
Abstract
Fibrin glue (FG) is used in a variety of clinical applications and in the laboratory for localized and sustained release of factors potentially important for tissue engineering. However, the effect of different fibrinogen concentrations on FG scaffold delivery of bioactive adeno-associated viruses (AAVs) has not been established. This study was performed to test the hypothesis that FG concentration alters AAV release profiles, which affect AAV bioavailability. Gene transfer efficiency of AAV-GFP released from FG was measured using HEK-293 cells. Bioactivity of AAV transforming growth factor-beta1 (TGF-β1) released from FG was assessed using the mink lung cell assay, and by measuring induction of cartilage-specific gene expression in human mesenchymal stem cells (hMSCs). Nondiluted FG had longer clotting times, smaller pore sizes, thicker fibers, and slower dissolution rate, resulting in reduced release of AAV. AAV release and gene transfer efficiency was higher with 25% and 50% FG than with the 75% and 100% FG. AAV-TGF-β1 released from dilute-FG transduced hMSCs, resulting in higher concentrations of bioactive TGF-β1 and greater upregulation of cartilage-specific gene expression compared with hMSC from undiluted FG. This study, showing improved release, transduction efficiency, and chondrogenic effect on hMSC of bioactive AAV-TGF-β1 released from diluted FG, provides information important to optimization of this clinically available scaffold for therapeutic gene delivery, both in cartilage regeneration and for other tissue engineering applications.
Introduction
Gene therapy can overcome these obstacles by delivering genes that encode chondrogenic growth factors or inhibitors of cartilage catabolites in various vector constructs. This also provides for a local delivery system that drives the expression of therapeutic molecules over an extended period and overcome the need for repeated administrations or interventions. Gene carriers include plasmids, nonviral vectors, and viral vectors. Although plasmid and nonviral vectors are less toxic, less immunogenic, and easier to prepare, their gene delivery efficiencies are significantly lower compared with viral vectors. 10 Hence, viral vectors are currently considered the most effective agents for in vivo gene transfer. 11
Viral vectors that have been used in the preclinical setting include adenovirus, adeno-associated virus (AAV), 12 and lentivirus. 13 AAV is derived from an endemic, nonpathogenic parvovirus. It has the following advantages over other viruses: sustained transgene expression over longer period, 14 reduced potential for host immune response, and the capacity to transduce both dividing and nondividing cells. 15 After transduction, the viral genome translocates into the target cell nucleus, and the DNA polymerase generates the transducing episome. Numerous serotypes of AAV have been identified, each having different preferential targets. Among the different serotypes, AAV serotype 2 (AAV2) is considered to have the best defined safety profile as it has already been used in clinical trials. 16
There are three general approaches for cartilage viral gene therapy applications: direct, indirect, and hybrid. The direct method involves the injection of viral vectors directly into the articular joint space to transduce local cells. Although long-term persistence of transgene expression has been observed with direct injection of AAV2, 14 most of the transduction occurs in soft tissues and it is difficult to localize the transduction to specific cell types. Further, rapid dispersion of viral particles from the joint space would prevent effective transduction of repair cells that are recruited to the defect site over time. Thus, the in vivo transduction efficiency remains low and nonspecific transduction of adjacent tissues and their transgene expression at undesired sites is usually observed. 15 The indirect approach involves the use of genetically modified cells, 12 and/or biodegradable scaffolds that release genetically modified cells.17,18 This leads to the site-specific release of bioactive molecules important in tissue regeneration, though temporary. In addition, this approach requires ex vivo manipulation of cultured cells, which faces high barriers with respect to clinical translation. The third hybrid approach involves implantation of biodegradable scaffolds embedded with genetic materials,19,20 leading to direct and localized in situ delivery of bioactive vectors to the host cells in proximity to the scaffold, as well as the sustained expression of the transgene. Further, scaffolds can act as support for injury-activated repair or progenitor cells, such as BM-derived mesenchymal stem cells (BM-MSCs), to migrate, attach, and differentiate at the defect site. Thus, the hybrid method offers the advantages of site-specific localization, direct transduction of host repair cells, and sustained transgene expression, while limiting the disadvantages of indiscriminate transduction and requirement of ex vivo manipulations.
Our group has previously shown that AAV2-transfroming growth factor-β1 (AAV2-TGF-β1) transduced human MSCs (hMSCs) implanted into a 1.5-mm-diameter osteochondral defect significantly improved cartilage regeneration over 12 weeks in vivo. 12 These results demonstrated that AAV2 is a suitable vector for gene delivery to improve the cartilage repair potential of the MSCs. However, although the ex vivo gene transfer is effective, the regulatory barriers for using genetically modified cultured human cells for therapeutic purposes clinically are extremely high. In addition, extraction and expansion of MSCs prolong the time before treatment can be applied and are very expensive. Therefore, we have explored the use of biodegradable scaffolds for release and delivery of bioactive substances to host MSCs within cartilage wound.21–24
Fibrin glue (FG) is a natural substance available for human use clinically as a commercial product or readily prepared from plasma for autologous applications. 15 These properties mean that laboratory findings may be more readily translated into potential human clinical applications than similar findings with synthetic polymers. Despite being widely used in clinical surgical practice as an adhesive and sealant for hemostasis, 25 only in recent years has the ability of FG to act as a delivery scaffold received attention. 26 Besides being commercially available for sterile applications, it is nonimmunogenic, biodegradable, and malleable so that it can be molded into irregular tissue defects. All of these advantages make FG an appealing scaffold for preclinical translational cartilage tissue engineering applications. It has been shown that diluted FG produces a more open fibrin network compared with undiluted FG scaffolds, 27 and that FG can act as an efficient scaffold for gene delivery.28–30
In the current study, we investigated the effect of different fibrinogen dilutions during the preparation of FG scaffold on the delivery of AAV2 and their early effects on human BM-MSC (hBM-MSC) chondrogenesis in vitro. The aim of this study was to test the hypotheses that diluted FG scaffolds will release more viral particles, result in higher transduction efficiency, and increase the chondrogenic potential of transduced hBM-MSCs.
Materials and Methods
Preparation of FG
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Twenty-four-well plates and 24-well plate inserts were purchased from BD Biosciences (San Jose, CA). Commercially available Tisseel® human fibrin sealant (Baxter, Deerfield, IL) was used to construct FG hydrogels. Fibrinogen (100% of the original material concentration, lyophilized human fibrinogen reconstituted in aprotinin fibrinolysis inhibitor) was prepared by serial dilution with phosphate-buffered saline (PBS; Gibco, Carlsbad, CA) into four different concentrations: 25%, 50%, 75%, and 100% fibrinogen. Different dilutions of fibrinogen were subsequently mixed with an equal volume (1:1 volume ratio) of thrombin solution provided in the Tisseel kit (human thrombin 400–625 IU/mL and calcium chloride 36–44 μmol/mL). The resultant FG constructs were named 25%, 50%, 75%, and 100% FG, according to the fibrinogen dilution before thrombin activation.
Characterization of different FG concentrations
Clotting time
100 μL of different fibrinogen dilutions was mixed with 100 μL Thrombin-CaCl2 in a 96-well plate to make 25%, 50%, 75%, and 100% FG scaffolds. Change in the turbidity of the FG scaffold solution was immediately measured by a VersaMax UV-Vis spectrophotometer (Molecular Devices, Sunnyvale, CA) at 550 nm wavelength. 31 The clotting time was defined as the time at which the maximum value was reached in the absorbance curve. Three replicates of each FG scaffold concentrations were examined in this study.
Scanning electron microscopy
Three FG scaffolds constructed from 200 μL of different fibrinogen dilutions and 200 μL Thrombin-CaCl2 were used for scanning electron microscopy (SEM) analysis. After fixation in 2.5% gluteraldehyde for 1 h and rinsing in PBS, FG scaffolds were dehydrated in increasing alcohol concentrations (30%, 50%, 70%, 90%, and 100%) for 15 min each, followed by hexamethyldisilazane for 1 h. Once hexamethyldisilazane was removed, the scaffolds were air-dried for an hour. Each FG scaffold was coated with an ultra-thin gold layer containing a gold sputter coater for 2 min at 25 mA and observed under an SEM (JSM-6335F; Jeol USA, Inc., Peabody, MA). Two separate images of each scaffold were captured. Pore size diameter and thickness of fibrin fibers were measured for each image using image analysis software, Metamorph 7.6.2.0 (Molecular Devices).
FG dissolution
Wet weight measurement: Different fibrinogen dilutions were mixed with an equal volume of thrombin-CaCl2 and injected into uncoated 24-well plate inserts with 8 μm pores at 200 μL FG/insert, and incubated at 37°C for 15 min to form hydrogels. The weight of each insert was measured before injection of the fibrinogen/thrombin solutions. Once the FG scaffolds had solidified, 500 μL PBS was added on top of each inserts, and the inserts were placed in a 24-well plate containing 1 mL PBS/well. After incubation at 37°C for 10 min, inserts were removed, PBS aspirated, surface PBS adsorbed by a filter paper, and the initial weight of the hydrogels (W0) was recorded. FG hydrogels were re-weighed (Wt) every 2 days to estimate the wet weight fraction, which was defined as (W0 − Wt)/W0. Five replicates of each FG concentration were examined in this study.
Protein dissolution: The conditioned PBS from each well, in which the FG hydrogels were incubated, was also collected at the time of hydrogel wet weight measurement. The amount of the protein dissolved and released into the PBS was measured by UV-Vis spectroscopy (NanoDrop, Wilmington, DE) at 278 nm, which measures the absorbance of the phenyl group of fibrinopeptide proteins. Fresh PBS was used as blank control.
AAV2-CMV-GFP in vitro transduction
AAV2-CMV-GFP-loaded FG preparation
Double-stranded AAV2-GFP with a cytomegalovirus promoter (dsAAV2-eGFP, hereunder referred to as AAV2-CMV-GFP) was prepared as previously described. 32 The AAV2 particles were packaged and purified using the adenovirus-free, triple-plasmid transfection method. The titer (vector genome per milliliter [vg/mL]) was determined by viral DNA dot blot method. 33 The stock AAV2-CMV-GFP titer was 3×1012 vg/mL.
The AAV2-CMV-GFP vector stock was added to the various fibrinogen preparations (25%, 50%, 75%, and 100%) to obtain a final concentration of 6.25×1010 vg/mL. After vortexing for 3 min, the AAV2-CMV-GFP-loaded fibrinogen solutions were mixed with an equal volume of thrombin-CaCl2 and injected into uncoated 24-well plate inserts with 8 μm pores, at 200 μL/well. After gelation at 37°C for 15 min, prewarmed 500 μL Dulbecco's modified Eagle's medium (DMEM; Gibco)–1% penicillin-streptomycin (Pen-strep; Gibco) was added to each insert. Three replicates of each AAV2-CMV-GFP-loaded FG concentrations were examined in this study.
AAV2-CMV-GFP released from the AAV2-CMV-GFP loaded FGs and their direct transduction of mammalian cells were assessed over 21 days. HEK-293 cells (ATCC, CRL-1573, Manassas, VA) were maintained in DMEM, 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), and 1% Pen-strep at 37°C and 5% CO2. Each day, ∼2.4×106 cells were seeded onto 24-well plates at a seeding density of 100,000 cells/well and incubated for 24 h, after which the medium was changed to serum-free DMEM–1% Pen-strep. Inserts containing AAV2-CMV-GFP-loaded FGs were placed into the wells of the 24-well plate seeded with HEK-293 cells and incubated for an additional 24 h at 37°C. The medium was placed below and in the inserts to allow migration of AAV2-CMV-GFP from the FG hydrogels to the seeded cells. Photomicrographs of GFP-expressing cells were taken daily using an Olympus DP71 camera (Olympus America, Center Valley, PA) and Nikon Eclipse TE2000-U microscope (Nikon Instruments Inc., Melville, NY). All GFP images presented in Figure 4C–F had the contrast increased by the same value in Adobe Photoshop CS4 (Adobe Systems, Inc., San Jose, CA) to improve the GFP signal for print.
AAV2-CMV-GFP release from FG
Each day, the conditioned serum free medium from each well was collected and preserved at −80°C until analysis. The amount of AAV2-CMV-GFP released into the conditioned medium from AAV2-CMV-GFP-loaded scaffolds was quantified by quantitative polymerase chain reaction (qPCR). All PCRs contained 0.5 μL of conditioned medium in a total volume of 10 μL using a commercially available pre-prepared 2× real-time TaqMan PCR master mix (Applied Biosystems, Foster City, CA). GFP-specific custom TaqMan forward (GTCCGCCCTGAGCAAAGA) and reverse (TCCAGCAGGACCATGTGATC) primers as well as FAM-labeled probes (CCCAACGAGAAGCG) were used. All qPCRs were performed with an ABI PRISM 7700 Sequence Detection System (Applied Biosystems) according to the following program: 12 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Three replicates of each experimental group were examined in this study.
In vitro transduction of AAV2-CMV-GFP released from FG
Transduction efficiency was quantified using flow cytometry and expressed as percentage of GFP-positive cells. Both AAV2-CMV-GFP-transduced and -nontransduced control cells were trypsinized, washed, and resuspended in 400 μL of PBS with 1% bovine serum albumin and 2% paraformaldehyde (both from Fischer Scientific, Fair Lawn, NJ). Ten thousand events were collected using a BD Biosciences LSR II (BD Biosciences, Franklin Lakes, NJ). The percentage of GFP-positive cells was determined by defining GFP-negative cells and gating out debris with FlowJo 7.6.1 software (Tree Star, Inc., Ashland, OR). Three replicates of each experimental group were examined in this study.
AAV2-CMV-TGF-β1 in vitro transduction
AAV2-CMV-TGF-β1-loaded FG preparation
Double-stranded AAV2-TGF-β1 with a cytomegalovirus promoter (dsAAV2-TGF-β1, hereunder referred to as AAV2-CMV-TGF-β1) was prepared as previously described 32 and purified as described in the section AAV2-CMV-GFP-loaded FG preparation. The stock AAV2-CMV-TGF-β1 titer was 2.5×1012 vg/mL.
The AAV2-CMV-GFP and AAV2-CMV-TGF-β1 vector stocks were separately added to 50% and 100% fibrinogen preparations to obtain a final concentration of 1.25×1011 vg/mL. After vortexing for 3 min, the AAV2-loaded fibrinogen solutions were mixed with an equal volume of thrombin-CaCl2 and injected into uncoated six-well plate inserts with 8 μm pores, at 1 mL/well. After gelation at 37°C for 15 min, prewarmed 3 mL chondrogenic medium–High-Glucose DMEM supplemented with 1% Pen-strep, 10–7 M dexamethasone, 50 μg/mL L-ascorbic acid-2-phosphate, 40 μg/mL proline (MP Biomedicals, Solon, OH), and 1% ITS+ Premix (BD, Franklin Lakes, NJ) was added to each insert. Three replicates of 50% and 100% AAV2-CMV-TGF-β1-loaded FGs were examined in this study against the 50% and 100% AAV2-CMV-GFP-loaded FGs.
AAV2-CMV-TGF-β1 released from the AAV2-CMV-TGF-β1-loaded FGs and their direct transductions of hBM-MSCs were assessed. The hBM-MSCs were obtained, maintained, and characterized as previously described.34,35 About 4.8×106 cells were seeded onto six-well plates at a seeding density of 200,000 cells per well and incubated for 24 h, after which the medium was changed to a chondrogenic medium. Inserts containing AAV2-CMV-GFP-loaded FGs and AAV2-CMV-TGF-β1-loaded FGs were placed into the wells of the 6-well plate seeded with hBM-MSCs and incubated for 24 h. The medium was placed below and in the inserts to allow migration of AAV2s from the FG hydrogels to the seeded cells. After the removal of the inserts, the cells were incubated for an additional 48 h at 37°C. The conditioned serum free medium and hBM-MSCs from each well were collected separately and preserved at −80°C until analysis.
In vitro transduction of AAV2-CMV-TGF-β1 released from FG
The level of TGF-β1 expression from hBM-MSCs was assessed by determining the level of TGF-β1 in cell culture supernatant using a commercially available ELISA (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer's description, and using a CCL-64 mink lung-epithelial growth inhibition assay as previously described with slight modification.36,37 Briefly, the CCL-64 cell line (ATCC, CRL-1573) was grown in DMEM, 10% fetal bovine serum, and 1% Pen-strep at 37°C in 5% CO2. CCL-64 cells were plated at 5,000 cell/well in 96-well plates and cultured in 100 μL of normal growth media for 3 h. The medium was then changed to 100 μL of serum-free DMEM-1% Pen-strep supplemented with 10% of conditioned media from hBM-MSC culture. The plates were incubated at 37°C in 5% CO2 for 24 h, and then 20 μL reconstituted XTT reagent from In Vitro Toxicology Assay Kit (Sigma-Aldrich) was added to each well for 2 h incubation. The absorbance at 450 nm was measured using VersaMax UV-Vis spectrophotometer (Molecular Devices).
Cartilage-specific gene expression was measured from hBM-MSCs by quantitative reverse transcription–polymerase chain reaction (qRT-PCR). Total RNA from the collected hBM-MSC samples was extracted using the RNeasy Mini Kit (Qiagen, Valencia CA). The predesigned human aggrecan and 18S TaqMan primers were purchased from Applied Biosystems. Human sox-9 custom forward (TGACCTATCCAAGCGCATTACCCA) and reverse (ATCATCCTCCACGCTTGCTCTGAA) primers as well as FAM-labeled probe (AGGCCAACCTTGGCTAAATGGAGCA) were purchased from Integrated DNA Technologies (IDT, Coralville, IA). All samples contained 1 μL of extracted RNA in a total volume of 10 μL using a commercially available pre-prepared 2×real-time TaqMan PCR master mix (Applied Biosystems). All qRT-PCRs were performed with an ABI PRISM 7700 Sequence Detection System (Applied Biosystems) according to the following program: 12 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Relative expression levels normalized to 18S were calculated using the 2−ΔCt method.
Statistical analysis
Data are expressed as mean±standard error of mean. Statistical analysis was performed using independent samples t-test for comparison between two samples (Fig. 5), and one-way independent analysis of variance or one-way repeated measures analysis of variance with post hoc Turkey for multiple comparisons (Figs. 1–4) using Statistical Package for Social Studies (SPSS) 17.0 for Windows (IBM, Chicago, IL). The significance level was set as p<0.05.
Results
Clotting time
After activation of the different fibrinogen dilutions with thrombin-CaCl2, there was an immediate change in the turbidity of the solution. The clotting time, or the time of maximal turbidity, was dependent on the fibrinogen concentration (p<0.001). Absorbance of the fibrin gel at 550 nm increased with time for all fibrinogen dilutions until it reached a plateau. The clotting time as a function of fibrinogen concentration is displayed in Figure 1. Clotting time of the 100% FG was significantly higher (p<0.05) than the diluted (25%, 50%, and 75%) FG scaffolds. Although there was no significant difference in clotting time between the dilute FG scaffolds, there was an increasing trend in clotting time with increasing FG concentrations.
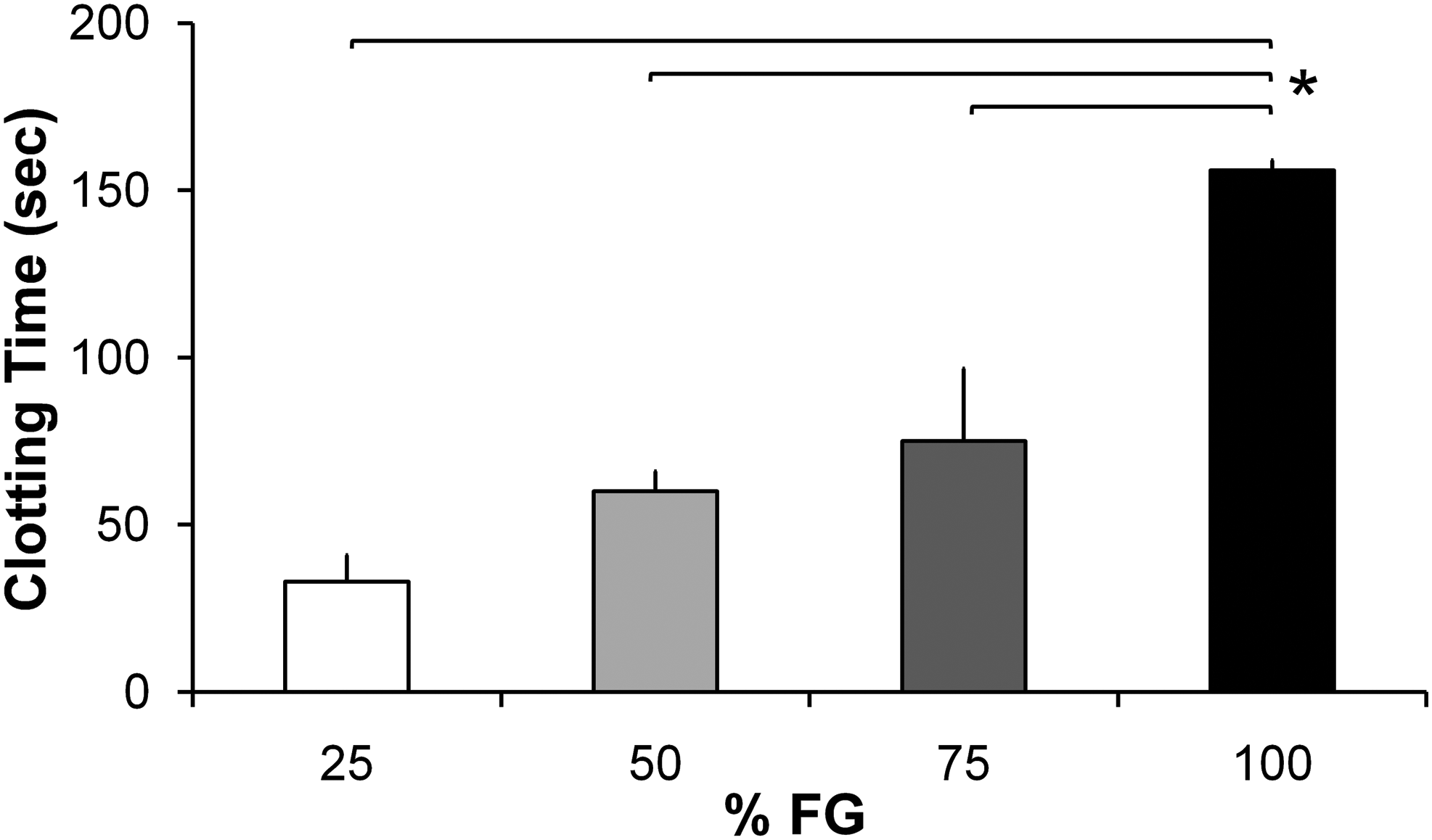
Clotting time showed an increasing trend with increasing fibrin glue (FG) concentrations. Clotting time of 100% FG was statistically higher than diluted FGs. Bars represent mean±standard error of mean; n=3. *p<0.05 compared with 100% FG.
SEM analysis of the FG hydrogel microstructures
SEM analysis of pore size diameter of the FG scaffolds showed that all four scaffolds had pore size diameters ranging 50–1200 nm. However, there was a denser, heterogeneous distribution of pores in the 100% FG (Fig. 2D) compared with the diluted FG hydrogels, which displayed a more homogenous and larger pore size diameters (Fig. 2A–C). The pore size of the 100% FG was significantly smaller in diameter (p<0.05) than those of the diluted FG scaffolds (Fig. 2E). The fiber thickness of the scaffolds, also assessed by SEM, increased with increasing FG concentrations (Fig. 2F). The increases in fiber thickness were significant between scaffolds with different fibrinogen concentrations (p<0.001).
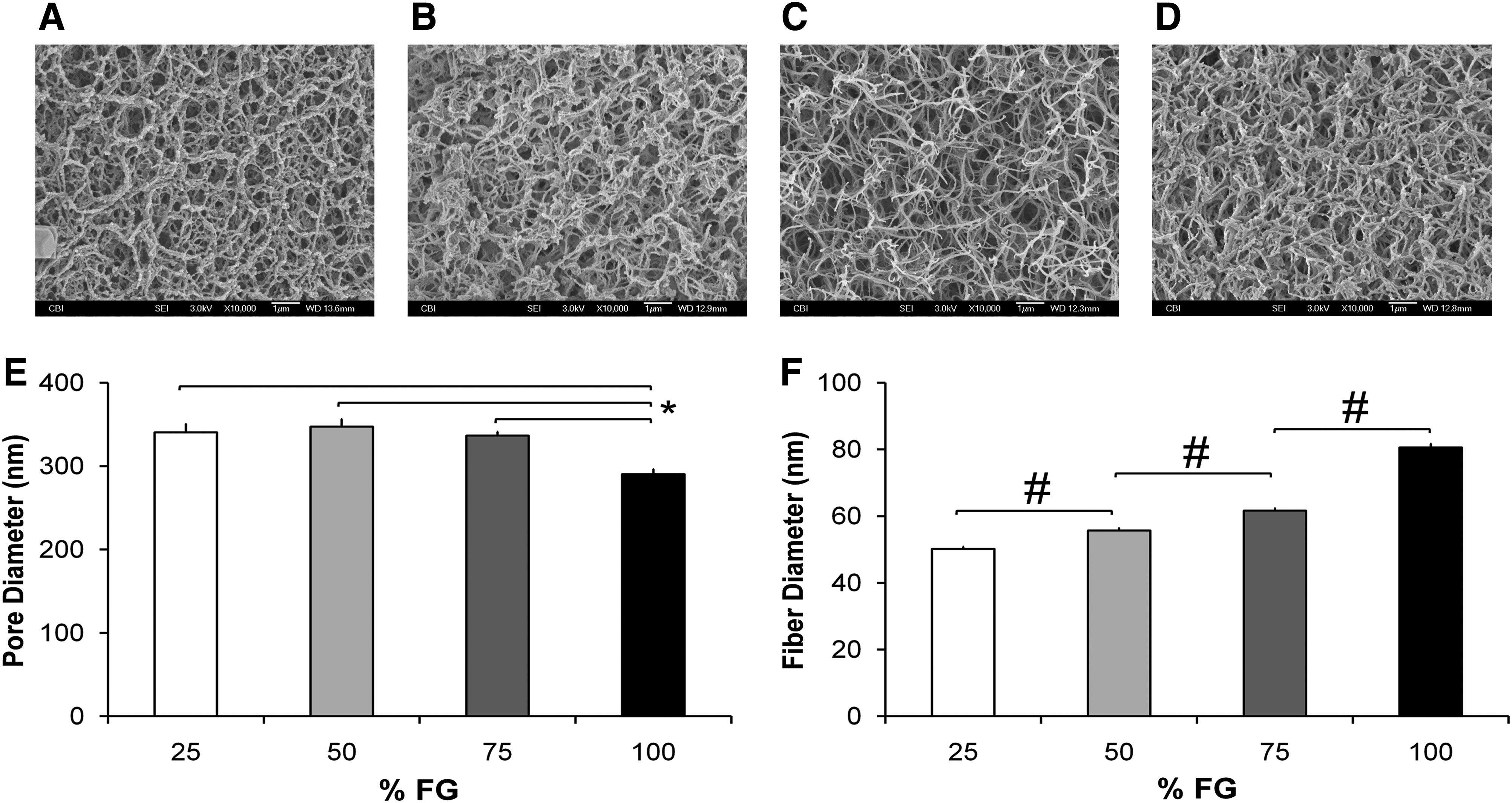
Scanning electron microscopy analyses of FG hydrogels.
In vitro dissolution of the FG hydrogels
Analysis of in vitro FG dissolution in PBS showed a biphasic pattern (Fig. 3A). Wet weight fraction result showed that the dissolution rate is significantly affected by different concentrations of FG scaffolds (p<0.001): 25% and 50% fibrinogen concentrations displayed a significantly higher rate of dissolution than 75% and 100% FG scaffolds. The first spike in dissolution occurred at day 2 for the 25% and 50% FG scaffolds, where the hydrogels lost ∼35% and 15% of their initial weight, respectively. This was followed by a second spike at day 8 when the hydrogels lost ∼80% of their initial weight. For the 75% and 100% FG scaffolds, the two spikes in dissolution occurred later at days 6 and 12, with loss of 20% and 90% of the initial weight for the respective time points. The pattern of protein release, observed by UV-vis spectroscopy (Fig. 3B), correlates well with the wet weight fraction finding, by being significantly affected by the percentage of fibrinogen (p<0.001). As well, the biphasic pattern of absorbance peaks that reflects most weight dissolution into the conditioned PBS can be noted.
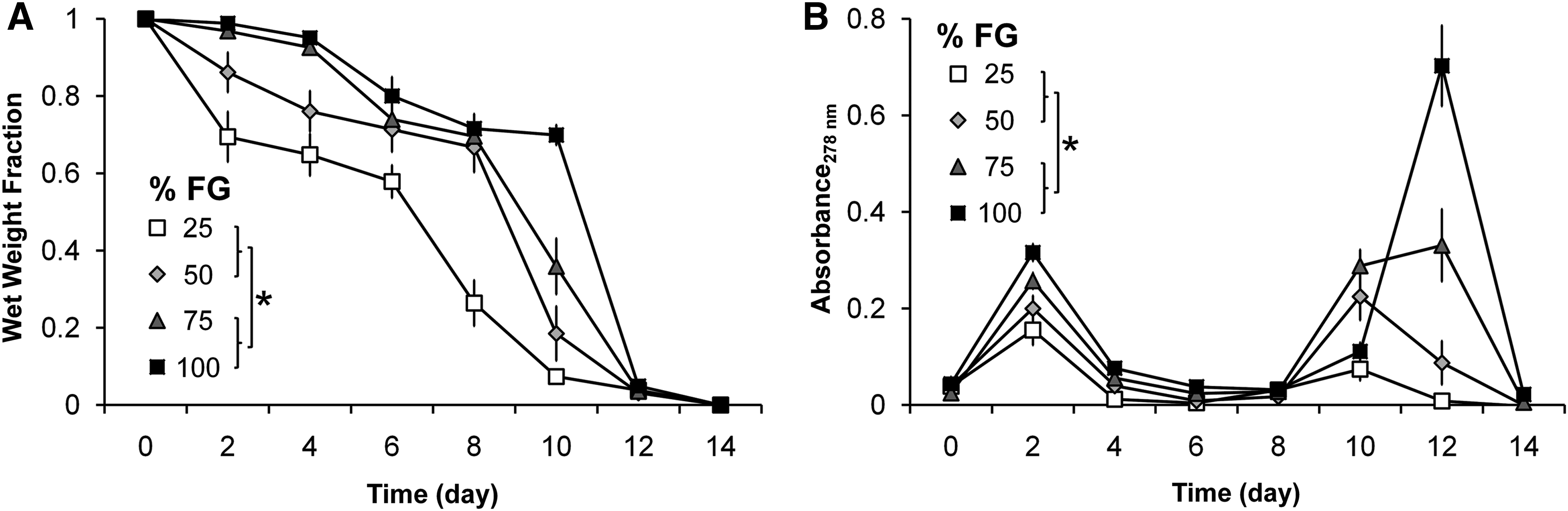
In vitro FG dissolution.
Quantification of the amount of released AAV2-CMV-GFP from FG hydrogels
The amount of AAV2-CMV-GFP released into the conditioned media from the AAV2-CMV-GFP-loaded FG scaffolds is significantly affected by different concentrations of FG scaffolds (p<0.001), and also displays a biphasic pattern (Fig. 4A). The spikes in the amount of AAV2-CMV-GFP released into the media occurred at day 2 and 9 for 25% and 50% FG scaffolds. In contrast, the spikes in the amount of AAV2-CMV-GFP released into the media occurred at day 2 and 15 for 75% and 100% FG scaffolds. Approximation of the area under the curves in Figure 4A suggests that the vector genomes of AAV2-CMV-GFP released into the media from 75% and 100% FG scaffolds were lower than that of 25% and 50% FG scaffolds.

In vitro characterization of adeno-associated viruses serotype 2 (AAV2)-CMV-GFP-loaded FG scaffolds.
Quantification of mammalian cell transduction efficiency of released AAV2-CMV-GFP
The percentage of GFP-positive cells from transduction of AAV2-CMV-GFP released into the conditioned media from the AAV2-CMV-GFP-loaded FG scaffolds is also significantly affected by different concentrations of FG scaffolds (p<0.001), and showed a biphasic pattern (Fig. 4B). This pattern is similar to those of the FG dissolution (Fig. 3) and AAV2-CMV-GFP release (Fig. 4A). FG scaffolds with 25% and 50% fibrinogen concentrations yielded significantly higher percentage of GFP-positive cells than the 75% and 100% FG scaffolds. There were spikes in the percentage of GFP-positive cells at days 1 and 8 for all FG scaffolds, which correlate with the biphasic release pattern of high amounts of AAV2-CMV-GFP at these time points (Fig. 4A). Although 75% and 100% FG scaffolds showed similar biphasic spikes in the percentage of GFP-positive cells, the response was significantly lower compared with 25% and 50% FG scaffolds. Photomicrographs of GFP-expressing cells taken daily confirmed quantitative findings by flow cytometry (Fig. 4C–F).
Quantification and functional analysis of TGF-β1 expression from hBM-MSCs transduced with released AAV2-CMV-TGF-β1
The amount of TGF-β1 synthesized by hBM-MSCs and released into the conditioned medium is significantly affected by different concentrations of FG scaffolds (Fig. 5A, B). Cells incubated with 50% FG loaded with AAV2-CMV-TGF-β1 made 2.5-fold more active-TGF-β1 than those incubated with 100% FG loaded with AAV2-CMV-TGF-β1, (p=0.017, Fig. 5A). As well, the conditioned medium from the hBM-MSC culture that was incubated with the 50% FG loaded with AAV2-CMV-TGF-β1 resulted in greater inhibition of CCL-64 cell proliferation (Fig. 5B), measured via mitochondrial activity (p=0.002). The different concentrations of AAV2-CMV-TGF-β1-loaded FG scaffolds also affected the cartilage-specific gene expression of the hBM-MSCs (Fig. 5C). Human sox-9 and aggrecan mRNA expressions were significantly higher for 50% FG versus 100% FG (p=0.049 and p=0.009, respectively). All the results were normalized against values from AAV2-CMV-GFP-loaded FG scaffolds of the same percentage fibrinogen concentration to account for the effects of viral transduction, FG, and endogenous TGF-β1 production by hBM-MSCs.
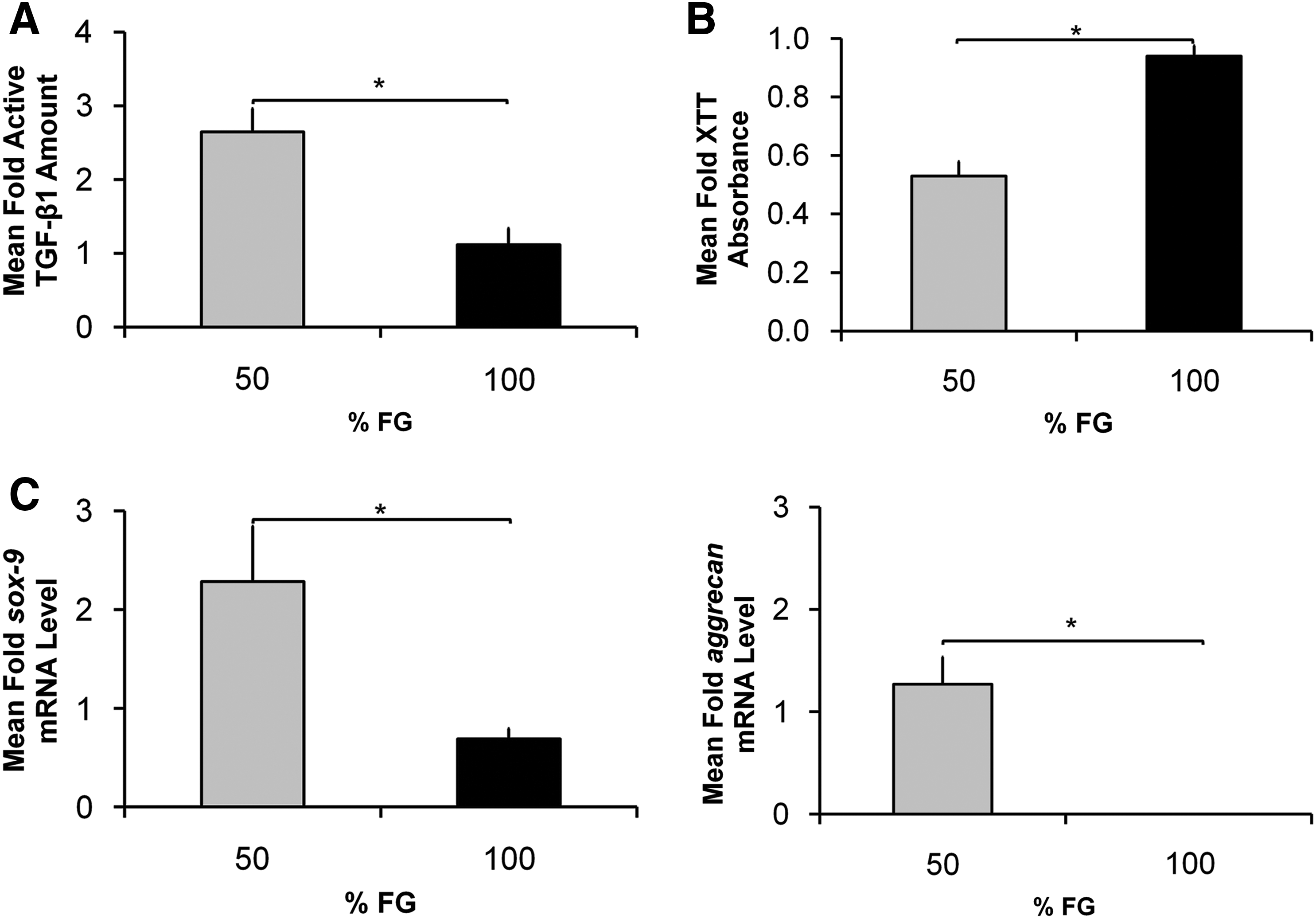
In vitro characterization of AAV2-CMV-TGF-β1-loaded FG scaffolds:
Discussion
Tissue engineering scaffolds allow for the delivery of cells or diffusible factors that can act locally to promote tissue regeneration.38,39 The combination of gene therapy and tissue engineering offers the potential to direct progenitor cells' proliferation and differentiation into functional tissue replacement. 15 We have previously shown that hMSCs transduced with AAV-TGF-β1 improved the in vivo cartilage repair when the ex vivo transduced cells were implanted into osteochondral defects of athymic rats. 12 However, in an effort to move beyond ex vivo gene transfer, the current focus of the project is on in situ delivery of therapeutic transgenes, such as TGF-β1, directly to host MSCs residing in cartilage wounds using biodegradable scaffolds. This strategy can be thought of as “augmenting” microfracture, which is an accepted human clinical standard for treatment of full-thickness cartilage defects. 40 The goal of this strategy is to induce localized exogenous transgene expression in repair/progenitor cells infiltrating into the scaffold for sustained release of these factors at the site of injury. Strategies that combine scaffolds, drug delivery technology, and gene therapy have the potential to provide more effective tissue regeneration with significant improvements on safety and cost issues compared to currently available therapies.
Although numerous novel synthetic and natural polymers have been investigated for cartilage tissue engineering studies,21,23,24 usage of an FDA-approved and autologous biomaterial as an AAV2-releasing scaffold can accelerate the transition of basic science research into human clinical studies. FG is a biomaterial widely used in clinical practice and tissue engineering applications. As well, dilute FG has been successfully used to treat cartilage wounds in a pilot human clinical study. 22 However, studies on its use as a delivery scaffold for gene vectors are limited, and only adenovirus delivery has been investigated for the delivery of viral vectors. 26 FG has been investigated as a delivery scaffold for controlled release of adenovirus in a rabbit ear ulcer model 29 and for perivenous adventitial gene transfer, 30 and was shown to provide enhanced in vivo transgene expression over adenovirus transduction without FG scaffold. For cartilage tissue engineering purposes, FG scaffolds with nonviral copolymer-protected polyethylenimine-DNA vectors achieved sustained release of the gene over 20-day period and successful in vitro transfection of human keratinocytes and rabbit articular chondrocytes. 28
Nonetheless, FG formulation for AAV2 delivery has not been optimized. The fibrinogen/thrombin concentration in the FG preparation has been shown to affect the structural properties of the scaffold, and in turn influence the proliferation rate and morphology of hMSCs. 27 Therefore, alterations in FG scaffold microstructure can influence its function as a delivery scaffold for AAV2. The aim of the current study was to assess, in vitro, the structural characteristics of FG hydrogels containing varying fibrinogen concentrations, their AAV2-particles release kinetics, and subsequent transgene expression in target cells for articular cartilage tissue engineering.
Undiluted 100% FG scaffold had longer gelation time compared with diluted scaffolds, which may be explained by data from SEM analysis, which showed that 100% FG scaffolds formed a denser scaffold with smaller pore sizes and thicker fibrin fibers than diluted FG scaffolds. In contrast, diluted FG scaffolds had a more open fibrin network, which was reflected by their shorter clotting times. Consistent with another finding, 27 diluted FGs had a more homogenous structure than undiluted 100% scaffold.
FG scaffolds dissolved over time in PBS, which was shown by the decrease in the wet weight over time and by the change in absorbance measurements of the phenyl group of the fibrinopeptide over time. Dissolution of the FG scaffolds coincided with the amount of AAV2-CMV-GFP released into the conditioned media. In addition, both the dissolution of the scaffold and the amount of AAV2-CMV-GFP released into the media occurred in a biphasic burst fashion, consistent with a typical hydrogel bulk-degradation pattern. The burst release pattern of AAV2-CMV-GFP was similar to the percentage of GFP-positive HEK-293 cells. There were more GFP-positive cells when a greater amount of AAV2-CMV-GFP was released into the media. Both the amount of AAV2-CMV-GFP released and the number of GFP-positive cells is higher in wells with 25% and 50% FG scaffolds than in those with 75% and 100% FG scaffolds. In addition to dissolution pattern of the FG scaffold, the more open fibrin network of the 25% and 50% FG scaffolds may contribute to greater release of AAV2-CMV-GFP and the higher percentage of GFP-positive cells compared with 75% and 50% FG scaffolds. These in vitro studies showed that commercial strength 100% as well as 75% FGs are less efficient at releasing AAV2 particles, and that dilute 25% and 50% FGs are more effective. Viral particles released from dilute FGs retained the ability to transduce HEK-293 cells up to 17 days in vitro.
In contrast to our findings with AAV2, a previous study investigating different concentrations of FG scaffold loaded with adenovirus had achieved optimal viral releasing properties with the higher fibrinogen concentration. 41 However, this was observed with adenovirus vector and with a diluted thrombin concentration. Compared with adenovirus, AAV is much smaller and has the advantages of improved safety profile and longer transgene persistence. 14 In addition, lower thrombin concentration decreases the rate of fibrin cross-linking. 26 Since in vivo FG decomposition will be accelerated via plasmin-mediated fibrin degradation in addition to diffusion-mediated dissolution, FGs prepared with undiluted thrombin are more likely to persist and release AAV2 particles longer for animal or human applications.
Although many more dilutions of FG at smaller increments could be studied to further assess the AAV2 release kinetics, especially with lower concentrations of FG, the four dilutions were chosen to support the goal of evaluating FG concentrations potentially useful for in vivo clinical applications. As well, FG dilutions of 25% approach the lower limit to achieve a clinically functional scaffold structure. 41 For this reason, 50% and 100% FGs were chosen for subsequent studies with AAV2-CMV-TGF-β1.
As our ultimate goal is to develop a localized AAV2 delivery system for cartilage tissue engineering, we further explored the concentrations of 50% and 100% FGs for delivery of AAV2 encoding for a therapeutic gene useful for cartilage regeneration. Transforming growth factor-β1 (TGF-β1) has long been shown to consistently induce BM-MSC chondrogenesis in vitro.34,42 We also have previously shown that implantation of AAV2-CMV-TGF-β1-transduced hMSCs into osteochondral defects has improved cartilage repair in vivo. 12 Therefore, we compared AAV2-CMV-TGF-β1 delivery from the 50% and 100% FGs to hBM-MSCs in vitro. The studies on FG scaffold delivery of therapeutic-AAVs, versus reporter gene-AAVs, especially for their chondrogenic effects, have been limited to date. 26 hBM-MSCs cultured with 50% FG-AAV2-CMV-TGF-β1 construct had higher concentration of active TGF-β1, which resulted in greater inhibition of CCL-64 cell proliferation, and higher cartilage-specific gene expression levels in transduced hBM-MSCs.
In summary, we have demonstrated that varying the fibrinogen concentrations in FG constructs changes the structural and functional characteristics of the scaffold for gene delivery. Diluting the fibrinogen concentration yields a scaffold with a network of larger pores and thinner fibers, which reduces entrapment and subsequently enhances the release of AAV2 particles. Lower concentration FG scaffolds have also been shown to promote greater proliferation for hMSCs, which are the principal cell types recruited for in situ chondrogenesis. 27 Therefore, implantation of diluted FG scaffolds containing bioactive AAV2 vectors have the potential to improve articular cartilage regeneration and be translated for clinical cartilage tissue engineering. Further in vivo studies are planned using small and large animal models to validate the enhanced repair potential of diluted FG scaffolds with therapeutic AAV2-TGF-β1 for localized in situ cartilage tissue engineering before translation into the clinical setting.
Footnotes
Acknowledgments
We thank Kimberlee K. Suter and Judong Lee for technical assistance. Funding for this work was provided by the National Institutes of Health RO1 AR051963 (C.R.C.) and RC2 AR058929 (C.R.C.). HHL receives partial support from T32 EB001026 (R.P.B.).
Disclosure Statement
No competing financial interests exist.