affymetrix DNA microarray analysis. Passage 3 SMCs (derived from three different animals/culture condition and passaged separately) were cultured for 7 days under the conditions specified above. Total RNA was prepared from the cell layer by RNeasy kit (Qiagen, Valencia, CA). Quality and purity of the RNA preparations were assessed by spectrophotometric determination of the ratio of absorbance at 260/280 nm and by quantification of the ratios of 28S:18S ribosomal RNA using a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA).
Synthesis of double-stranded cDNA, in vitro transcription of biotin-labeled cRNA targets, and fragmentation of target cRNA were performed as outlined by Affymetrix protocols (Affymetrix, Santa Clara, CA) and as described previously. 22 Fragmented cRNA samples were hybridized overnight at 45°C to Affymetrix Rat Expression 230 GeneChips. Posthybridization washing and phycoerythrin-streptavidin staining and fluorescence scanning were performed using Affymetrix instrumentation in accordance with manufacturer's protocols.
Gene hybridization intensities were normalized using Robust Multichip Average and detection scores obtained by MAS5 algorithm, both implemented with Expression Console software (Affymetrix). Probe sets scoring “Present” in fewer than three of the samples were excluded from further analysis. Differentially expressed genes were then identified by SAM 23 multiclass analysis with delta adjusted to limit false discovery rate to <5%. Expression data were imported into dChip (2005) for graphical and clustering analysis. 24 Genes presented in Figure 8 were selected after visual inspection of hierarchical clustering data for the differentially expressed genes.
Results
Aneurysm progression and SMC phenotype
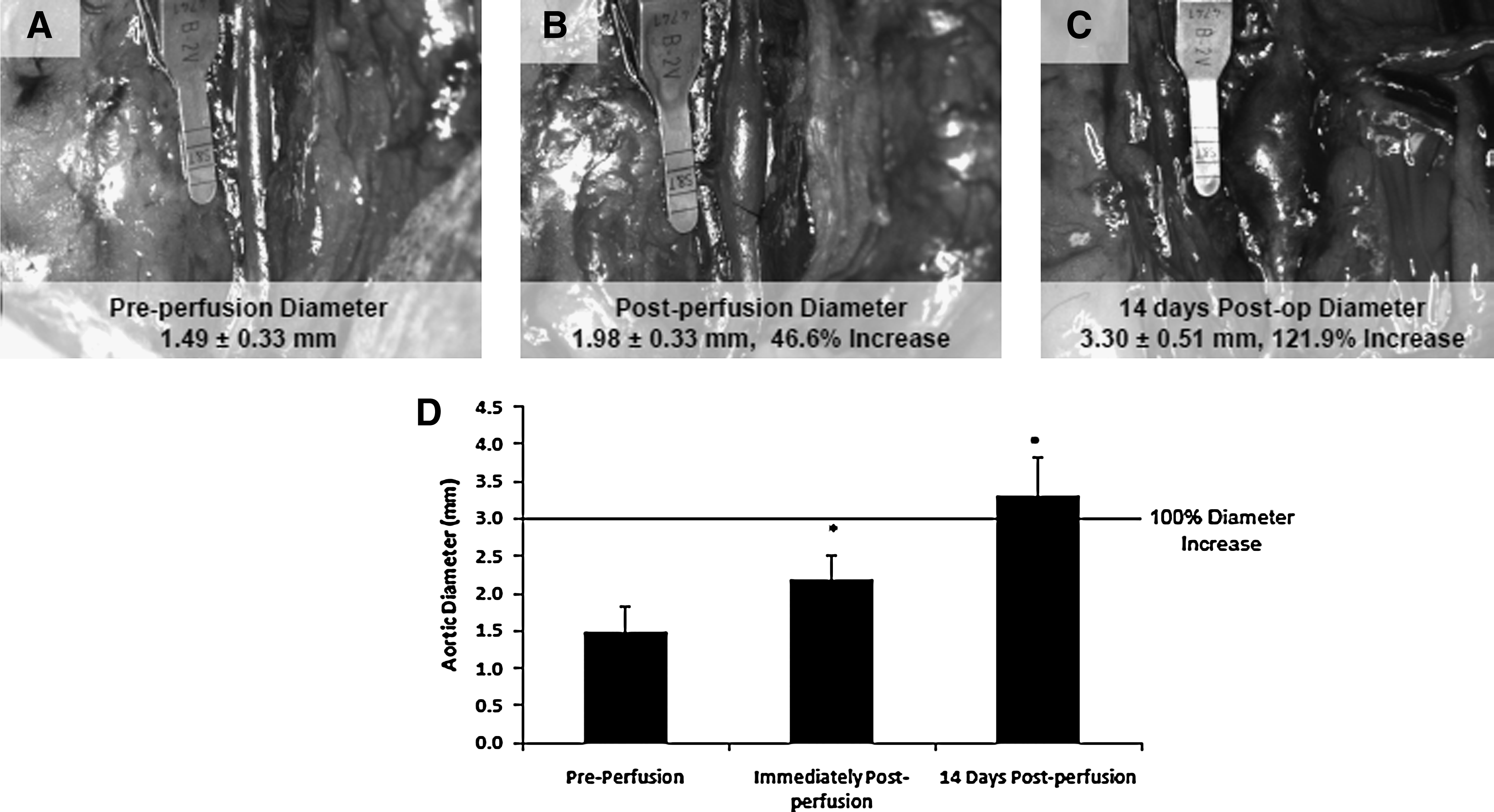
A local 122% increase in rat aortic diameter, typical of a late-stage aneurysmal expansion, was induced over a 14-day period after elastase perfusion (Fig. 2A–D). SEM and histological assessment (H&E and VVG) of the elastase-perfused aortae revealed a flattened lumen and disruption in the elastic medial layer of the vessel wall compared to the no treatment control and the treatment sham (saline perfusion) (Fig. 3A). Immunofluorescence labeling indicated the involvement of macrophages (green; Fig. 3B) in the etiology of matrix breakdown within the expansion. Assessment of sham-treated aortae revealed mild disruption and disorganization of the elastic structures relative to healthy, control aortae (Fig. 3A), but which were very minimal compared to elastase-perfused aortae, as also previously seen.25–27 SEM showed no significant calcific deposition within elastase-perfused aortae.

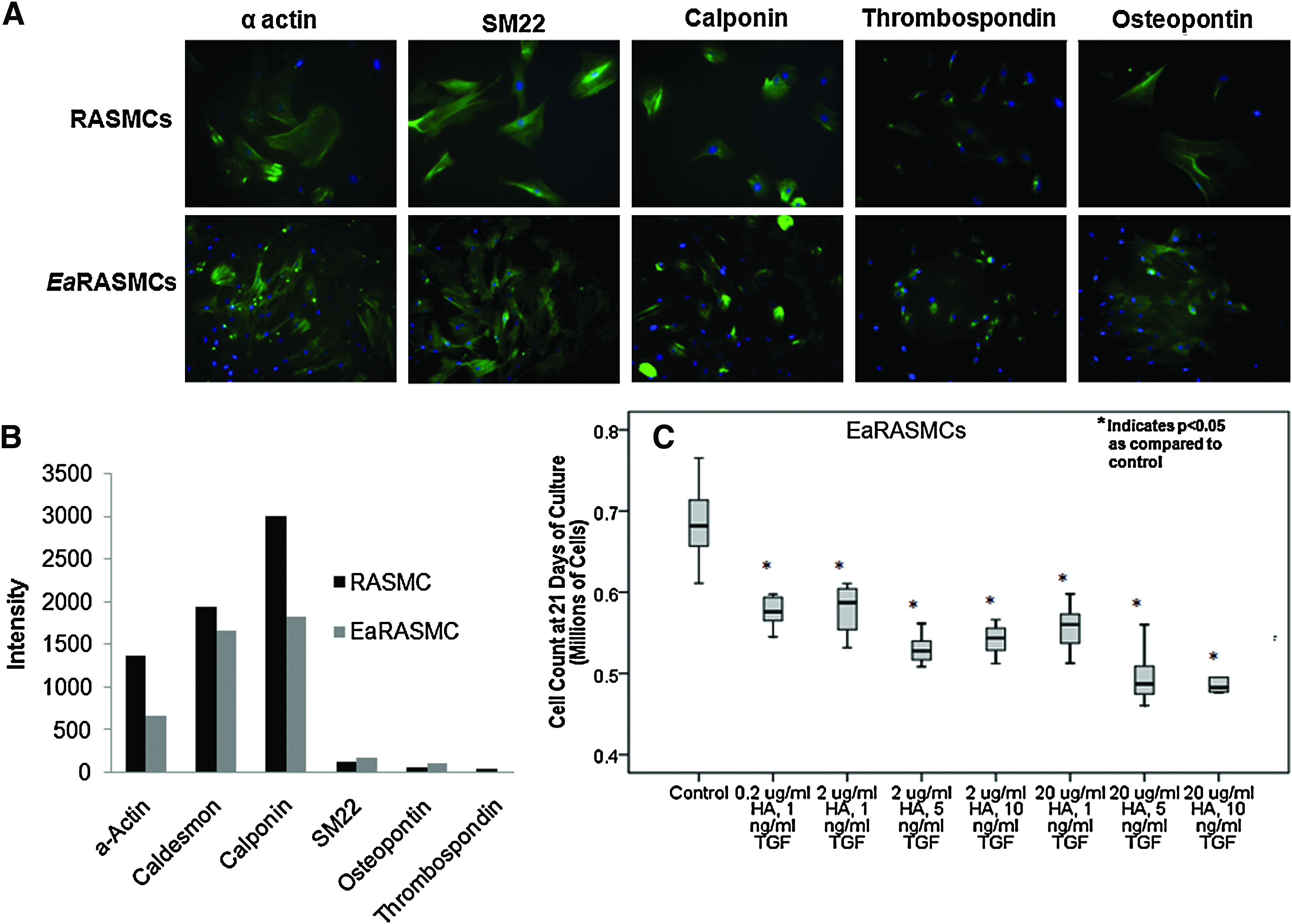
Morphologically, EaRASMCs derived from elastase perfusion-injured aortae appeared smaller and less spread than healthy RASMCs. Phenotypic analysis using immunofluorescence revealed the EaRASMCs to exhibit slightly decreased expression of contractile markers, most significantly, calponin, as compared to healthy RASMCs, as illustrated in Figure 4A. The EaRASMCs also exhibited increased expression of osteopontin. Flow cytometry analysis (total intensity of marker expression per 100 cells; Fig. 4B) indicated that there was a decrease in α-actin, caldesmon, and calponin contractile marker expression as compared to healthy RASMCs; there was no notable difference in SM22, osteopontin, and thrombospondin expression as compared to RASMCs (Fig. 4B).
EaRASMC proliferation
Figure 4C shows the count of EaRASMCs cultured with different dose combinations of TGF-β and HA-o. Relative to healthy, passage-matched RASMCs that proliferated 12.2±1.1-fold over 21 days of culture, 20 factor-untreated EaRASMCs proliferated more rapidly with their ultimate count at 21 days postseeding being 2.8±0.2 times that of the healthy RASMCs. Culture of EaRASMCs with TGF-β and HA-o significantly suppressed their proliferation, with maximum attenuation observed for doses of 20 μg/mL HA-o and 10 ng/mL TGF-β (count at 21 days representing 75% of that in untreated control EaRASMC cultures; p=0.0001 vs. control).
Matrix synthesis
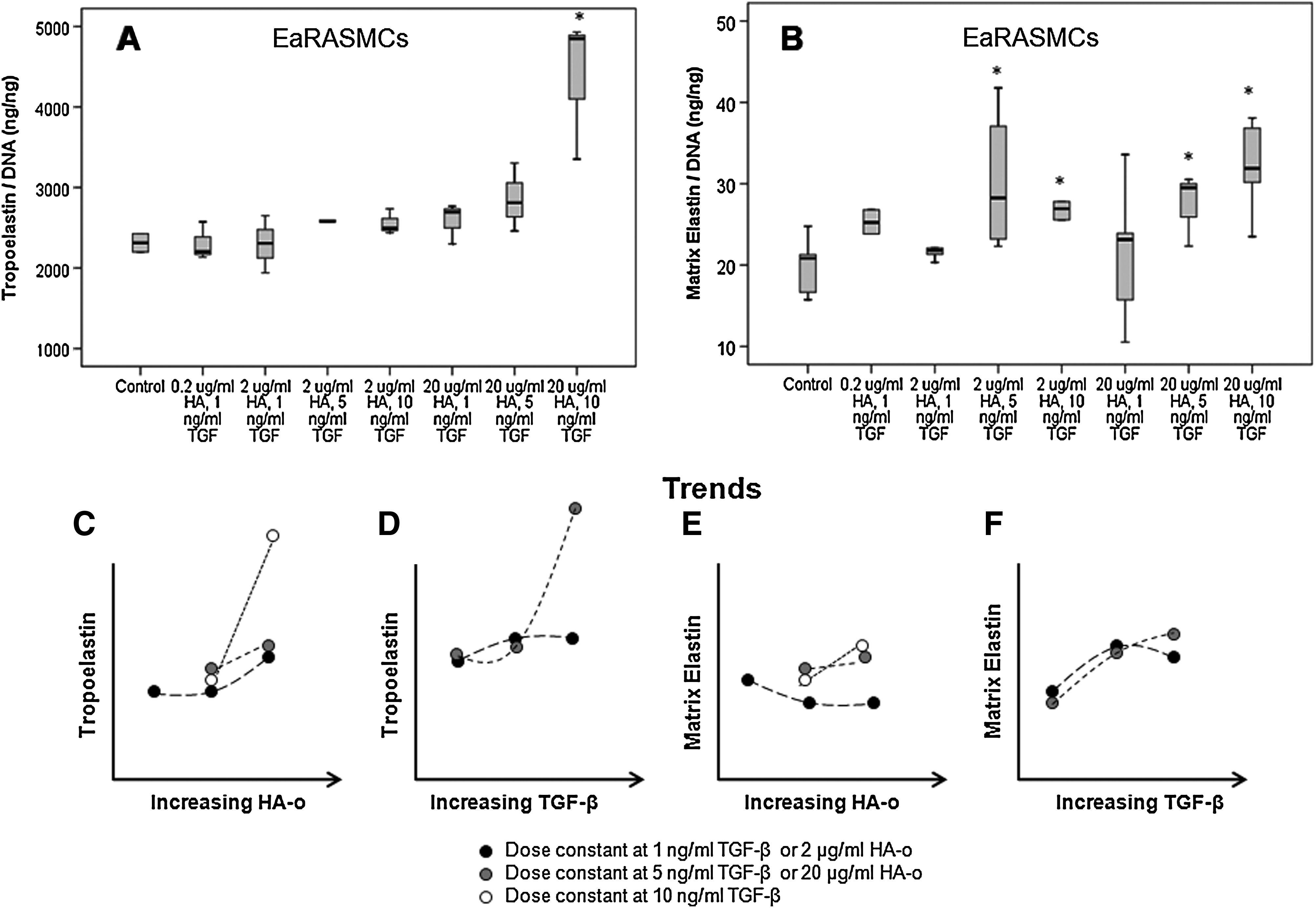
Total elastin production by EaRASMCs (10.1 mg/well) was much lower compared to healthy, passage-matched RASMCs (33.3 mg/well). 20 As shown in Figure 5A, a high dose combination of 20 μg/mL HA-o and 10 ng/mL TGF-β induced a significant increase in cellular tropoelastin production by EaRASMCs (1.89-fold increase; p=0.05 vs. control), which translated into increased matrix elastin formation (1.49-fold increase; p=0.006). At moderate doses of 2 μg/mL HA-o and 5 ng/mL TGF-β, 2 μg/mL HA-o and 10 ng/mL TGF-β, and 20 μg/mL HA-o and 1 ng/mL TGF-β there was no significant increase in tropoelastin, yet there was an increase in elastin crosslinked into the matrix form. As shown in Figure 5B, when HA-o and TGF-β were provided to aRASMCs at the lowest dose determined to provide elastogenic benefit, that is, 2 μg/mL HA-o and 5 ng/mL TGF-β, a significant increase in matrix elastin (including both highly cross-linked, alkali-insoluble structural elastin and an alkali-soluble fraction) was observed, relative to control cultures (1.40-fold increase; p=0.049). At this lowest elastogenic dose, the absolute amounts of matrix elastin and of tropoelastin protein produced by EaRASMC cultures was 112% and 25%, respectively, of that produced by healthy, factor-untreated RASMCs. At the factor dose deemed most elastogenic (20 μg/mL of HA-o and 10 ng/mL of TGF-β), the corresponding figures were 106% and 40%.
Effects of HA-o and TGF-β factor supplementation on tropoelastin
Ultrastructure of matrix elastin
Representative images of control EaRASMC layers and those treated with the HA-o/TGF-β dose (20 μg/mL and 10 ng/mL, respectively) deemed most elastogenic are shown in Figure 6. The images clearly indicate that treatment with the factors significantly enhances fibrous elastic matrix deposition over control cultures, which contained a few clumps of amorphous elastin and sporadic elastin fibrils.
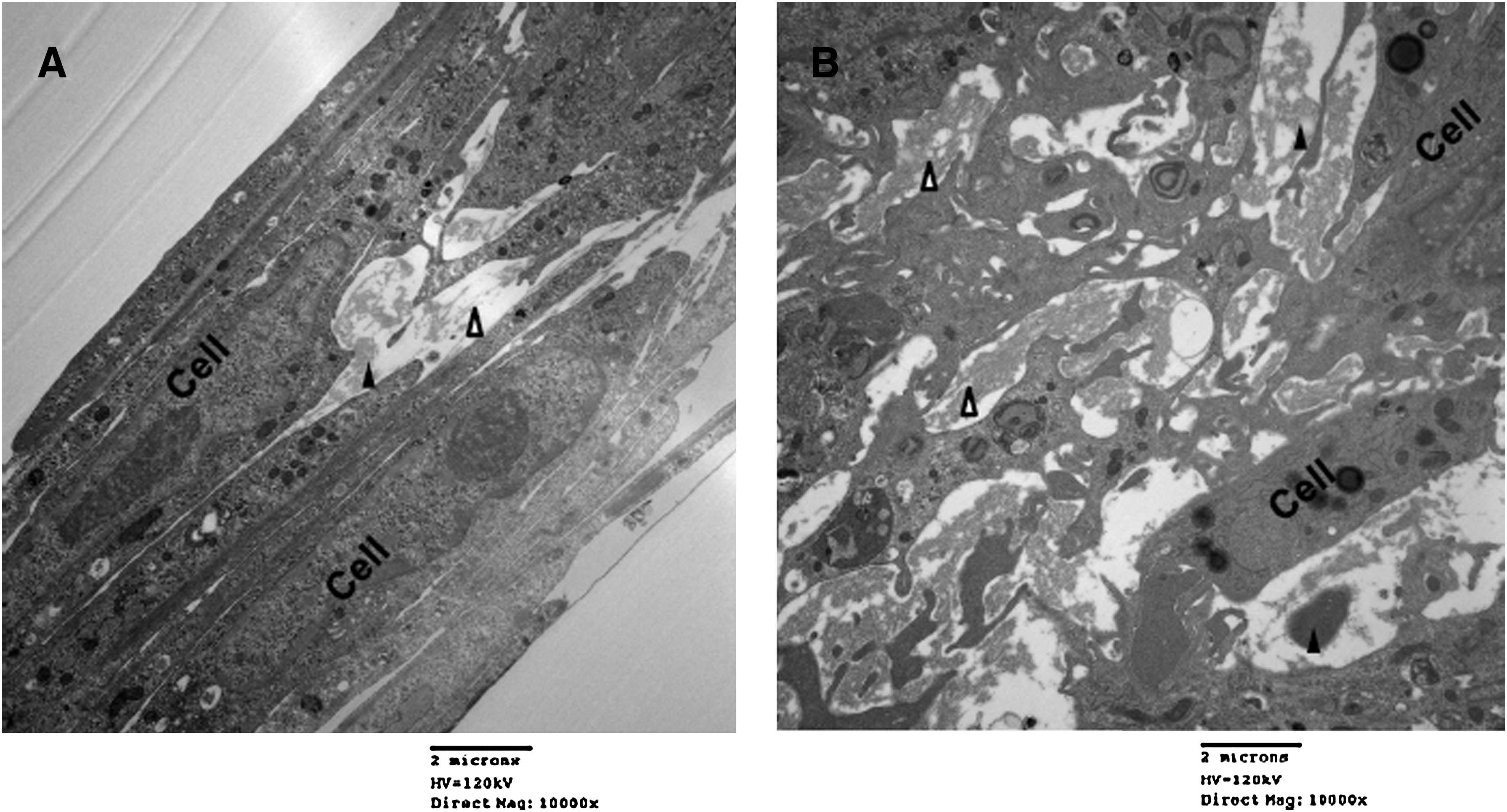
Representative transmission electron microscopic images of 21-day old EaRASMC layers cultured with
Von Kossa staining
In all culture groups, calcium deposits were absent or minimal, indicating that there was no/minimal calcific deposition, typically due to enhanced uptake of Ca2+ from culture medium, or due to possible induction of an osteogenic phenotype in the EaRASMCs by the provided doses of TGF-β.
Activity of proteolytic enzymes
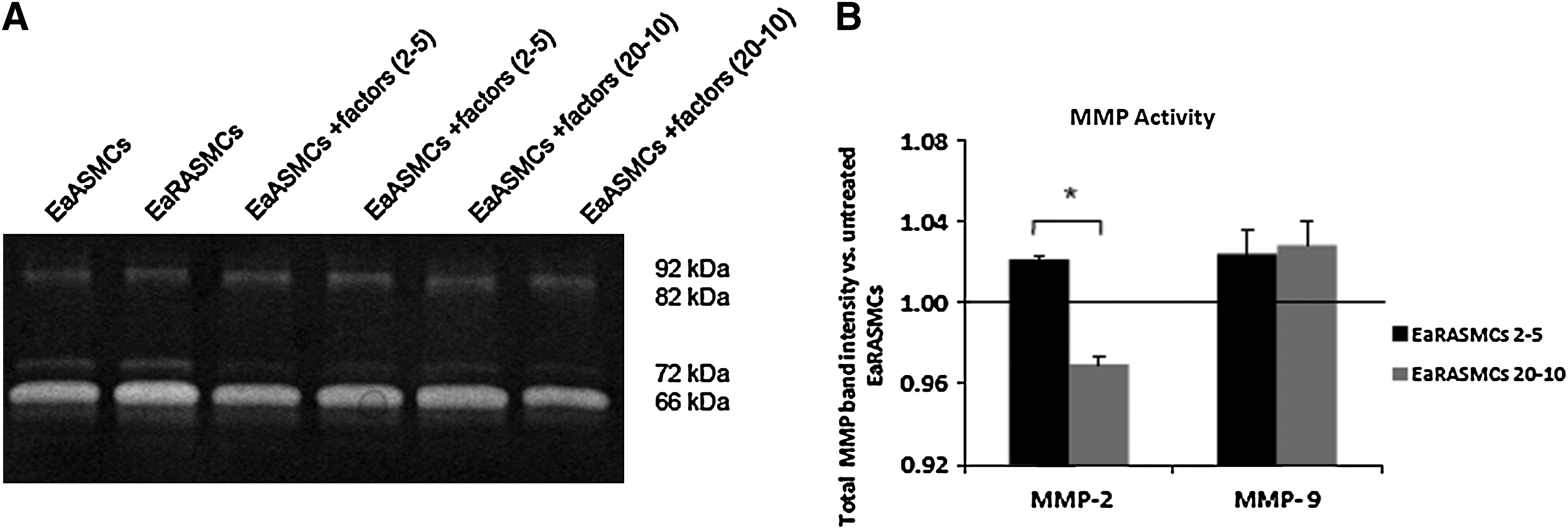
Gelatin zymography revealed that total (i.e., of active and zymogen forms) MMP-2 activity and that specifically of active MMP-2 were increased by 1.93±0.34-fold and 2.54±0.42-fold in aortal tissues that were perfused with elastase, over untreated control aortae (Fig. 3C, D). MMP2 and 9 activities within EaRASMC cultures were not affected by addition of the factors, at any dose (Fig. 7). The total activity of MMP-2 generated by EaRASMC cultures was attenuated only in the presence of 20 μg/mL of HA-o and 10 ng/mL of TGF-β. MMP9 activity was unaffected by factor dose (Fig. 7).
DNA microarray
Gene expression levels in RASMCs, EaRASMCs, and EaRASMCs treated with HA-o and TGF-β were examined by DNA microarray analysis. Between the treatment groups, numerous genes were differentially expressed (2361 genes, with false discovery rate <5%; not shown). However, a subset of these genes showed a characteristic expression profile in which the expression level in EaRASMCs differed from the healthy RASMCs, but then returned to nearly healthy levels in the EaRASMCs treated with HA-o and TGF-β (Fig. 8).
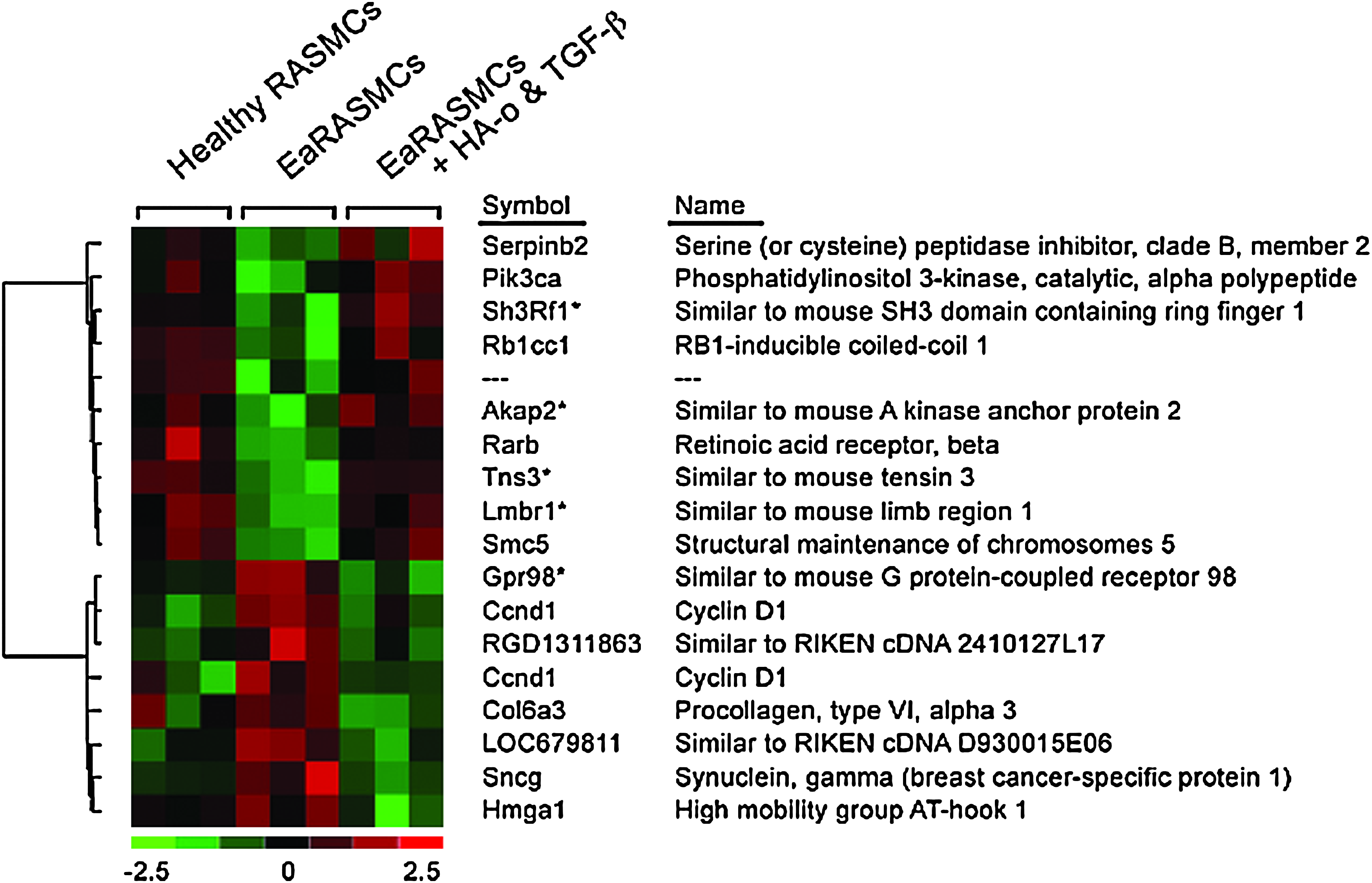
Heat map showing differential gene expression between cultures of EaRASMCs and healthy RASMCs. Only a select sub-set of genes that exhibited such differences are shown. Both cell types were evaluated at passage 3. Red represents upregulation and green represents downregulation of genes in each set of cultures relative to the other. Also seen is the impact of HA-o and TGF-β factors on gene expression by EaRASMCs. Shown specifically are genes whose expression were returned to “healthy” levels after 7 days of culture with factors (2 μg/mL of HA-o and 5 ng/mL of TGF-β). Differences between treatment groups was defined by significance analysis of microarrays, multiclass analysis, with delta adjusted for false discovery rate <5%. *Genes (probe sets) on the microarray that did not have an assigned identity but which were subsequently conditionally identified based on homology searches against the mouse genome.
Discussion
Promising nonsurgical approaches to AAA treatment include pharmacological inhibition of MMP release/activity within AAAs by tissue inhibitors of matrix metalloproteases (TIMPs) 7 or modified tetracyclines 8 (e.g., doxycycline). A limitation common to these approaches is that they suppress only two of the three factors that contribute to AAA growth, namely, infiltration of inflammatory cells into the aortal wall, and their enhanced release of active, proteolytic MMPs; the third and an equally important factor, that is, elastic matrix degradation, and lack of its auto-regenerative repair cannot be addressed by these approaches. With the absence of any means to actively regenerate new elastic matrix structures within the AAA, active regression of the condition is not possible. Moreover, recent studies that explored the utility of doxycycline and MMP inhibitor in stabilizing AAAs also suggest that it impedes accumulation of even the very limited amounts of new elastic matrix by poorly elastogenic adult vascular SMCs, virtually eliminating any potential for regenerative elastic matrix repair within such stabilized AAAs, leading to their regression. In such cases, additional impetus in terms of elastogenic factors can potentially enhance net accumulation of new elastic matrix.
In previously published work, we showed that healthy RASMCs can be elastogenically stimulated in vitro with HA-o and TGF-β1 factors. 28 More recently, we demonstrated that SMCs isolated from rat aortal expansions created by peri-adventitial caustic injury, which resemble early stage AAAs, (a) continue to exhibit an abnormal, activated phenotype when passaged in vitro culture, (b) are much more elastogenically challenged relative to healthy, passage-matched RASMCs, and can (c) be elastogenically stimulated in vitro with HA-o and TGF-β factors. 16 The relevance of the present study lies in the fact that AAAs are often only diagnosed at advanced stages of development, when matrix deterioration is severe. Since cell behavior is influenced by their microenvironment, there is a need for evidence that SMCs from such severely afflicted tissues can also respond favorably to elastogenic stimulation, and if so, if the effective doses of elastogenic factors differ significantly from that we ascertained useful with healthy RASMCs. 28
In the current study, at 2 weeks postinjury, elastase-perfused rat abdominal aortae generated many of the outcomes typical of human AAAs, such as medial elastic matrix disruption, increased activity of proteolytic MMP-2, but not MMP-9, presence of macrophages, and an ∼120% increase in aortic diameter, similar to more advanced human AAAs. Despite these similarities, the AAAs so generated also differed somewhat from human AAAs in not being associated with calcific deposits and thrombi as many (but not all) human AAAs tend to be. Thus, SMCs within elastase-perfusion-induced rat AAAs experience a slightly different microenvironment than do human AAA SMCs and quite possibly fewer pathological stimuli. Despite this, in the absence of any small animal model of AAAs that replicates each and every possible facet of AAA pathology, the current choice of an AAA model is pertinent in its ability to mimic several key aspects of SMC activation within human AAAs.
Since phenotypic characterization of RASMCs and assessment of their elastogenic potential (innate and induced) requires sufficient cell numbers for study, which cannot be obtained from primary tissue sources, passaging and number expansion of primary cells in culture is inevitable. Morphologically, we found early passage EaRASMCs exhibited decreased volume/spreading as compared to healthy RASMCs, an observation that has been reported with other diseased/activated SMC types, including human (unpublished data). Phenotypic analysis using immunofluorescence and flow cytometry revealed a significant number of EaRASMCs to exhibit decreased α-actin, caldesmon, and calponin expression as compared to healthy RASMCs. Caldesmon plays a key role in SMC contraction by binding to acto-myosin contractile units like actin, tropomyosin, myosin, and calmodulin 29 and may have a regulatory role in the contractile apparatus; it has been reported to inhibit the Mg2+-ATP-ase activity and hence inhibit smooth muscle contractility. 30 Calponin, the expression of which was even more significantly reduced, binds with actin light chains depending on the presence of Ca2+, thereby regulating the contractile apparatus of SMCs31,32; due to this binding, it has also shown to inhibit actin-activated Mg2+-ATP-ase activity in vitro. 33 Together, these suggest a loss of contractile phenotype in EaRASMCs. In addition, the EaRASMCs also exhibited an increased expression of osteopontin, a multifunctional pro-inflammatory cytokine implicated with vascular disease and enhanced inflammatory response 34 ; osteopontin has been shown to induce chemotaxis of macrophages and monocytes, which stimulates calcification 35 and enhances MMP activity, thereby promoting vascular wall deterioration. 36 In tandem with these phenotypic marker alterations, elastase-perfused aortae sections (Fig. 3) exhibited enhanced MMP activity over saline-perfused control aortae, suggesting activated cell phenotypes. However, Von Kossa staining showed no visible matrix calcification in either the elastase-perfused aortal segments or in cultures of isolated EaRASMCs, indicating that the treatment does not induce an osteogenic phenotype so much as to lead to such severe outcomes.
In general, EaRASMCs proliferated much faster than did healthy RASMCs, a response that we have noted with healthy and aneurysmal human aortal SMCs as well, in a separate study pending publication. A comparison of the effects of the factors on EaRASMCs revealed that HA-o and TGF-β suppressed cell proliferation significantly, relative to untreated EaRASMCs. In terms of absolute amounts, elastin synthesis was much lower (30%) in cultures of untreated EaRASMCs relative to healthy RASMCs. In EaRASMC cultures, unlike in healthy RASMC cultures, low dose combinations of HA-o (<20 μg/mL) and TGF-β (<10 ng/mL) had no effect on tropoelastin production; this agrees well with our prior findings that diseased SMCs require higher doses of these factors for elastogenic stimulation. 16 Also, as seen in our trend curves (Fig. 5C), the dependence of tropoelastin synthesis on HA-o dose is enhanced at higher doses of concurrently delivered TGF-β. This might possibly be due to sensitization of SMC receptors for HA-o (e.g., CD44 37 ) by TGF-β, leading to enhanced downstream intracellular outcomes of HA-o/receptor interactions and increased tropoelastin synthesis. The trend curve in Figure 5D on the other hand shows that at the lowest HA-o dose (2 μg/mL), TGF-β effects on tropoelastin synthesis dominate, as apparent from the classic but weakly biphasic TGF-β dose response at HA-o doses; at higher HA-o doses, this kind of biphasic response to TGF-β was not observed within the studied TGF-β dose range.
It is also apparent from our results that HA-o and TGF-β, in general, appear to more sensitively influence the elastin crosslinking/matrix assembly machinery than cellular tropoelastin synthesis, which, as discussed above, is increased only at the highest dose combination (20 μg/mL of HA-o and 10 ng/mL of TGF-β). Also, it appears that the predominant effects of the factors lie in the enhancement of elastic matrix deposition, which agrees with inferences we have made in our previous studies on elastogenic upregulation of healthy SMCs,28,38 and SMCs isolated from CaCl2-induced rat aortal expansions 16 and human prerupture AAAs (yet unpublished data). The increase in elastic matrix deposition without parallel increase in tropoelastin synthesis at the lower HA-o/TGF-β dose combinations suggests that at these doses, the factors only influence the elastin crosslinking machinery. In support of these outcomes, our prior studies have shown that TGF-β and HA-o together enhance both mRNA expression, protein amounts, and activity of the elastin crosslinking enzyme, lysyl oxidase (LOX). In addition, it is noted that increases in tropoelastin and matrix elastin synthesis with increasing TGF-β dose are more pronounced at higher HA-o doses.
MMP-2 (but not MMP-9) activity was attenuated in EaRASMC cultures that received the maximum (and most elastogenic) factor dose tested (20 μg/mL HA-o and 10 ng/mL TGF-β) but was not impacted by lower doses of the factors, such as at 2 μg/mL HA-o and 5 ng/mL TGF-β, which induced comparable levels of elastic matrix synthesis. This implies a possible need to deliver higher factor dose equivalents to AAAs to sufficiently stabilize the elastic matrix against MMP-mediated breakdown. However, a potentially more useful approach might be to co-deliver the factors with any of several available specific or nonspecific MMP inhibitors (e.g., doxycycline) to AAAs to inhibit endogenous MMPs to (a) protect pre-existing elastic fibers against deterioration and thus enhance the quantity and quality of new elastic fiber deposition, and (b) permit accumulation of newly regenerated elastic matrix. Such co-delivery of elastogenic factors would provide the benefit of stimulating intrinsically poor elastic matrix regeneration within AAA tissues as discussed earlier in this section, and in parallel enhance the outcomes of such induced regeneration by locally attenuating inflammatory cell-mediated proteolysis.
At the transcriptional level, DNA microarray analysis provided a plethora of valuable information that merits further investigation. Our preliminary observations from this data are multifold. First, elastase perfusion affects the expression of many genes in cultured EaRASMCs relative to RASMCs (Fig. 8, and not shown). It is interesting that this differential gene expression is maintained ex vivo and suggests that SMCs derived from an AAA maintain an altered gene expression in vitro culture, at least until the third passage. At this time, we have not further investigated the differentially expressed genes between the EaRASMCs and RASMCs; we have instead focused our initial efforts on understanding the effects of HA-o and TGF-β1 on EaRASMCs. Of particular interest, BMP-4 expression was decreased in EaRASMCs relative to healthy RASMCs; HA-o and TGF-β appear to have no effect on that expression level. This supports our observations that there were no calcific deposits found within elastase-perfused aortae or in low passage cultures of isolated primary EaRASMCs. Interestingly, elastin mRNA expression was decreased in EaRASMC cultures relative to healthy RASMCs and the factors (2 μg/mL HA-o and 5 ng/mL of TGF-β) appeared to further suppress the same. This result agrees with that of biochemical assays (see section Matrix synthesis), which showed tropoelastin synthesis by EaRASMCs to be quite deficient relative to healthy RASMCs, and a slight decrease observed in tropoelastin synthesis by EaRASMCs cultured with the above dose of factors (25% of healthy RASMC levels) relative to untreated EaRASMCs (30% of healthy RASMC levels). This result, however, sharply contrasts with the 1.5-fold increase in elastin mRNA expression we previously observed in factor-supplemented healthy RASMC cultures (also passage 3), relative to untreated RASMCs, 28 which translated to an eightfold increase in tropoelastin production by the cells. 28 While this result emphasizes differences between aneurysmal and healthy RASMCs in the context of elastic matrix synthesis, it also suggests that HA-o and TGF-β, at least at the low dose combinations tested (<20 μg/mL of HA-o and <10 ng/mL of TGF-β) have mildly suppressive effect on elastin gene transcription. Our findings of increased tropoelastin synthesis by EaRASMCs when treated with the highest dose of the factors (20 μg/mL of HA-o and 10 ng/mL of TGF-β) also suggest that we might expect elastin mRNA expression to be enhanced in cells receiving that treatment; this is yet to be confirmed. If so ascertained, it might serve as an additional testament to the biphasic, elastogenic factor dose-dependent effects of TGF-β. Our hypothesis as to the ability of HA-o and TGF-β to induce elastin mRNA expression, if provided at high enough doses is also peripherally supported by findings in our parallel (yet unpublished) study that anuerysmal human aortic SMC cultures enhance synthesis of tropoelastin by fivefold in response to the factors (2 μg/mL of HA-o and 5 ng/mL of TGF-β).
The microarray results also revealed decreased expression of specific MMPs (e.g., MMP-12) and increased expression of specific tissue inhibitors of MMP (e.g., TIMP-3) in EaRASMC cultures that received the factors. This observation is promising and points to the possibility that the factors are “protecting” the elastic matrix from enzymatic breakdown. These data are extremely interesting, yet the design of future experiments to further assess the mechanistic role of HA-o and TGF-β1 on elastin matrix formation in AAA models is necessary.
A final note pertains to the extrapolation of the present data to human AAA SMCs. Human AAAs do not exhibit a uniform set of pathological characteristics, but rather represent a diverse subset of features that are determined by their initiating cause and site of localization, and which in turn uniquely determine the progression of the disease. Thus said, since there is no single animal model of AAAs that can replicate the plethora of pathological characteristics exhibited by human AAAs, we have selected one which exhibits several if not all common pathological aspects, that is, an elastase perfusion model of rat AAAs. Importantly, this model replicates the inflammatory and significantly elastin-disrupted matrix microenvironment typical of all advanced human AAAs. Thus, it may be presumed that AAA RASMC behavior including their response to the factors might be generally evocative of how human AAA SMCs might respond, though outcomes with the latter cell type may differ somewhat if sourced from human AAAs exhibiting calcific stiffening of the aortal wall and/or thrombus, which our model does not replicate. Supporting this hypothesis, results of a parallel, yet unpublished study we conducted showed that SMCs from thrombus-engorged prerupture human AAA can also be elastogenically induced with HA-o and TGF-β, just as we demonstrate here, and that the effective factor doses overlap with the factor dose range shown useful in this study (2–20 μg/mL of Ha-o and 5–10 ng/mL of TGF-β). However, different from EaRASMCs, whose production of tropoelastin was enhanced only at the highest tested dose (20 mg/mL of HA-o and 10 ng/mL of TGF-β), tropoelastin synthesis by aHASMCs was enhanced even by lower doses of factors (2 mg/mL of Ha-o and 5 ng/mL of TGF-β). Also, identical doses of the factors stimulated much greater fold-increases in tropoelastin and matrix elastin synthesis in aHASMCs cultures over factor-free control cultures, relative to EaRASMC cultures (e.g., 5.8±0.76-fold and 4.7±0.6-fold increases in DNA-normalized amounts of tropoelastin and matrix elastin synthesed by aHASMCs versus 1.9±0.25-fold and 1.5±0.4-fold for EaRASMCs at 20 mg/mL of HA-o and 10 ng/mL of TGF-β). These findings suggest that aneurysmal SMCs isolated from elastase perfusion induced rat AAAs resemble human aneurysmal SMCs in their ability to be elastogenically stimulated, and that factor doses for such stimulation for the two cell types, overlap. However, the preliminary comparisons between the two cell types also suggest that aHASMCs respond more sensitively to elastogenic factors than do EaRASMCs; this needs to be confirmed with a more rigorous cnad controlled study with SMCs isolated from multiple source human tissues that represent the diversity of pathological aspects associated with human AAAs.
Conclusions
The present study shows that primary SMCs isolated from elastase-perfusion induced AAAs can be passaged and cultured in vitro with retention of a diseased phenotype characterized by hyper-proliferation, enhanced expression of markers of synthetic activity and of the activity of elastolytic MMPs-2 and 9, relative to healthy RASMCs and distinctly different patterns of gene expression as indicated by DNA microarray analysis. In the context of the typical diagnosis of AAAs at only advanced stages of development, the present study also provides evidence that diseased SMCs within such AAAs can be coaxed to enhance elastin synthesis and regenerate fibrous elastic matrix to possibly regress the condition. Similar to outcomes we observed previously with healthy RASMCs and SMCs isolated from very early-stage induced rat AAAs (i.e., by periadventitial injury with CaCl2), here too we showed that HA-o and TGF-β can induce EaRASMCs to increase their synthesis of tropoelastin precursors, to improve elastic matrix amounts to levels comparable to that synthesized by healthy RASMCs, and further encourage elastic fiber formation over deposition of formless amorphous elastin clumps. Our study however indicates that much higher doses of HA-o and TGF-β are required to elastogenically induce SMCs from late-stage AAAs than are required for healthy SMCs or early stage AAA SMCs. Moreover, an important deduction from this study is that in light of the limited effects of the elastogenic factors on MMP activity, codelivery of pharmacologic inhibitors of MMPs together with elastogenic factors might be necessary to stabilize the AAA microenvironment to preserve the existing elastic matrix framework and allow newly regenerated elastic matrix to accumulate. Finally, the study indicates that the elastase perfusion-injury model of AAAs may be suitable as a surrogate in the context of elastin regeneration within advanced human AAAs.
Footnotes
Acknowledgments
This study was funded by the National Institutes of Health R21EB006078-01A1, RO1HL0092051-01A1, and R01HL0092051-01S (Ramamurthi A). Gacchina C was supported by a National Institutes of Health predoctoral award T32 HL007260. The authors would like to acknowledge Dr. John Curci and Terri Ennis at Washington University in St. Louis for providing training on the elastase perfusion procedure on mice and Victor M. Fresco and the MUSC Proteogenomics Facility for assistance with the DNA microarray analysis.
Disclosure Statement
No competing financial interests exist.