
Abstract
To adequately and permanently restore organ function after grafting, human tissue-engineered skin substitutes (TESs) must ultimately contain and preserve functional epithelial stem cells (SCs). It is therefore essential that a maximum of SCs be preserved during each in vitro step leading to the production of TESs such as the culture process and the elaboration of a skin cell bank by cryopreservation. To investigate the presence and functionality of epithelial SCs within the human TESs made by the self-assembly approach, slow-cycling cells were identified using 5′-bromo-2′-deoxyuridine (BrdU) in the three-dimensional construct. A subset of basal epithelial cells retained the BrdU label and was positive for the SC-associated marker keratin 19 within TESs after a chase of 21 days in culture post-BrdU labeling. Moreover, keratinocytes harvested from TESs gave rise to SC-like colonies in secondary monolayer subcultures, indicating that SCs were preserved within TESs. To evaluate the effect of cryopreservation with dimethyl sulfoxide and storage in liquid nitrogen on SCs, human epithelial cells were extracted from skin samples, amplified in culture, and used to produce TESs, before cryopreservation as well as after thawing. We found that the proportion and the growth potential of epithelial SCs in monolayer culture and in TESs remained constant before and after cryopreservation. Further, the functionality of these substitutes was demonstrated by successfully grafting human TESs on athymic mice for 6 months. We conclude that human epithelial skin SCs are adequately preserved upon human tissue reconstruction. Thus, these TESs produced by the self-assembly approach are suitable for clinical applications.
Introduction
The homeostasis of a self-renewing tissue is dependent on stem cells (SCs), which can become mitotically active to regenerate the epidermis or after injury. 7 Since their presence is required to ensure the long-term renewal and wound healing of the epithelium, SCs are at the forefront in tissue engineering of self-renewing tissues such as skin. To adequately and permanently restore organ function after grafting, tissue-engineered skin substitutes (TESs) must ultimately contain and preserve functional epithelial SCs. They reside in specific locations, referred to as niches, which allow the maintenance of their stemness.8–10 For permanent skin replacement, the culture methods leading to the production of living substitutes must preserve SCs, and the tridimensional environment must recapitulate the niche to be able to sustain their potency. Otherwise, the epithelium will be lost after grafting as a consequence of proliferation arrest, terminal differentiation, and desquamation of all the epithelial cells. Keratinocytes can be maintained and propagated in vitro under adequate culture conditions.2,3,11,12 The presence of epithelial SCs that form holoclones allows long-term proliferation.12–14 Compared to newborn skin, cultures from adult donors have a lower expansion potential and contain less SCs. 15 Therefore, it is important to carefully validate that, especially for adult donors, SCs persist within human skin substitutes dedicated to clinical purposes.
Skin epithelial SCs, known as label-retaining cells (LRCs) based on their slow-cycling nature, have been localized in the basal layer of the epidermis and in the bulge area of hair follicles.16–18 The identification of molecular markers for epidermal SCs is still a matter of debate. However, the combination of molecular markers with the analysis of clonal growth potential and slow-cycling properties is useful for SC studies.19–21
It is acknowledged that the three-dimensional environment (niche) contributes to tissue organization and functionality.22,23 The importance of an appropriate underlying dermal component is probably required for epithelial SC maintenance through the niche.24–27 Many bilayered skin substitutes comprising a dermal substrate in addition to the epidermis have been proposed, but preservation of SCs varies between models.15,28
Another aspect to consider in the development of tissue replacement strategies for the treatment of large cutaneous defects is the limited tissue availability. Conventional keratinocytes cultured on plastic substrate with an irradiated fibroblast feeder layer is an effective technique to expand keratinocytes in vitro from a skin biopsy of a few square centimeters in size. Under those conditions, the feeder layer extends the lifetime of cultured epithelial cells that are maintained in a low differentiation status.2,11,29–31 Human fibroblasts can also be expanded in vitro from a small skin biopsy. 32 Combined with the self-assembly approach of tissue engineering,33–36 these culture methods allow the production of sufficient TESs to cover the entire adult body surface from a small biopsy of intact skin harvested. This approach of tissue engineering allows the fabrication of completely autologous TESs without the addition of any exogenous material, a criterion that is not to be neglected when the goal of the product is to allow the permanent integration of the tissue in the host.
The establishment of cryopreserved cell banks has been a significant progress for both experimental and clinical applications. 37 Indeed, cryopreserved allogenic or autologous epithelial cells may be thawed and cultured in vitro to produce living substitutes suitable for grafting patients suffering from chronic or extended skin wounds, such as ulcers and severe burns.2,34–37 Cryopreservation methods are routinely employed to establish skin cell banks before their utilization in monolayer culture or to produce engineered tissues in vitro. The persistence of SCs in epithelial cell cultures after thawing and culturing on plastic for the production of autologous cultured epithelium or allogeneic skin construct has been reported.1,38,39 However, little is known about the effect of cryopreservation on the epithelial SC content of tridimensional tissue-engineered skin constructs produced from adult donors for autologous grafting.
The objective of this study was to evaluate whether epithelial SCs are adequately preserved upon tissue reconstruction with the self-assembly approach of tissue engineering and if the use of cryopreserved epithelial cell bank for tissue reconstruction allows the maintenance of the SC content within these constructs. Using a combination of molecular and functional analyses, we showed that the TESs elaborated by the self-assembly approach of tissue engineering provide a microenvironment supporting epithelial SCs and represent a high-quality TES model suitable for in vitro and preclinical studies as well as for clinical applications.
Materials and Methods
This study was conducted in accordance with our institution's guidelines and the Declaration of Helsinki. The protocols were approved by the institutional ethics review board for the Protection of Human Subjects of the Centre hospitalier affilié universitaire de Québec.
Tissue and cell culture
Human cells were obtained from healthy skin specimens originating from surgical procedures. Keratinocytes were isolated from skin specimens harvested from different anatomical regions [scalp, chest, and periauricular (preauricular and postauricular)] from adult donors between 15 and 68 years of age, as well as from newborn foreskin from donors between 12 h and 17 days, by the two-step thermolysin and trypsin method previously described.1,40 The strains used for each experiment and figures are reported in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/tea). Briefly, for keratinocyte isolation, skin fragments were digested with 500 μg/mL thermolysin (Sigma Chemicals, St-Louis, MO) in the HEPES buffer [10 mM 4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid (MP Biomedicals Inc., Montreal, QC, Canada), 6.7 mM potassium chloride, 142 mM sodium chloride, and 1 mM calcium chloride] overnight at 4°C. Then, the dermis was separated from the epidermis with forceps. Keratinocytes were dissociated from the epidermis in a trypsin/ethylenediaminetetraacetic acid (EDTA) solution [0.05% trypsin 1–500 (Intergen, Toronto, ON, Canada) and 0.01% EDTA/disodium salt prepared in phosphate-buffered saline (PBS)] for 15 min at 37°C. Keratinocytes were then collected by centrifugation and were either analyzed directly or plated for primary cell culture (P0) as follows: 26,666 keratinocytes/cm2 were seeded on a lethally irradiated S3T3 fibroblast (i3T3) feeder layer (20,000 cells/cm2) in a keratinocyte culture medium consisting of a 3:1 mixture of the Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 medium (Invitrogen, Burlington, ON, Canada) supplemented with 5% Fetal Clone II serum (HyClone, Logan, UT), insulin, hydrocortisone, cholera toxin, epidermal growth factor (EGF), and antibiotics, and cultured as previously described.8,33 Trypsinized cells at P0 were either seeded for culture [passage (P) 1 and P2] or cryopreserved (see the Cryopreservation and thawing of epithelial cells section) for subsequent thawing and culture.
For the production of TESs, human dermal fibroblasts were obtained from three different (18, 21, and 23 years old) adult healthy skin donors and cultured in a fibroblast medium [DMEM supplemented with 10% fetal bovine serum (FBS; Hyclone)], as described.36,41
Cryopreservation and thawing of epithelial cells
Cells were cryopreserved according to the following slow-rate cooling method: primary cultures of freshly extracted keratinocytes were trypsinized, counted, and centrifugated. Cell pellets were kept on ice until resuspension at a density of 1×106 or 2×106 cells/mL in a cold (4°C) freezing medium consisting of 90% fetal calf serum and 10% dimethyl sulfoxide (DMSO; Sigma Chemicals) and distributed in cryogenic vials (Nalgen Labware, Rochester, NY). Cryogenic vials were then placed in a freezing container (Nalgene Labware) that has been filled with 99% ethanol and precooled at 4°C, and placed at −80°C for 24 h to allow a freezing rate of −1°C/min. Cells were stored in liquid nitrogen until use. When needed (one month to one year later), cryogenic vials were rapidly thawed in a 37°C water bath, and cells were suspended in a cooled keratinocyte medium. Cells were then centrifuged, washed once in a 37°C keratinocyte medium, centrifuged, and cultured as described previously.
TES production and identification of LRCs
TESs were produced as described elsewhere.34,36 Briefly, 8,000 human fibroblasts/cm2 at P2 to P5 were cultured for 21–28 days in a fibroblast medium supplemented with 50 μg/mL ascorbic acid (Sigma Chemicals). 42 After peeling off from the culture flask, two (for in vitro experiments) or three (for in vivo experiments) fibroblast sheets were superimposed to produce the tissue-engineered dermis. One week later, cultured keratinocytes at P2 that were either never cryopreserved or previously cryopreserved and thawed, were seeded at a density of 200,000 cells/cm2 on the tissue-engineered dermis and cultured in a keratinocyte medium supplemented with 50 μg/mL ascorbic acid. After 7 days of culture submerged in the medium, TESs were raised at the air–liquid interface to allow adequate epidermal differentiation, and cultured in the same medium without EGF. For in vivo experiments, TESs were cultured 8 days submerged in the medium before grafting on athymic mice. In some experiments, TESs were incubated in vitro with 5′-bromo-2′-deoxyuridine (BrdU; Sigma Chemicals) to identify slow-cycling cells. Two pulse-labeling strategies were assessed. Ten micromoles of BrdU was either added to the culture medium of TESs for the first 7 days of culture under submerged condition or between the 10th and the 17th day of culture at the air–liquid interface. The surface area of each TESs was 5 cm2, except when stated otherwise. Biopsies were taken or keratinocytes were harvested (see the Keratinocytes isolation from TES section) 1 day or 21 days after the end of the BrdU pulse. Controls consisted of TESs cultured in parallel for the same duration without BrdU.
Keratinocyte isolation from TESs
TES fragments were digested with 250 μg/mL thermolysin in the HEPES buffer for 3 h at 4°C. Then, the dermis was separated from the epidermis with forceps. Keratinocytes were dissociated from the epidermis with the trypsin/EDTA solution for 20 min at 37°C. Keratinocytes harvested from 4 to 12 TESs were pooled. Cells were either seeded at 3,000–60,000 cells/cm2 on an iS3T3 feeder layer and cultured as described above, or fixed in 70% cold ethanol for fluorescence-activated cell-sorting (FACS) analysis (see the Flow cytometry analysis section).
Population doublings
Cumulative population doubling is the sum of population doublings from P0 until a given passage. Population doublings were calculated at each passage using the number of cells obtained at the end of culture and the number of cells seeded. It was calculated as follows:
Histological and immunofluorescence analyses
For histological analysis, biopsies were fixed in Histochoice (Amresco, Solon, OH) and embedded in paraffin. Microtome sections (5-μm thick) were stained with Masson's trichrome 43 using Weigert's hematoxylin, fuchsin-ponceau, and aniline blue stains. For immunodetection, biopsies were embedded in Tissue-Tek OCT Compound (Sakura Finetek, Torrance, CA) and frozen in liquid nitrogen.
Immunofluorescence assays were performed on 5-μm-thick cryosections permeabilized with acetone (10 min at −20°C) as previously described. 18 The cell nuclei were counterstained with Hoechst reagent 33258 (Sigma Chemicals). The antibodies used were as follows: guinea pig polyclonal anti-Keratin (K) 15 clone gp15.1 (ARP, Belmont, MA), mouse monoclonal anti-K10 clone RKSE60 (Cedarlane, Burlington, ON, Canada), anti-human filaggrin (BTI, Stoughton, MA), anti-human involucrin (Sigma Chemicals), anti-α3-integrin clone HB-8530 (ATCC, Manassas, VA) conjugated to Alexa fluor 488 (Invitrogen), anti-laminin-5 (α3-subunit) conjugated to fluorescein isothiocyanate (FITC) (a gift from P. Rousselle, IBCP, Lyon, France), anti-human Ki67 (BD Biosciences, Mississauga, ON, Canada), anti-human K19 clone A53-B/A2 44 (gift from U. Karsten, Institute of Biological Sciences, University of Rostock, Germany) conjugated to Alexafluor 488 (Invitrogen), and anti-human collagen IV (a gift from J.A. Grimaud, Pasteur Institute, Lyon, France). The following secondary antibodies were used: rhodamine-conjugated goat anti-mouse (Millipore, Billerica, MA), Alexa-488-conjugated rabbit anti-mouse (Invitrogen), and DTAF-conjugated goat anti-guinea pig (Accurate Chemicals & Scientific Corporation, Westbury, NY). For immunofluorescence on TESs grafted on nude mice, cryosections were sequentially fixed with formaldehyde (3.7%, 10 min) and methanol (10 min at −20°C) for HLA [mouse monoclonal anti-HLA-ABC Common (Clone 22.63.4); Chemicon, Temecula, CA] conjugated to FITC or fixed only with acetone (10 min at −20°C) for human K19 and human filaggrin detection. The Vector® M.O.M. Basic Kit (ID Labs, London, Canada) was used to reduce nonspecific staining arising from endogenous immunoglobulins in the tissue.
To detect both BrdU and K19, cryosections were subsequently incubated in formaldehyde (3.7%, 10 min), in cold methanol (100%, −20°C, 10 min), and in sodium hydroxide (0.07 N, 15 s). Then, the tissue sections were incubated with a mouse monoclonal anti-BrdU antibody (BD Biosciences). After PBS washes, tissue sections were incubated with a goat anti-mouse rhodamine-conjugated secondary antibody (Millipore). The mouse monoclonal anti-human K19 antibody conjugated to Alexa fluor 488 (Invitrogen) was subsequently added. Sections were processed as for immunofluorescence assays.
For indirect immunofluorescence, negative controls consisted in the omission of the primary antibody during the labeling reaction were used, and while for direct labeling, appropriate isotypic controls were used.
Tissue sections were observed under a Nikon Eclipse E600 microscope (Nikon, Montreal, QC, Canada) or a Zeiss Axio Imager (Zeiss, Toronto, ON, Canada) and photographed with a CoolSNAP, Sensys, or Axiocam digital camera. Images were processed with Adobe Photoshop 9.0 software (Adobe System, San Jose, CA).
Flow cytometry analysis
Flow cytometry analyses were carried out as previously reported. 18 For detection of K19, cells were washed in PBS containing 1% bovine serum albumin (BSA; Sigma Chemicals) and incubated with anti-K19 conjugated to Alexa fluor 488 or its isotype control (BD Biosciences). For the detection of BrdU-labeled cells, fixed cells were washed in PBS–1% BSA and permeabilized with 0.2 mg pepsin (Sigma Chemicals) per mL of 2 N HCL. After 30 min, the pepsin enzymatic reaction was stopped with 0.1 M Borax (Sigma Chemicals) at pH 8.5. Cells were incubated with the anti-BrdU antibody conjugated to phycoerythrine (PE; BD Biosciences) or its isotypic control (BD Biosciences). For double-labeling experiments, cells were first incubated with the mouse anti-K19 antibody conjugated to Alexa fluor 488 or its isotype control. After PBS–1% BSA containing 0.5% Tween-20 (Biorad, Hercules, CA) washes, cells were incubated with the anti-BrdU antibody conjugated to PE or its isotypic control (BD Biosciences).
Samples were analyzed with a fluorescence-activated cell sorter (BD Biosciences), and a number of 10,000 (for simple labeling) or 15,000 (for double labeling of K19 and BrdU) events were acquired for each sample. The cell population was gated to exclude false-positive differentiated cells and cell debris as previously described. 18 Each pool of cells was analyzed at least three times.
Colony-forming efficiency
Newborn and adult keratinocytes were seeded at 3,333 cells (P0) or at 800 cells (P1 and P2) in 60-mm culture Petri dishes and cultured for 12 days as previously described. 19 Cell colonies were fixed with a 3.7% buffered formaldehyde solution and stained with 1% rhodanile blue. Colonies distinguished by their diameter [>4 mm (holoclones), 2–4 mm, and <2 mm] were counted, and the percentage of holoclones was calculated as follows: number of colonies >4 mm formed divided by the total number of colonies formed multiplied by 100. 19 Five Petri dishes were seeded per condition.
TES grafting on nude mice
Twelve adult male athymic nu/nu mice (42 days old) purchased from Charles River Laboratories of Canada (Lasalle, Quebec) were maintained under sterile housing conditions. Forty-eight and 24 h before surgery, the mice were injected with ceftazidine (3 mg/mouse; Glaxo, Toronto, Canada) to prevent infections. Antibiotics [100 IU/mL penicillin G and 25 μg/mL gentamicin (Sigma, Mississauga, Canada)] were also added to the sterile drinking water. The TESs [produced with adult (six TESs) or newborn (six TESs) keratinocytes at P3 that had been cryopreserved] were grafted on the dorsal muscular bed as previously described. 41 The surface area of each TES was 3.1 cm2. Mice were inspected regularly for visual signs of necrosis. The graft take was assessed visually by an experienced surgeon. Four mice (two grafted with newborn and two with adult TESs) were killed 4 days, 21 days, and 6 months after grafting, and biopsies were harvested for histological and immunofluorescence analyses.
Statistical analysis
Data are represented as mean±SEM. For statistical comparison, the bilateral Mann–Whitney U-test for unpaired samples was used, while the bilateral Wilcoxon signed-rank or the bilateral Student's t-test was used for paired samples using GraphPad Prism4 software (GraphPad Software, Inc., San Diego, CA). The confidence interval was set at 95% (p≤0.05).
Results
Identification of BrdU-LRCs in TESs
Human TESs were produced with the commonly used newborn epithelial cells as well as with adult epithelial cells. Since these latter are not frequently reported, adult results are shown, and newborn are only added when different. Histological and immunohistological analyses of TESs at different stages of the TES production revealed a fully differentiated epidermis at 10 days of culture at the air–liquid interface. Indeed, the four layers of human epidermis (stratum basale, spinosum, granulosum, and corneum) were observed on TES histology (Fig. 1A, B). Basal cells were cuboidal (Fig. 1B) and expressed K15 (Fig. 2A, I), K14 (data not shown), and α3-integrin (Fig. 2B, J), whereas K10 expression was restricted to the suprabasal layers (Fig. 2C, K). The apparent number of basal layers that seems >1 (Fig. 2A–C, I–K) is due to the oblique angle of sectioning as reported previously (Fig. 2S). 45 Cornified envelope-associated proteins such as filaggrin (Fig. 2D, L), involucrin, and transglutaminase (data not shown) were expressed in the suprabasal layers. Laminin-5 and collagen IV (Fig. 2E, M, and data not shown), two basement membrane components, formed a continuous line at the dermoepidermal junction. The proliferation marker Ki67 (Fig. 2F, N) was also detected in the nucleus of many basal cells, and K19 was identified in a small subset of basal keratinocytes in TESs produced with keratinocytes from newborn as well as adult donors (Fig. 2G, H, O, and P). K19+ cells were more abundant in TESs made from newborn than adult human keratinocytes (Fig. 2G, O and Fig. 2H, P, respectively). Taken together, these results showed that the epidermis of the TESs shares many features with native epidermis after 10 days of culture at the air–liquid interface.
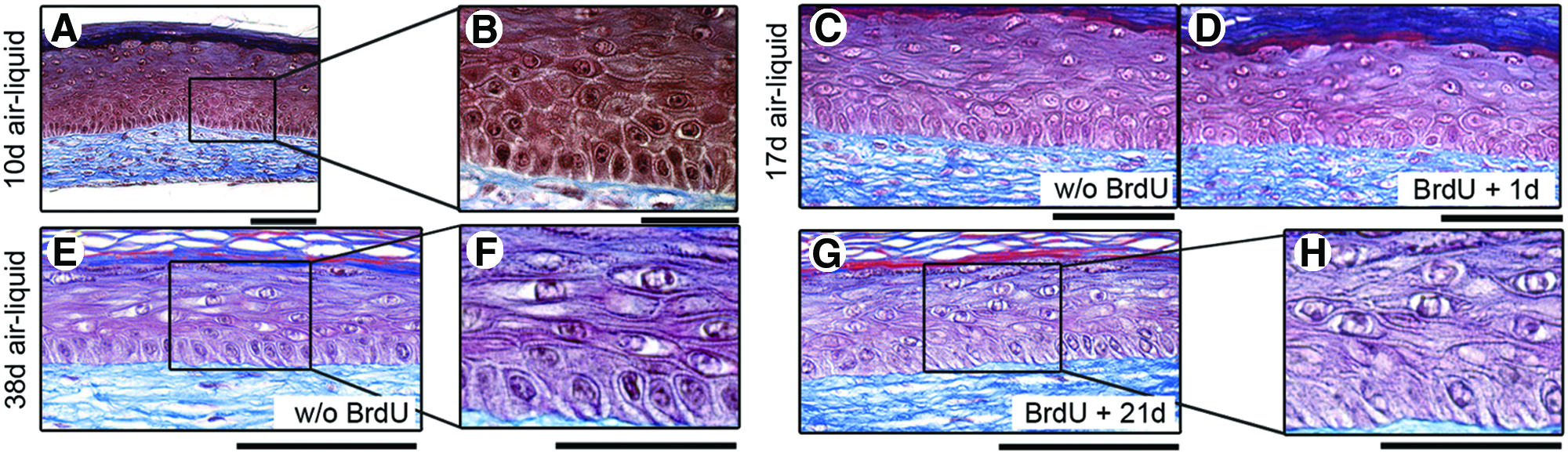
Histological analysis (Masson’ trichrome staining) of the tissue-engineered skin substitutes (TESs) treated or not with 5′-bromo-2′-deoxyuridine (BrdU).
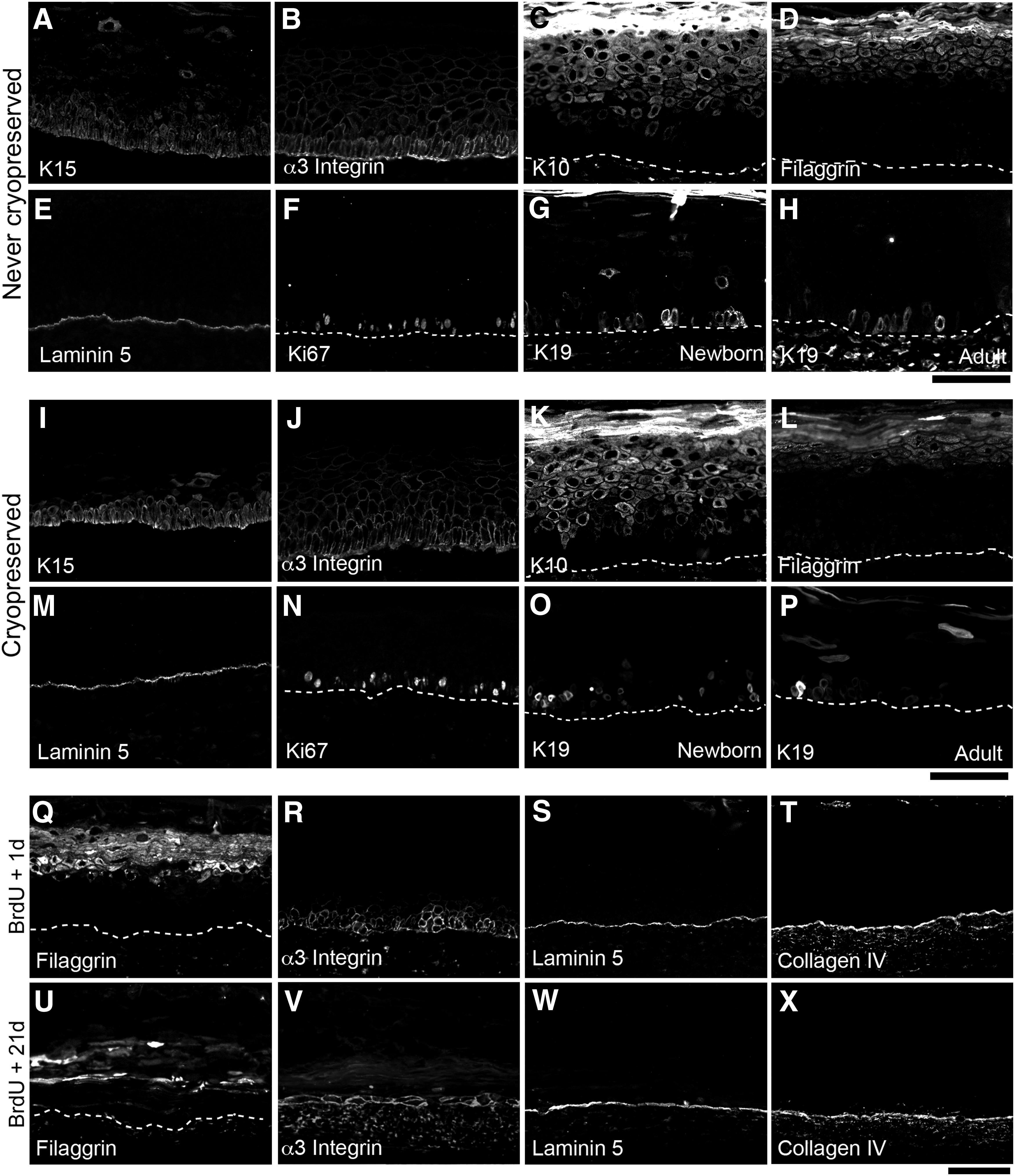
Immunohistological assessment of the TESs made with adult cells cryopreserved or not, and treated
The slow-cycling nature of skin SCs in an adequate undamaged microenvironment, also called a niche, is a functional criterion that led to their identification in skin.16–18,46 Aiming to track SCs in TESs, two BrdU-labeling strategies were investigated. The first consisted in the addition of BrdU to the culture medium of TESs during the submerged culture period for 7 days (Supplementary Fig. S1). One day after the removal of BrdU, most epithelial cells were BrdU labeled (data not shown), while after a chase period of 21 days without BrdU, all keratinocytes had lost their BrdU despite the maintenance of a well-stratified epidermis (data not shown). We interpreted that during the first days of submerged culture, SCs were in an active proliferation phase to reconstitute the epidermis and had diluted the BrdU. Thus, they could not be identified with BrdU labeling later on.
Considering that the previous results let believe that the homeostasis of the epidermis is reached at the 10th day of culture at the air–liquid interface (Fig. 1; see also Dubé et al. 47 for numerous time points of TES maturation), the second BrdU-labeling strategy in which BrdU was added in the culture medium of TESs from the 10th day to the 17th day of culture at the air–liquid interface was thus developed. One day or 21 days after the BrdU pulse, the epidermis of BrdU-labeled TESs (Fig. 1D, 1G, and high magnification 1H) maintained histological integrity similar to control TESs (without BrdU) (Fig. 1C, E, and F). The expected expression of filaggrin (Fig. 2Q, U), α3-integrin (Fig. 2R, V), laminin-5 (Fig. 2S, W), collagen IV (Fig. 2T, 2X), and K14 and K15 (data not shown) in adult (Fig. 2) and newborn (data not shown) TESs indicated that the BrdU treatment had not interfered with the normal differentiation program of keratinocytes within TESs.
Then, BrdU incorporation in TESs was evaluated by immunofluorescence. Immediately after the labeling period (one day later), a large number of basal and suprabasal keratinocytes were positive for BrdU in newborn (Fig. 3B) and adult (Fig. 3D) TESs. The number of BrdU-labeled cells greatly decreased after the 21-day chase period, but the persistence of LRCs was observed in the basal layer of newborn and adult TESs (Fig. 3F and H, respectively, arrows). The expression of K19 was further investigated within newborn and adult TESs. As expected, the number of K19+ cells was higher in newborn than in adult TESs. Many LRCs also expressed K19 (Fig. 3F, H, arrows). In some cases, unexpected LRC-K19-positive cells (BrdU+K19+) were present in suprabasal layers, suggesting that a loss of contact between SCs and the basement membrane had occurred. We also observed some LRCs negative for K19 (most of them were in the suprabasal layers, suggesting that they were differentiating) and alternatively, K19+ cells negative for BrdU. These latter could be SCs that were not dividing during the period of BrdU labeling. Collectively, these results indicate that the TESs made by the self-assembly approach provide a microenvironment supporting the establishment of SCs in a relatively quiescent state.

Immunostaining and quantification of the stem cells (SCs) in TESs.
Quantification of BrdU-LRCs and K19 expression by flow cytometry
To quantify the percentage of BrdU-labeled cells and K19+ cells in adult and newborn TESs, keratinocytes were harvested from TESs and processed for FACS analysis. Within the same age group (newborn or adult), no significant difference was observed in the percentage of K19+ cells between the cell suspension used to seed TESs, the cells not labeled with BrdU from TESs cultured 10 days at the air–liquid interface, and the cells from TESs 1 day and 21 days after the end of the BrdU-labeling period (Fig. 3Q). However, as expected, a higher proportion of K19+ cells was observed in newborn TESs compared to adult TESs having received or not the BrdU (Fig. 3Q). The percentage of BrdU-labeled cells was similar between newborn and adult TESs, either 1 day after the end of the labeling period or after the 21-day chase period (Fig. 3R). The double labeling of K19 and BrdU revealed that 10%±3% (n=5) of epidermal cells dissociated from newborn TESs expressed K19 and retained the BrdU label after the 21-day chase period (Fig. 3S). Comparing the percentage of LRCs (BrdU+ cells, Fig. 3R) with the percentage of K19+BrdU+ cells (Fig. 3S) within newborn and adult TESs after the 21-day chase period demonstrated that about half of LRCs expressed K19 when keratinocytes originated from newborns, and a third when they originated from adults. Although both cultured newborn keratinocytes and TESs engineered with newborn cells contained significantly more K19+ cells compared to those made from adult cells (Fig. 3Q), the number of LRCs (Fig. 3R) and the number of BrdU+K19+ cells (Fig. 3S) within TESs after 38 days of culture at the air–liquid interface were higher in newborn, but not statistically different compared with adult TESs.
Collectively, these results show that SCs, identified by their slow-cycling properties, were maintained in TESs produced with the self-assembly approach of tissue engineering. They also indicated that a significant percentage of these LRCs was also expressing the SC marker K19, yet not all K19+ keratinocytes were LRCs in TESs.
Keratinocytes harvested from TESs give rise to SC-like colonies in secondary monolayer subcultures
To assess the proliferation potential of keratinocytes after their culture in the tridimensional environment provided by the TESs, cells were dissociated from the epidermal portion of TESs at the end of the culture period. Isolated cells were then grown in monolayer culture with an i3T3 feeder layer. Under these conditions, keratinocytes dissociated from TESs that had been cultured at the air–liquid interface for a period of 10 days (Fig. 4A, B), 17 days (Fig. 4C, D, and I), or 38 days (Fig. 4E–H) formed holoclones, SC-like colonies (Fig. 4I). Moreover, keratinocytes harvested from newborn TESs cultured for 38 days at the air–liquid interface were passaged up to seven times for a total of more than 39 population doublings (Fig. 4J) and were still able to form colonies (Fig. 4F–H). These results confirm that epithelial cells with a high proliferative potential were preserved in TESs.
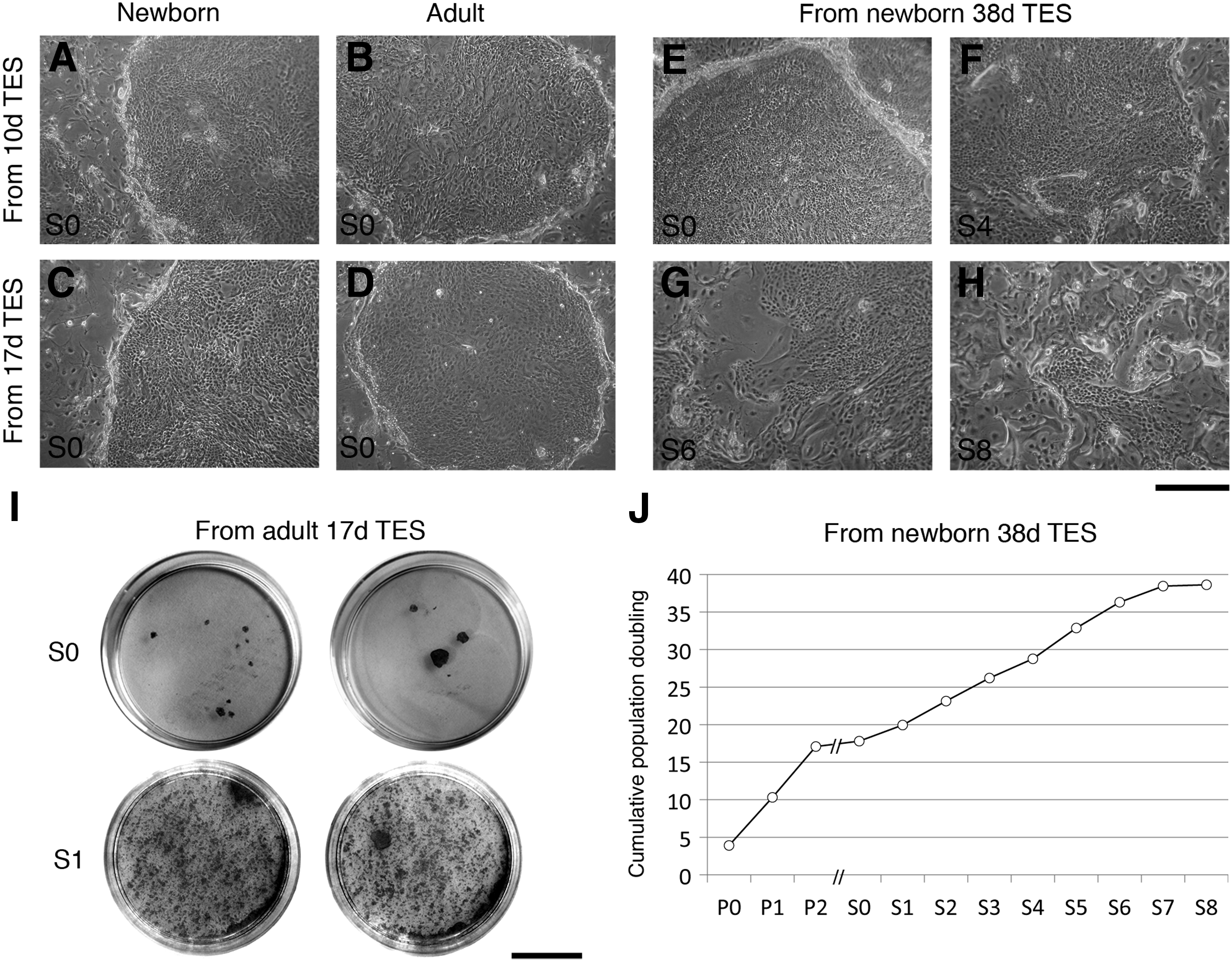
Morphological and proliferative analysis of TES-dissociated keratinocytes.
Preservation of epithelial SCs in monolayer culture after cryopreservation
To determine the influence of cryopreservation on SCs, the percentage of K19+ cells within keratinocyte populations harvested from newborn and from adult skin was determined by flow cytometry before (referred to as never cryopreserved) and after one cycle of cryopreservation and thawing (referred to as cryopreserved). Immediately after extraction or after primary culture (P0), the percentage of K19+ cells was significantly higher in keratinocyte populations from newborn (16%±4%, n=9) than adult (3%±1%, n=8) skin. Variation in the number of cells expressing K19 upon passages was then evaluated. To normalize interindividual differences, the percentage of K19 at P0, P1, or P2 was compared to the percentage of K19 at the extraction for each strain, and expressed as fold change. For newborn keratinocytes, there was no significant difference in the percentage of K19+ cells at P0, P1, and P2 (Fig. 5A). For adult keratinocytes, a significant increase in the percentage of K19+ cells at P0 compared to extraction was noticed (Fig. 5B). It remained stable in P1 and P2. We next compared the percentage of K19+ (Fig. 5C, D) and α3-integrin+ (Fig. 5E, F) cells within cryopreserved and never-cryopreserved keratinocyte populations. No significant difference was observed at P1 or P2, for both newborn and adult cells (Fig. 5C–F). Thus, these results suggest that our cryopreservation process did not affect the skin epithelial SC recovery.
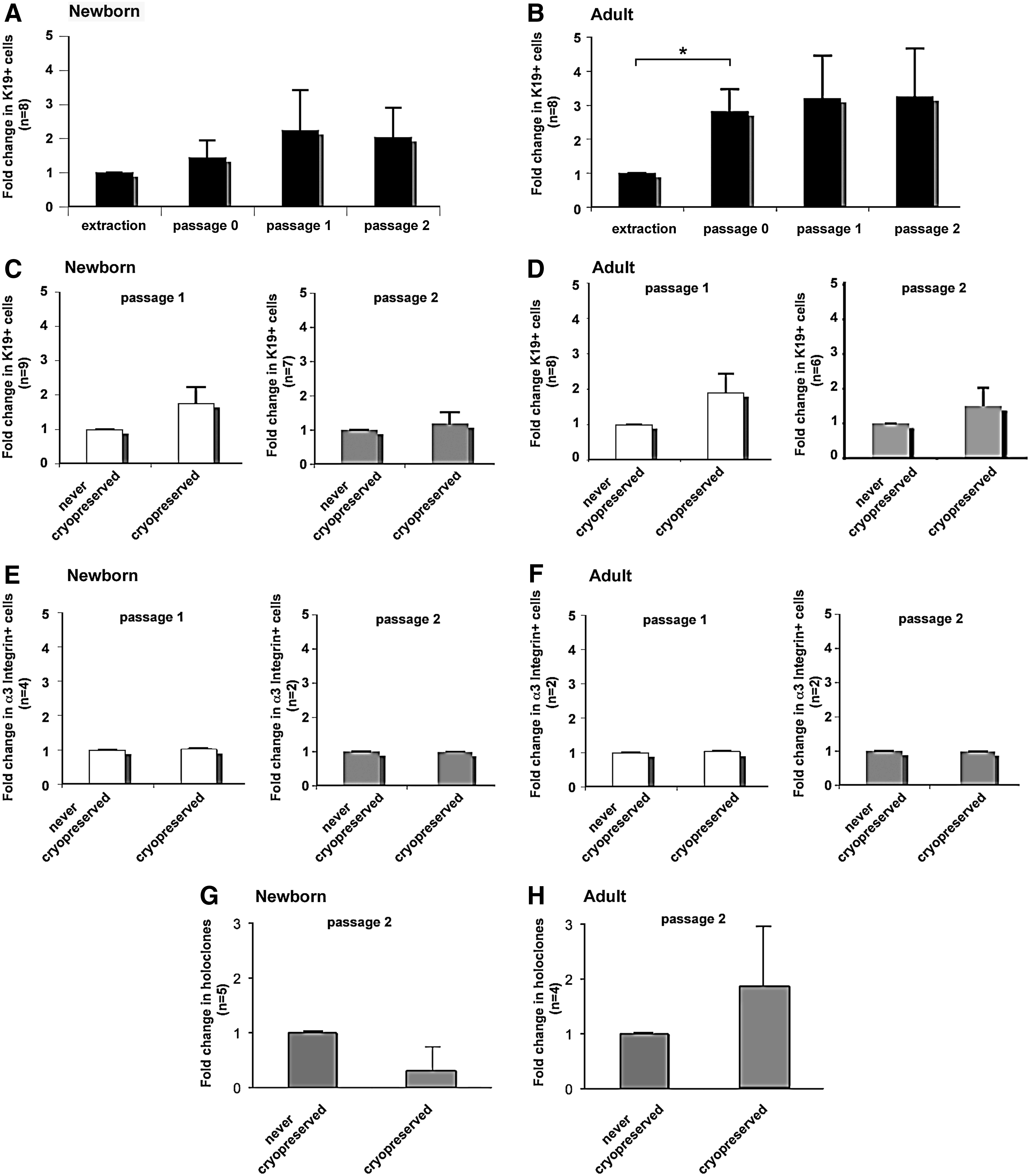
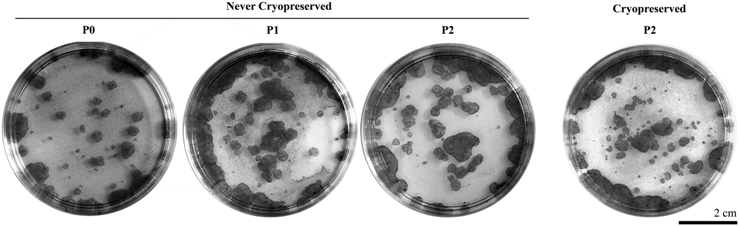
Flow cytometry analysis and holoclone formation of keratinocytes in continuous conventional culture on plastic that were never cryopreserved or keratinocytes that have been cryopreserved, thawed, and cultured.
To investigate the effect of cryopreservation on the proliferation capacity of cultured newborn and adult keratinocytes, we compared the percentage of holoclones for cryopreserved cells and never-cryopreserved cells. The average number of holoclones formed by cultured newborn and adult keratinocytes is presented in Table 1, and the macroscopic view of the colonies is in Table 2. The number of meroclones and paraclones is presented in the Supplementary Table S2. No significant difference in the percentage of holoclones was observed between cryopreserved and never-cryopreserved keratinocytes, for both newborn and adult keratinocytes (Tables 1 and 2; Fig. 5G, H).
The bilateral Student's t-test for paired samples was applied to compare newborn to adult cells, as well as cells at passage 2 (P2) that were previously cryopreserved or not, and no significant difference was obtained.
Data represent mean±SEM.
The number of independent experiment (n) performed for each experimental condition is specified.
The persistence of SCs within TESs was not affected by the use of cryopreserved cells
Given that the TESs made by the self-assembly approach provide a microenvironment supporting the SC retention and integration, we evaluated the potential of epithelial SCs cryopreserved or not to settle within the TESs and to reconstruct a fully differentiated epidermis in vitro. Tissue-engineered skin sections stained with Masson's trichrome revealed similar histological characteristics between TESs produced with either cryopreserved keratinocytes or never-cryopreserved keratinocytes (Fig. 3A, C, E, and G compared with Fig. 3I, K, M, and O, respectively). Phenotypic characterization of TESs produced with cryopreserved keratinocytes (Fig. 2I–P) using classic markers of epidermal basal/SCs, proliferation, and differentiation, including K19, K15, K14, K10, filaggrin, involucrin, transglutaminase, integrins (α3- and β1-subunits), laminins (α3- and γ2-chains), and Ki67 revealed their proper expression similar to TESs produced with noncryopreserved keratinocytes (Fig. 2A–H). Moreover, the distribution of K19+ as well as BrdU-labeled cells was also similar (Fig. 3B, D, F, and H compared with Fig. 3J, L, N, and P, respectively).
To quantify the overall percentage of SCs that were preserved in the TESs, keratinocytes forming the reconstructed epidermis were dissociated and subsequently analyzed by flow cytometry. No significant difference was observed between the percentage of BrdU+K19+ cells present in TESs reconstructed with never-cryopreserved keratinocytes and those reconstructed with cryopreserved keratinocytes, whether the cells were obtained from newborn (Fig. 3T) or adult (Fig. 3U) TESs. These results showed that cryopreserved keratinocytes have maintained their capacity to produce a well-structured epidermis. Moreover, TESs made with cryopreserved cells contained a similar amount of SCs, as demonstrated by the detection of BrdU+K19+ cells after the 21-day chase period.
TES grafting on nude mice
To evaluate SC functionality and the long-term effect after grafting, TESs produced with cryopreserved adult and newborn human keratinocytes were grafted on athymic mice. Macroscopic evaluation demonstrated a good graft take 4 days (not shown), 21 days (Fig. 6A), and 6 months (Fig. 6D) post-grafting. HLA detection on 6 months post-graft (Fig. 6I) confirmed the human origin of epithelial cells, whereas histology and filaggrin staining indicated a normal differentiation pattern (Fig. 6B, C, E, F, and K). TESs presented a well-organized epidermis with all four characteristic layers (Fig. 6B, C, E, and F). Moreover, at 6 months post-grafting, the tissue-engineered dermis was homogeneous, and the initial fibroblasts cell sheets merged completely to form a dense extracellular matrix (Fig. 6E). The persistence of the graft after 6 months and the detection of K19+ cells (Fig. 6G), at their specific basal localization, confirm the maintenance of functional SCs within TESs. Taken together, these results indicate the long-term stability of TESs in vivo.

Long-term effects of TES transplantation in vivo: macroscopic and histological analysis of the TESs after grafting on athymic mice for 21 days
Discussion
When skin damages are deep and extend over the epithelial SC reservoir of hair follicles, the regeneration of the epithelial barrier is only possible by the migration of intact epithelial cells surrounding the wounds. In the case of large wounds, the regeneration of the epidermis in a natural fashion is often compromised. In the worst scenario, the cutaneous tissue is destroyed in its full thickness, and the wound closure ultimately depends on skin grafts. Advances in tissue engineering now offer new hopes for patients with burns affecting even up to 90% of their total body surface area. The ideal skin substitute for clinical purposes should be nontoxic, have no antigenicity, be immunologically compatible, not transmit disease, and capable to self-renew after grafting. Living skin substitutes can be elaborated in vitro from the patient's own cells, using the self-assembly approach of tissue engineering, in the absence of an artificial scaffold. However, skin SCs play a central role in the therapeutic success of such autologous transplants. Indeed, SCs are necessary for the persistence and functional regeneration of the epithelium throughout the patient's lifespan. The main objective of this study was to confirm the presence and the functionality of epithelial SCs within the TESs made by the self-assembly approach, especially those produced with keratinocytes from adult donors. The demonstration that our TESs contain SCs and that they persist after grafting on athymic mice (current study), combined with previous results,33,34 indicates that this skin substitute possesses all the sought-after characteristics of a permanent autologous skin graft.
Bilayered skin substitutes comprising both living fibroblasts and keratinocytes present many advantages. Fibroblasts produce growth factors and the extracellular matrix, which promote the formation of an optimal microenvironment to support the differentiation of keratinocytes in a well-structured epidermis.48–56 Fibroblasts secrete many components of the basement membrane such as collagens IV and VII, laminin-5, and nidogen. 57 Cytokines released by fibroblasts also stimulate the secretion of collagens IV and VII by keratinocytes.58,59 The early formation of the basement membrane at the dermoepidermal junction is an important factor in the success of healing. Indeed, the basement membrane ensures the cohesion between the dermis and the epidermis, and also participates to SC regulation and anchoring. Keratinocytes presenting SC properties are associated with a high expression level of α2β1- and α3β1-integrins, two collagen IV receptors.60–62 β1-Integrin mediates adhesion to the extracellular matrix, and its expression diminishes during epidermal differentiation. 63 The skin SCs also express the α6β4-integrin, a laminin receptor concentrated in hemidesmosomal structures and distributed at the basal pole of keratinocytes from the basal layer.64–66 Interestingly, in contrast with collagen-gel-based skin substitutes,15,28 keratinocytes presenting SC properties within the TESs presented here stayed confined in the basal layer of the epidermis in their expected position during the extended culture duration, emphasizing the importance of the stromal compartment composition and stability. This is consistent with a previous study showing that LRCs are preserved in hyalograft-scaffold-based, but not in collagen-based, organotypic cultures, due to the high-matrix metalloproteinases present in the latter. 28 However, the epidermis can be peeled off from the dermis without prior treatment in these organotypic cultures. In TESs produced by the self-assembly approach, however, the epidermis is tightly bound to the dermis, and enzymatic digestion is necessary to separate it from the underlying dermis. This is consistent with the well-organized basement membrane observed under the epithelial tissue by transmission electron microscopy on these TESs cultured in vitro or transplanted into a mouse model.30,31,67 This mechanical stability of the epidermis is important for substitutes produced for transplantation.
Despite many years of research, it remains difficult to identify human skin SCs due to the lack of an exclusive marker. The various proteins associated with SCs, such as K15, 67 K19,18,46 p63, 68 high expression level of β1- or α6-integrin subunits,61,64,69 or melanoma chondroitin sulfate proteoglycan, 70 stain clusters of cells within the epidermal basal layer or a broader range of cells than the expected SC population. For K15, 67 different antibodies against this protein present distinct labeling, and C8/144b is the only one that labels specifically a small subset of the basal cells.39,67,71 Another anti-K15 was used in the present study; the basal cell labeling is similar to that reported previously.39,46 Concerning K19, it was found to be present in a group of basal cells in foreskin as well as in the bulge of human scalp, and in slow-cycling cells of murine pelage and vibrissa follicles.14,46 Small clusters or single K19+ basal cells were identified in TESs made from fibroblasts and keratinocytes from human or murine origin (this study and Refs.35,46), as well as in the skin substitute generated from adipose-derived stromal cells, 72 in organotypic, 39 and in the hyalograft® three-dimensional organotypic culture model. 28 The greater percentage of K19+ cells in newborn TESs compared to adult TESs reflects the different content of K19+ cells present in the initial keratinocyte population seeded on tissue-engineered dermis.18,19 Although the percentage was higher in newborn compared to adult, there was no significant difference between the number of LRCs in newborn and adult TESs cultured for 38 days at the air–liquid interface (see Fig. 3). In our TES model, the expression of the cytoskeletal intermediate filament K19 could persist after SCs had exited their niche, explaining why K19+ cells were occasionally observed in TES's suprabasal layers (see Fig. 3). The persistence of K19-BrdU double-stained cells in the basal layer throughout TES production indicates that SCs were preserved in a suitable position.
In the fabrication of TESs for clinical purposes, long-term storage of epithelial cells provides the possibility to build cell banks, which could later be used to produce tissues. Cryopreservation methods allow storage and transportation of cells, therefore facilitating their use. However, the cryopreservation process must be tightly controlled to avoid a reduction in the number of SCs harvested after extraction and subsequent cultures. From the several studies assessing the different cryopreservation techniques, the slow-rate method using DMSO as a cryoprotectant proved to be appropriate for many cell types.73–76 However, cryopreservation of SCs appears more arduous, depending both on the SC type and on the freezing technique employed.73–80 The cooling rate and the choice of cryoprotectant are determinants in the success of a cryopreservation procedure, especially for SCs, for which the viability after thawing is of great importance. While a standard cryopreservation procedure (slow-rate freezing and rapid thawing) in a freezing medium (DMEM) containing 10% DMSO and 50% FBS has proven to be efficient for murine embryonic SCs, 81 human embryonic SCs have rather demonstrated a poor cell recovery after similar slow-cooling cryopreservation using 90% FBS and 10% DMSO. 82 Vitrification, a technique in which a high concentration of cryoprotectant (usually 20% DMSO) and rapid cooling allow a glass-like solidification of the freezing medium that prevents intracellular ice crystal formation, has demonstrated a great efficacy for cryopreservation of human embryonic SCs.80,82 However, vitrification requires special laboratory equipment and is more time consuming compared to conventional cryopreservation methods. The high concentration of DMSO used for vitrification may also cause cell toxicity. Compared to these strategies, the cryopreservation procedure presented here offers the advantage to be quick and to require no specialized equipment.
In the present study, we demonstrated that the proportion of K19+ LRCs in TESs remained constant in never-cryopreserved and cryopreserved keratinocytes from adult as well as newborn donors, suggesting that functional SCs were preserved during the cryopreservation process. Altogether, these findings support the use of cryopreserved keratinocytes for the fabrication of TESs and alleviate the concern about the loss of SCs during the freezing and thawing process even in strains with a lower number of SCs such as those from adult donors.
In conclusion, detection of LRCs in the TESs in vitro, combined with the high proliferation potential of the TES-dissociated epithelial cells in subculture, and the persistence of human TESs after grafting on athymic mice demonstrate that functional SCs were preserved within TESs regardless of the age of the donor. Moreover, our work adds evidence that an appropriate tridimensional environment is a key element for SC preservation. Our results also indicate that human TESs with functional SCs can be produced by the self-assembly approach with cryopreserved skin epithelial cells. Thus, we conclude that this approach is appropriate for the production of TESs dedicated to a clinical application requiring SC replenishment, and that the autologous TESs made by the self-assembly approach is a promising skin substitute for the permanent closure of full-thickness skin injuries.
Footnotes
Acknowledgments
The authors gratefully thank Drs. Alphonse Roy, Félix-André Têtu, and Maurice Bouchard for providing skin biopsies; Drs. P. Rousselle [Institut de biologie et de chimie des protéines (IBCP), Lyon, France], U. Karsten (Institute of Biological Sciences, University of Rostock, Germany), and J.A. Grimaud (Institut Pasteur de Lyon, France) for providing antibodies; as well as Francis Bisson and Israël Martel for expert technical assistance. This work was supported by the Canadian Institutes for Health Research (CIHR), Fondation des Pompiers du Québec pour les Grands Brûlés (FPQGB), the Fonds de Recherche du Québec-Santé (FRQS), and the Réseau ThéCell du FRQS. C.F. received studentship and C.P. a postdoctoral fellowship from the FRQS. J.F. is a recipient of a FRQS Career Award from the FRQS. L.G. was the holder of the Canadian Research Chair on Stem Cells and Tissue Engineering from the CIHR.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.