
Abstract
Introduction:
For the regeneration of large volume tissue defects, the interaction between angiogenesis and osteogenesis is a crucial prerequisite. The surgically induced angiogenesis by means of an arteriovenous loop (AVL), is a powerful methodology to enhance vascularization of osteogenic matrices. Moreover, the AVL increases oxygen and nutrition supply, thereby supporting cell survival as well as tissue formation. Adipose-derived stem cells (ADSCs) are interesting cell sources because of their simple isolation, expansion, and their osteogenic potential. This study targets to investigate the coimplantation of human ADSCs after osteogenic differentiation and human umbilical vein endothelial cells (HUVECs), embedded in a vascularized osteogenic matrix of hydroxyapatite (HAp) ceramic for bone tissue engineering.
Materials and Methods:
An osteogenic matrix consisting of HAp granules and fibrin has been vascularized by means of an AVL. Trials in experimental groups of four settings were performed. Control experiments without any cells (A) and three cell-loaded groups using HUVECs (B), ADSCs (C), as well as the combination of ADSCs and HUVECs (D) were performed. The scaffolds were implanted in a porous titanium chamber, fixed subcutaneously in the hind leg of immunodeficient Rowett Nude rats and explanted after 6 weeks.
Results:
In all groups, the osteogenic matrix was strongly vascularized. Moreover, remodeling processes and bone formation in the cell-containing groups with more bone in the coimplantation group were proved successful.
Conclusion:
Vascularization and bone formation of osteogenic matrices consisting of ADSCs and HUVECs in the rat AVL model could be demonstrated successfully for the first time. Hence, the coimplantation of differentiated ADSCs with HUVECs may therefore be considered as a promising approach for bone tissue engineering.
Impact statement
Due to their low donor-site morbidity, osteogenic potential and their good cell expansion, human adipose-derived stem cell is an interesting cell source. The goal of this study is to transfer the in vitro results of increasing osteogenic potential upon cocultivation with human umbilical vein endothelial cell, to the rat arteriovenous loop model within the next step. Without using growth factors, the biomaterials, fibrin and HAp, which have already been successfully applied in clinical practice, are supposed to support bone formation by the implanted cells. The results demonstrated that vascularized bone tissue was formed in the cell-containing groups after 6 weeks.
Introduction
Extensive bone tissue defects, caused by neoplasm, trauma, infection, or surgical resection, represent a challenge in therapy due to the structural bone integrity and vascularization to regain function.1,2 Today, autologous cancellous bone or vascularized bone grafts, which may be associated with a higher donor-site morbidity, represent the gold standard for bone reconstruction. In this regard, biomaterials with osteogenic properties may be used to reconstruct large-volume bone defects. 2–5 Fibrin is a hydrogel which is often successfully applied in tissue engineering applications and showcases many properties of capable biomaterials.3,4,6 Besides a good biocompatibility and biodegradation, fibrin provides the cells with a comparative milieu as the extracellular matrix.7,8 These, and other properties make up excellent conditions for a balance between biodegradation and tissue regeneration.
In addition to fibrin, we used granular hydroxyapatite (HAp) to generate an osteogenic scaffold. HAp is the main component of the inorganic substance in bones and teeth. Natural or synthetically produced HAp has already been widely used for bone and dental defect reconstructions. The granular form, porosity, and mechanical strength of the material stabilizes the scaffold, facilitates construct vascularization, and supports cell migration.9–11 Not only the characteristics of the biomaterial but also the nutrient and oxygen supply are essential for cell survival and bone formation of the scaffold. In this regard, the most challenging part in the fields of bone tissue engineering is to sufficiently vascularize the cell-loaded osteogenic scaffolds. Extrinsic vascularization from the surrounding tissue will lead to hypoxic cell death of the centrally located osteogenic cells in bone tissue engineering constructs due to a limited diffusion range of 200 μm.
A promising strategy is the surgically induced angiogenesis or so-called prefabrication. More than four decades ago, Erol and Sira described the microsurgical interposition of a vein graft between the saphenous vein and artery creating an arteriovenous loop (AVL). 12 The AVL enables intrinsic vascularization of manifold biomaterials such as spider silk, 5 alginate dialdehyde crosslinked with gelatin 13 or 45S5 bioactive glass. 3 The combination of the intrinsic (by means of an AVL) and extrinsic vascularization (by a porous titanium chamber) mode enhances total vascularization, cell survival, and provides good conditions for bone tissue formation.1,14
Furthermore, the implantation of endothelial cells, such as human umbilical vein endothelial cells (HUVECs), provides another opportunity to facilitate the creation of a new vascular network because of their angiogenic potential.15–17 For bone tissue engineering applications, the coimplantation of HUVECs and mesenchymal stem cells is a promising approach. The literature describes enhanced proliferation and osteogenic differentiation by cocultivation of both cell types.18,19 Human adipose-derived stem cells (ADSCs) were used because of their simple isolation with a low donor-site morbidity, their good ex vivo cell expansion and their osteogenic potential.20–22
This study targets to investigate the impact of HUVEC on vascularization and bone formation by ADSC-loaded osteogenic scaffolds in the AVL model.
Materials and Methods
Cell culture
ADSCs were obtained with the informed consent of the patients, according to hospital's Ethics Committee Guidelines [AZ: 126_16]. ADSCs were isolated from excess subcutaneous fat of abdominal free flaps during autologous breast reconstructions or abdominoplasty according to an established protocol. 20 The one male and two female donors had an average age of 50 ± 15.87 years. HUVECs were purchased from PromoCell (Heidelberg, Germany). The cells were cultured in a culture flask (Greiner Bio-One, Kremsmünster, Österreich) at a density of 5000/cm2 in a humidified atmosphere at 37°C and 5% carbon dioxide. ADSCs were cultured in minimum essential medium alpha (MEMα) (Life Technologies Limited, Paisley, UK) cell culture medium, enriched with 10% fetal calf serum (FCS) (Biochrom, Berlin, Germany) and 1% penicillin/streptomycin (Sigma-Aldrich, Steinheim, Germany). For HUVECs, endothelial cell growth medium (ECGM) (PromoCell) was used containing endothelial basal medium, 10% FCS, 1% penicillin/streptomycin (Sigma-Aldrich), and supplements. Both cell types were raised until passage 4.
Before implantation, ADSCs were grown under osteogenic conditions over 14 days with ECGM, supplemented with 50 μg/mL L-ascorbic acid, 10 mM glycerophosphate, 1 × 10–8 M dexamethasone, and 0.01 μM 1.25-dihydroxyvitamine D3 (all supplements purchased from Sigma) according to an established protocol. 14 Osteogenic differentiation was proofed and validated by an Alizarin Red assay and alkaline phosphatase (ALP) assay.
Fluorescence-activated cell-sorting analysis
The mesenchymal stem cell characteristics of the isolated ADSCs were analyzed by fluorescence-activated cell sorting (FACS). ADSCs were prepared in Dulbecco's phosphate-buffered saline (PBS) (Sigma-Aldrich) in a concentration of 1000 cells/μL. ADSCs were marked with fluorescent conjugated antibodies. The labeled ADSCs were passed through a laser light beam and the fluorescence intensity was measured by an electronic detector. The cells were analyzed on a FACScan® with CellQuest® software (Becton Dickinson, Franklin Lakes, NJ) as described previously. 23
Alizarin Red staining and assay
Matrix mineralization was assessed with Alizarin Red S Staining Quantification Assay Kit according to the manufacturer's specifications (ScienCell, Carlsbad, CA). ADSCs growing under osteogenic conditions (ADSC OC) were, fixed, washed, and the Alizarin Red staining solution was added. ADSCs being cultured in ECGM cell culture medium without osteogenic supplements served as a control group. Both groups were sown in six-well plates in replicates of five with a density of 5000 cells/cm2. Afterward, images were made with an Olympus IX81 microscope (Olympus, Hamburg, Germany) and processed with CellSens Dimension V1.5 software (Olympus). Acetic acid (ScienCell) was added, cells collected using a cell scraper, and the samples heated at 85°C for 10 min. After a cooling and centrifugation step the supernatant was collected, neutralized with 10% ammonium hydroxide (ScienCell), and absorbance measured at 405 nm.
ALP assay
ADSCs cultured in ECGM with (ADSC OC) and without (control group) osteogenic supplements were sown in replicates of five in six-well plates, with a density of 5000 cells/cm2. The osteogenic differentiation was determined by using an ALP Assay Kit (Abcam, Cambridge, GB). According to the manufacturer's protocol, cells were washed, lyzed, and pNPP solution added. After the ALP converted the pNPP substrate, the reaction was quenched and the absorbance measured at 405 nm to this end. The ALP activity was normalized to the protein content by the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA) according to a standard protocol.
Matrix preparation
HAp granules
Hemispherical shaped HAp granules were fabricated by a ceramic injection molding (CIM) process, which is usually for complex structures. The HAp feedstock consisted of 56.5 Vol% hydrophobized, calcined HAp powder (Sigma-Aldrich Corp., St. Louis, MO), 39.1 Vol% paraffin (Granopent P; Carl Roth GmbH, Karlsruhe, Germany), and 4.4 Vol% carnauba wax (naturfarben; Carl Roth GmbH). For the casting mold manufacturing, a negative replica technique was used. Positive granular casting molds with a granular size of 1.5 × 1.5 × 0.75 mm were three-dimensional (3D)-printed with a z-resolution of 20 μm using a stereolithography (SLA) 3D-printer (Digitalwax® 028J; DWS S.r.l. Zanè, Italy) and afterward molded with a polydimethylsiloxane (PDMS) elastomer (Elastosil M 4643 A/B; Wacker Chemie AG, München, Germany). Possible surface roughness was transferred to the surface of the granules according to the selected resolution. In contrast to direct 3D-printing (powder bed, robocasting), indirect printing can be used to produce smoother surfaces due to the higher z-resolution by a factor of 6–8. The feedstock was injection molded into the negative PDMS forms at 120°C. The as-fabricated granules were debinded at 500°C in air and afterward sintered at 1150°C for 2 h. A detailed description of the HAp injection mold process can be found elsewhere.9,24,25
The microstructure of the sintered HAp granules was analyzed with an environmental scanning electron microscope (ESEM) (Quanta 200 FEG; FEI Company, Hillsboro, OR). The pore sizes were determined by image analysis of the scanning electron microscope (SEM) micrographs analyzing a minimum area of 200 × 200 μm2 (∼4500 pores). The total porosity was geometrically determined on cylindrical reference samples with dimensions of 2.9 × 2.9 × 1.7 mm. Additionally, the 3D-pore structure and related pore network was analyzed by microcomputed tomography (μCT) using a Skyscan 1172 (Skyscan, Kontich, Belgium) equipped with a tungsten tube (λ = 0.024 nm), an 11 MP detector operating at 80 kV and 100 mA. The topography and surface quality were determined by confocal laser scanning microscopy (VK-X100; Keyence Corporation, Osaka, Japan).
Fibrin matrix
The fibrin was prepared as stated in fabricator's protocol of the Tisseel Kit (Baxter AG, Vienna, Austria). The fibrinogen was mixed with aprotinin and the thrombin diluted in calcium chloride. The fibrinogen and thrombin solutions were diluted in PBS (Sigma-Aldrich) in a 1:4 and 1:10 ratio, respectively. The solutions were kept in separate 1.0-mL Luer-Lock syringes and mixed during application in the Tisseel applicator (Baxter). The fibrin clotted after 4 to 5 s in a fibrinogen: thrombin ratio of 1:1. Before cell implantation, ADSCs and HUVECs were detached with accutase (Sigma-Aldrich) from the cell culture flasks (Greiner Bio-One), counted, centrifuged, and mixed with the diluted fibrinogen. The cells were implanted in a concentration of 2 × 106 cells per construct. HUVECs and ADSCs were implanted in equal parts (1 × 106 cells each) for group D. The cell-containing fibrinogen was filled into the Luer-Lock syringe of the Tisseel applicator and the fibrin application performed as previously described.
AVL implantation
The animal experiments were approved by the Animal Care Committee of the University of Erlangen and the Government of Mittelfranken, Germany (55.2–2532.1–53/14). For the study 32 male, T cell-deficient Rowett Nude (RNU) rats (Charles River Laboratories, Sulzfeld, Germany) were used. Four experimental groups were performed with eight animals per group. The rats aged between 7 and 9 months and had a body weight between 320 and 450 g. The microsurgery part of the AVL operation was carried out with a Carl Zeiss operating microscope (Oberkochen, Germany). With isoflurane (op-pharma; Burgdorf, Germany) the rats were anesthetized and the operation was performed according to a well-established protocol. 26 Briefly described, the saphenous vein and artery of the left hind limb were microsurgically anastomosed with a saphenous vein graft of the right hind limb. The titanium chamber had the dimension of 1.0 × 1.0 × 0.8 cm with pores facilitating extrinsic vascularization. 27 To prevent AVL dislocation, the chamber was prepared with four green pins (Braun, Melsungen, Germany). The chamber was then placed in the left groin and filled with a first layer of HAp granules. The diluted thrombin and fibrinogen (Baxter AG) were distributed around the granules. Fibrinogen directly converted to fibrin after 4 to 5 s. The AVL was placed around the pins and a second layer of fibrin (Baxter AG) was utilized to surround the vessel surface (Fig. 1). The cells in groups B–D were evenly distributed in the constructs. Finally, the chamber was filled completely with granules and fibrin, closed with the lid and fixed subcutaneously. The skin was closed, and the rats were precisely and accurately monitored during the implantation time of 6 weeks.
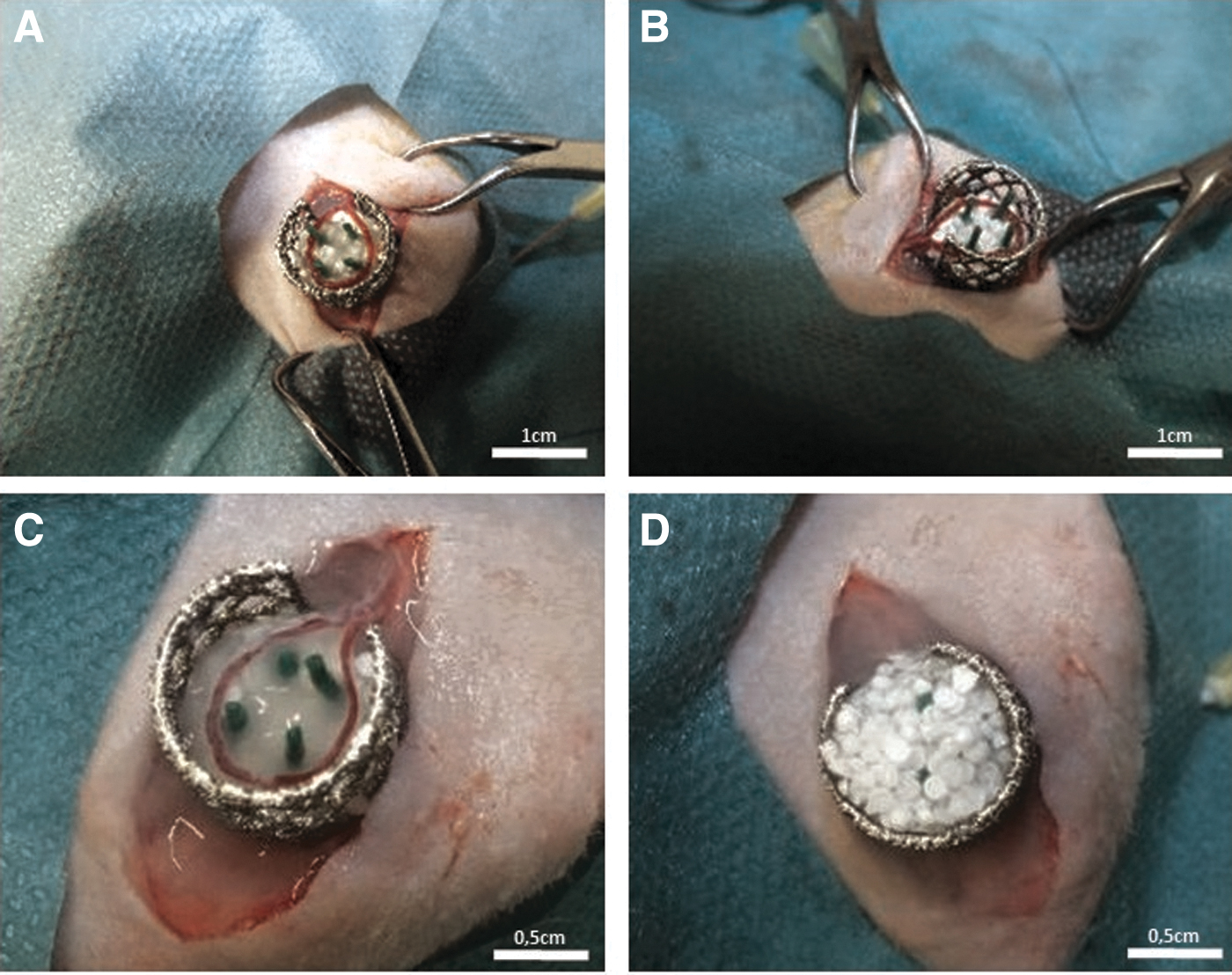
AVL operation. After the microsurgical anastomosis was successfully completed, the titanium chamber was fixed in proper position
Explantation procedure
After 6 weeks the rats were perfused with Microfil® (Flow Tech, Inc., Carver, MA). The abdominal approach was carried out through a median laparotomy. After dissection of the caval vein and abdominal aorta, the aorta was cannulated with a G21 cannula (Braun). The caval vein was cut and the vascular system was flushed with 100 mL of Ringer heparin solution (100 IU/mL) (Braun). Twenty milliliters of Microfil MV-122 solution with 1 mL curing agent (Flow Tech, Inc.) were applied and the caudal caval vein and aorta were ligated. After storage at 4°C overnight, the constructs were explanted, weighed, and fixed in Roti®-Histofix 4% (Carl Roth GmbH) for 24 h. The constructs were transferred in 20% ethylene diamine tetraacetic acid (EDTA) (Sigma-Aldrich) for decalcification. The constructs were decalcified up to 14 weeks.
Histological staining and analysis
After complete decalcification, the constructs were cut into two equal pieces and embedded in paraffin for histological staining. Three μm cross sections from the central construct part, vertical to the AVL, were performed with a microtome. For each histological staining two central cross-sections were used. Hematoxylin and Eosin (H&E) as well as Masson's Trichrome staining were carried out following standard protocols. For a better visualization of the newly formed vessels, α-smooth muscle actin staining (α-SMA), specific for human and murine smooth muscle cells, was performed. Briefly described, the deparaffinized and rehydrated histological slices were prepared with pronase and blocking solution (Zytomed Systems GmbH, Berlin, Germany). Afterward, the diluted (1:350) anti-α-SMA antibody was added overnight. Thereafter, a second ALP-labeled antibody (AP-Polymer) and Fast Red TR/Naphthol AS (Sigma) substrate was applied. Haemalaun was used for counterstaining.
To detect the implanted human cells after explantation, an immunofluorescence staining with the cluster of differentiation markers 31 and 105 was carried out. According to the manufacturer's instructions, a polyclonal human CD105 antibody (R&D Systems, Wiesbaden, Germany) combined with the fluorescent Alexa Fluor 488 (Thermo Fisher Scientific)-conjugated secondary immunoglobulin G (IgG) antibody was used for the detection of implanted ADSCs. The CD105 primary antibody had <0.2% crossreactivity with murine Endoglin and the Alexa Fluor 488-conjugated secondary IgG antibody was pretreated with a blocking solution to eliminate crossreactivity. Implanted HUVECs were identified using a monoclonal CD31 antibody (Dako Agilent, Santa Clara, CA), without rat crossreactivity (as declared by the manufacturer), combined with the NorthernLights™ NL557-conjugated secondary IgG antibody (R&D Systems). The latter one was pretreated with a blocking solution to eliminate crossreactivity. The images were taken with an Olympus IX81 microscope (Olympus) and processed with the software from cellSens Dimension V1.5 (Olympus).
To analyze the parameters vessel count, vessel distribution, and bone area, Microsoft Paint 3D (version 6.1), ImageJ (version 2) with the processing package Fiji and GNU image manipulation program (GIMP) (version 2.10) was used. Vessels were counted manually with Paint 3D. The JPG files of the H&E-stained slices were marked with one black dot in the size of 10 pxl for each vessel cross-section. After turning the file into 8-bit RGB the 10 pxl dots were measured automatically by ImageJ. With the help of Fiji each dot was attached to one x- and y-coordinate in the file. To analyze the vessel distribution, the mean of the distance between the coordinates of each vessel to the AVL was calculated. The bone area in the histological slices was determined by five independent unblinded viewers and marked in GIMP and Paint 3D using the “intelligent scissors” tool. This tool marked the bone areas with color differences between adjacent pixels, which further strengthened the objectivity of the evaluation. Total bone area was automatically calculated with ImageJ. To determine the area of the newly formed tissue, the total construct size was measured using ImageJ.
Statistical analysis
GraphPad Prism 8.00 (GraphPad Software, San Diego, CA) was used for statistical analysis. p-Values ≤0.05 were considered as statistically significant.
Results
FACS analysis
The ADSC isolations were tested for certain mesenchymal stem cell markers. 28 The cells were positive for CD90 and CD73 (99.93 ± 0.06%), CD105 (99.70 ± 0.1%), and negative for CD31 (99.30 ± 0.7%). Osteogenic differentiation did not influence the mesenchymal stem cell properties. These ADSCs were positive for CD90 and CD73 (97.33 ± 2.9%), CD105 (73.01 ± 16.26%), and negative for CD31 (99.37 ± 0.1%).
Osteogenic differentiation
The matrix mineralization of the ADSC cell culture was evaluated by Alizarin Red staining and Alizarin Red assay after 14 days of cultivation under osteogenic conditions. Compared with the control group, the differentiated ADSCs (ADSC OC) had an increased deposit of extracellular calcium corresponding to the red coloring in the cell culture (Fig. 2A). The measurement of the absorption at 405 nm demonstrated a statistically significant difference between both groups in the Alizarin Red assay (0.12 ± 0.03 vs. 1.52 ± 1.36; p ≤ 0.001) (Fig. 2B).
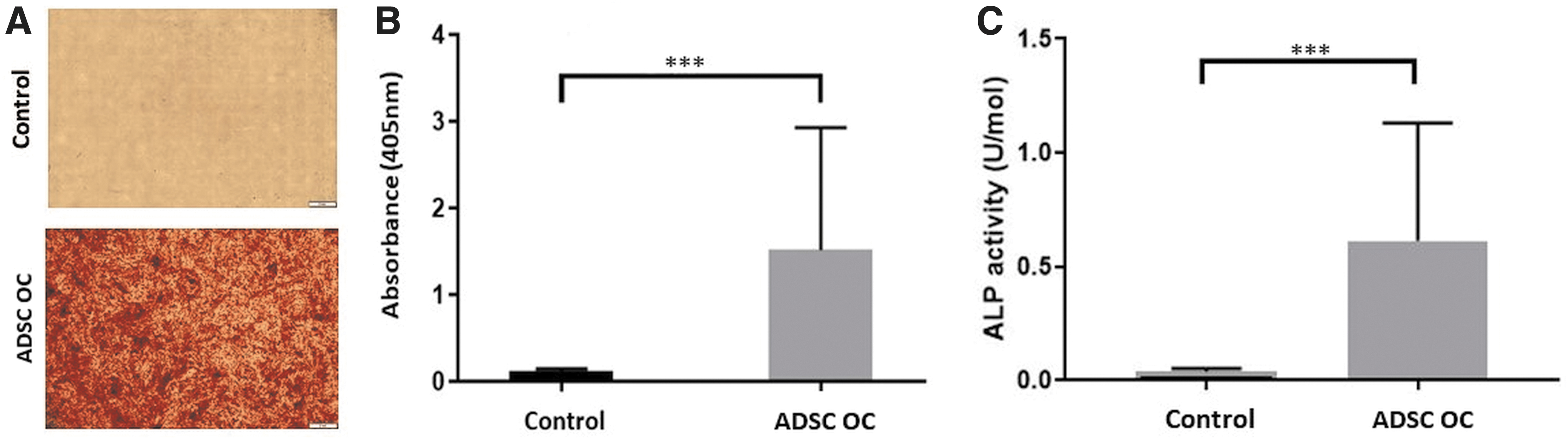
Osteogenic differentiation. The Alizarin Red Assay depicts matrix mineralization of undifferentiated ADSCs (control) and the ADSCs growing under osteogenic conditions (ADSC OC)
The osteogenic differentiation was further determined with the ALP assay. Compared with the control, the ADSC OC demonstrated a significant higher activity of the ALP (0.04 U/mol ±0.01 vs. 0.61 U/mol ±0.51; p ≤ 0.001) (Fig. 2C). Both assays indicated the potential of the ADSCs to differentiate into osteogenic cells and initiate matrix mineralization.
Microstructure of the HAp granules
Hemispherical HAp granules were successfully fabricated combining CIM with 3D-printing and silicon molding. CIM is a cost-effective, near-net shape manufacturing technique providing high-dimensional accuracies even for submillimeter components with complex shapes.25,29 The high-dimensional and shape accuracy of the fabricated HAp granules is displayed in the SEM micrographs and topography (Fig. 3A, B).

Granule ultrastructure. SEM-micrograph of a sintered HAp granule showing the rough surface and the print layers of the 3D-printed preforms, scale bar = 200 μm
The layerwise 3D-printing process of spherical shapes generates a rough surface with a gradual topography. The microstructure of the HAp granules was analyzed by SEM and μCT (Fig. 3C, D) and is characterized by a homogenous distributed residual sintering porosity of 13.4 ± 2.0% with a mean pore size of 0.78 ± 0.40 μm.
Surgical outcome and macroscopic appearance
One animal died 5 days after implantation, resulting in only 31 constructs being explanted. In the postmortem examination no pathological changes were found. Besides, no major complications such as suture dehiscences or hematomas were found at all. Not only the surgical procedures, but also the implanted biomaterial was well tolerated by the RNU rats. The titanium chambers were fully surrounded by connective tissue after 6 weeks. The surface of the constructs was yellow colored by Microfil suggesting incipient vascularization (Fig. 4A). After dissecting the osteogenic matrices from the titanium chambers, we measured the construct weight. Interestingly, all cell-containing groups (group B 2.13 ± 0.46 g, group C 1.80 ± 0.40 g, group D 1.93 ± 0.10 g) had a significant higher construct weight compared with the control group (1.50 ± 0.06 g) (Fig. 4B).
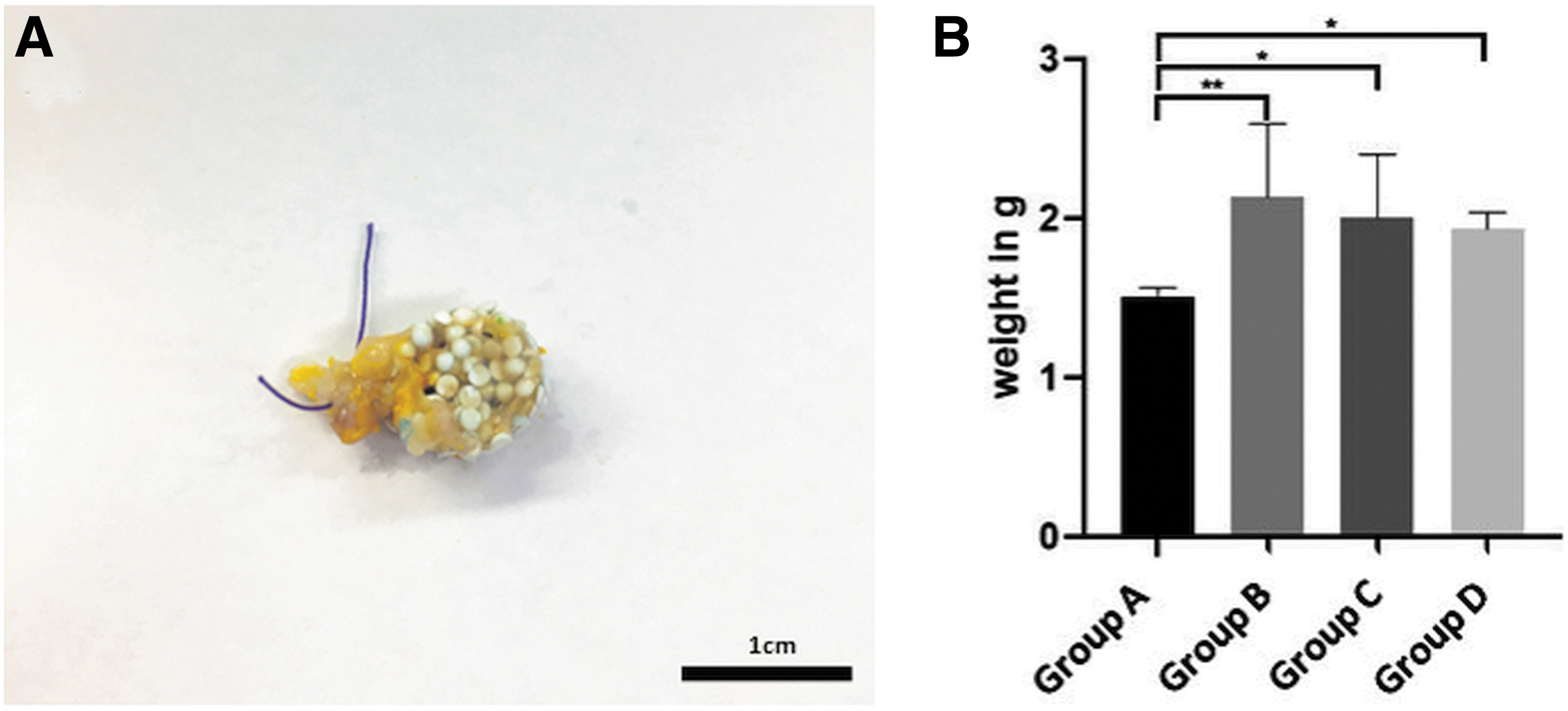
Macroscopic appearance and construct weight. The constructs were detached from the titanium chamber. At the surface of the constructs yellow-colored vessels, filled with Microfil®, are visible
Histological analysis
We found 26 out of 31 patent AVLs (84%). In each group, there was one thrombosis of the AVL except in group A. In group A there were two thrombosed AVLs. For the histological and statistical analysis, we excluded the five constructs with an AVL thrombosis.
In all groups, fibrovascular tissue was found between the HAp granules. Moreover, the constructs were highly vascularized (Figs. 5 and 6). The α-SMA staining demonstrated that most of the newly formed vessels contained human and rat smooth muscle cells in their media layer. Moreover, Microfil was only intraluminal indicating intima integrity as well as a certain vessel maturation (Fig. 6).
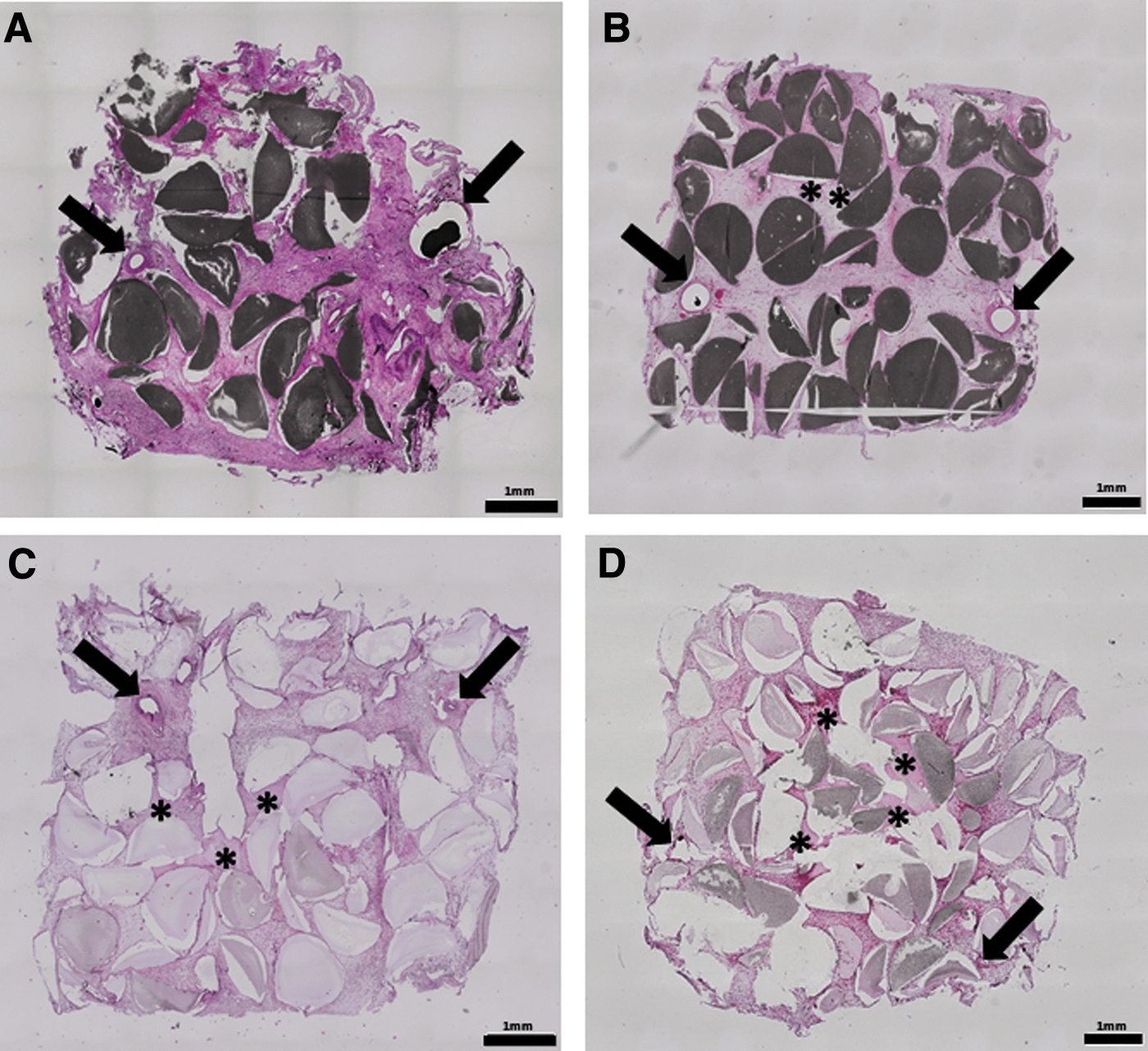
Histological overview images. The histological slices of group A
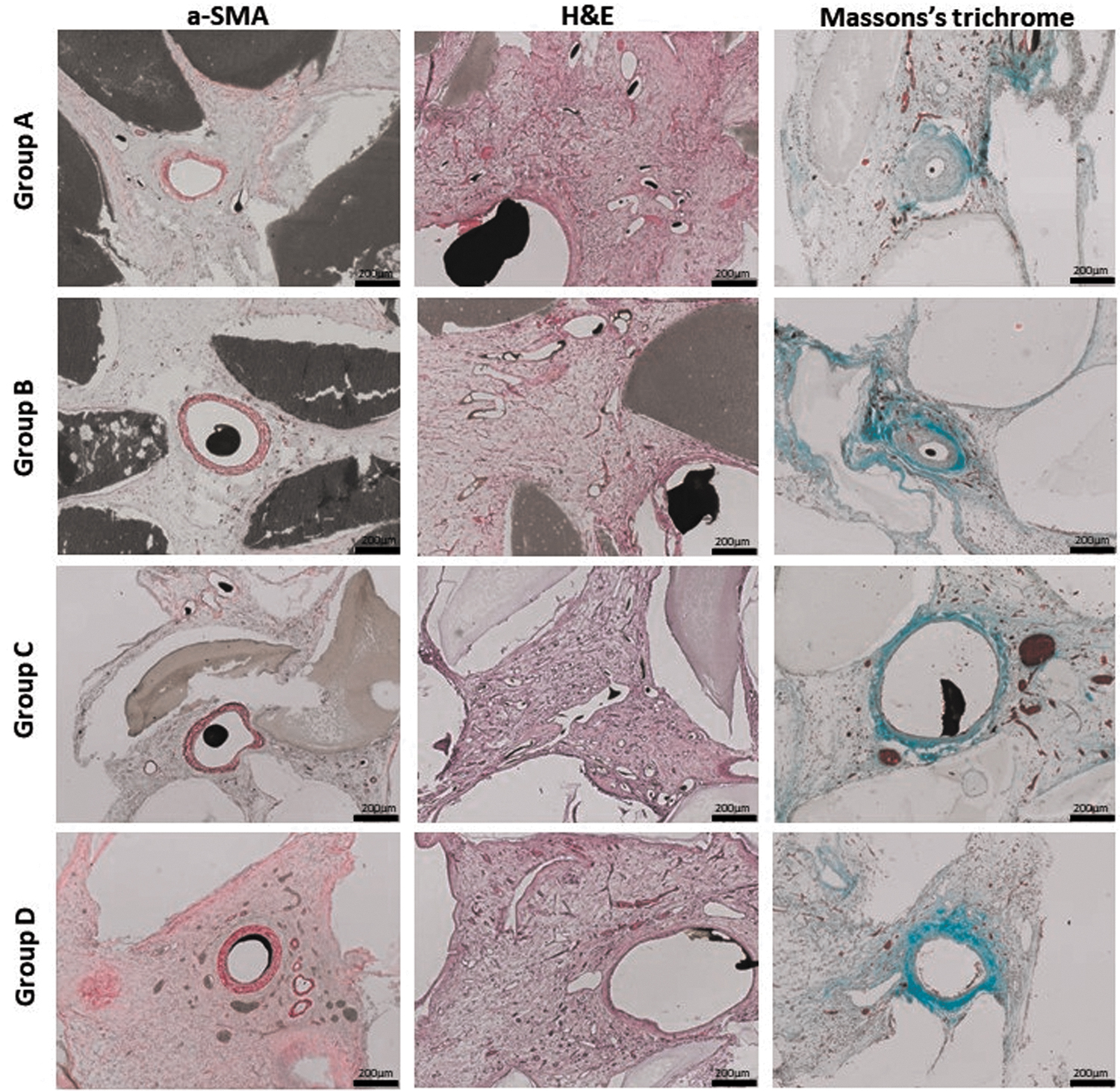
Detailed view on vascularization. The histological slices were stained with α-SMA, H&E and Masson's Trichrome staining. The newly formed fibrovascular tissue in the surrounding of the AVL is depicted in the histological cross sections. The α-SMA staining dyes the smooth muscle cells in the vessel walls. The Microfil appears as a black area inside the vascular lumen and the HAp granules appear gray. The H&E staining dyes the cell nuclei dark and the cytoplasm light pink. The Masson's Trichrome staining dyes reticular and collagen fibers light green, cytoplasm, muscle cells and erythrocytes red and nuclei brown. Scale bar = 200 μm. α-SMA, α-smooth muscle actin staining.
The vessel number per cross-section was calculated as a surrogate parameter for vascularization. Although being not significant for statistic reasons, the highest vessel number was detected in the HUVEC-containing group (group B) (Fig. 7). In this regard, we counted 1392 ± 518 vessels (group B), 1034 ± 453 vessels (group C), 916 ± 546 vessels (group A), and 908 ± 416 vessels (group D). Calculating the mean distance between the coordinates of each vessel to the AVL, the vessel distribution was quantified. Although a higher number of vessels reached the peripheral construct parts in groups C and D, a statistically significant difference compared with the control group A could not be proved herein. Interestingly, a significantly shorter distance of the newly formed vessels to the AVL in group B than in group D (3.02 ± 0.28 vs. 3.99 ± 0.44 mm; p ≤ 0.05) (Fig. 7) could be found. By means of the fluorescence staining with hCD105 and hCD31, the implanted cells were detected in the specimen (Fig. 8).
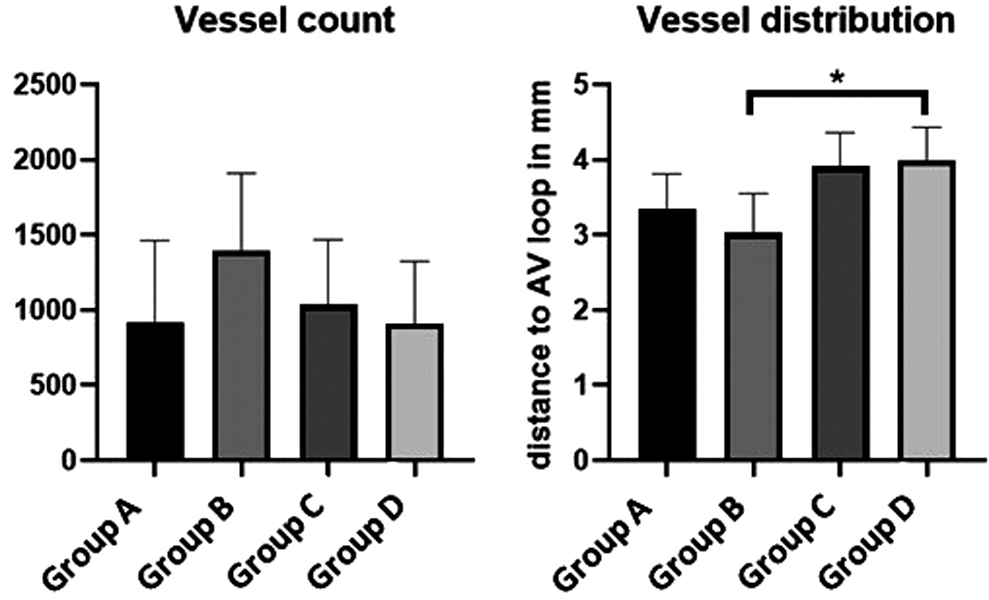
Quantification of vascularization and vessel distribution. The diagram on the left depicts the total vessel number per histological cross-section. The right-sided diagram represents the mean vessel distribution. A higher distance of the vessels to the AVL in group D opposite to group B could be demonstrated (*p ≤ 0.05).
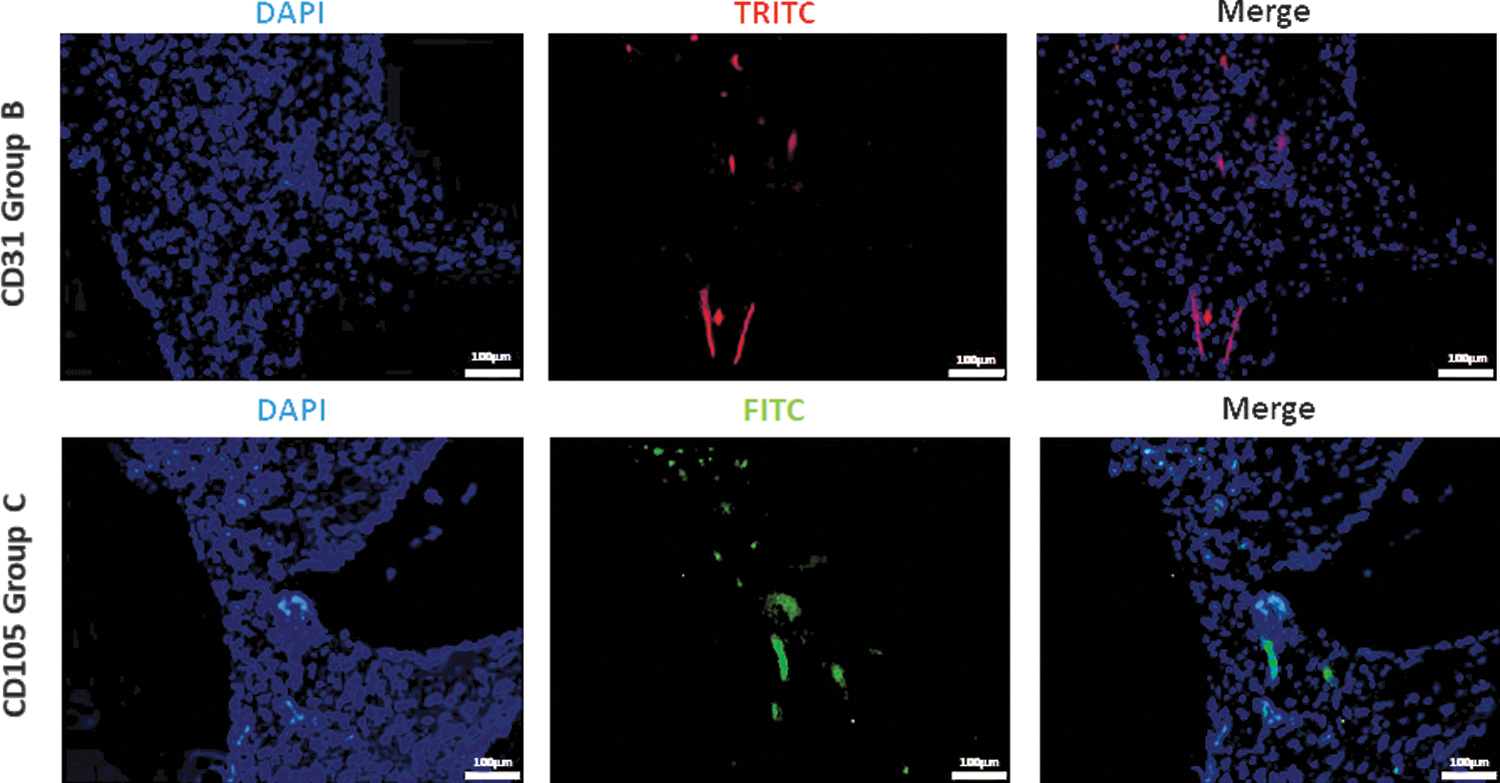
Immunofluorescence staining. HUVECs (group B), labeled with CD31 and NorthernLights™ NL557-conjugated secondary antibody, appeared red in TRITC channel. The CD105 and Alexa Fluor 488-conjugated secondary antibody labeled ADSCs (group C) green in FITC channel. The nuclei were stained with DAPI (blue). Scale bar = 100 μm. DAPI, 4′,6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine.
In the cell containing constructs (groups B–D), we were able to prove newly formed bone tissue (Figs. 5B–D and 10). The quantification of the bone area resulted in a clear trend toward the groups C and D. In this context, a statistically significant larger bone area in group D compared with groups B and C (1.88 ± 0.43 vs. 0.59 ± 0.48 mm2; p ≤ 0.01 and 1.88 ± 0.43 vs. 1.06 ± 0.62 mm2; p ≤ 0.05) (Fig. 9) could be observed. The newly formed bone tissue was predominately located in the center of the constructs. As previously mentioned, no bone tissue was found in the control group without cells (group A).
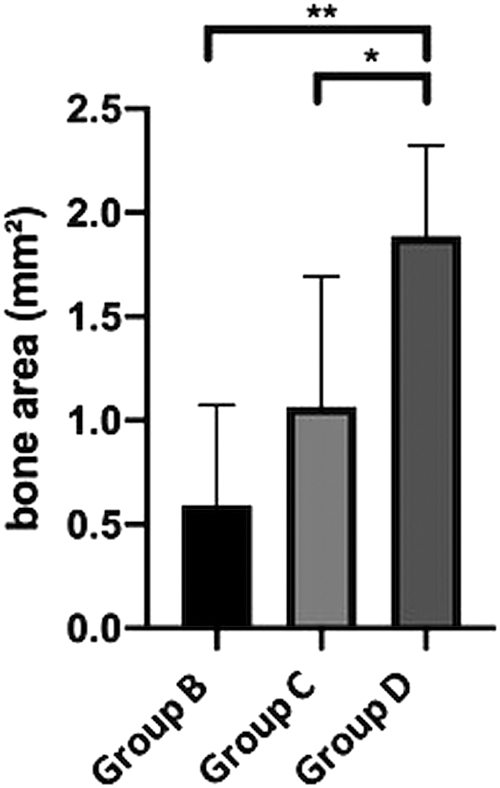
Quantification of bone area. The total bone area was higher in the ADSC-containing groups (C and D) compared with the HUVEC group (B). Moreover, the highest bone content was found in the ADSC and HUVEC coimplantation group (D). Statistically significant differences are indicated for *p ≤ 0.05 and **p ≤ 0.01.
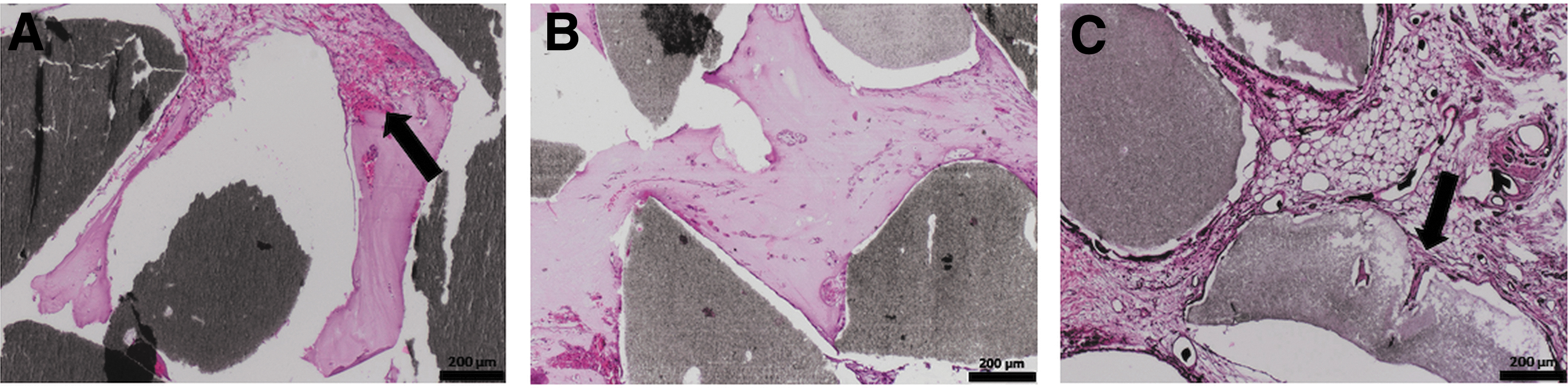
Detailed view on bone formation.
Furthermore, within the histological sections especially the cell containing groups displayed remodeling processes of the HAp granules. The remodeling processes were particularly evident in the peripheral areas of the granules (Fig. 10).
Discussion
The interaction of angiogenesis and osteogenesis is an indispensable parameter in the generation of bioartificial bone tissue. As extrinsic vascularization of tissue-engineered bone scaffolds always bears the risk of apoptosis of implanted cells and/or lower bone tissue formation, intrinsic vascularization seems to be a promising strategy.19,30 The AVL model representing an angiogenesis model, serves in a perfect manner for the investigation of biomaterial vascularization and osteogenesis, due to enhanced oxygen and nutrient supply of the implanted cells. Besides the AVL model, other angiogenesis models such as the arteriovenous (AV) bundle were also applied for bone tissue engineering applications. 31 A direct comparison between the AV bundle and the AVL by Tanaka et al. demonstrated that the AVL model displays a higher vascularization potential. 32
This study is based on the hypothesis that bone formation is supported by the choice of appropriate biomaterials, such as fibrin and HAp,3,10 in combination with bone-forming cells. As being well described in the pertinent literature HUVECs support osteogenic differentiation, HUVECs and human ADSCs were used to increase bone formation in the AVL model.18,19,33,34 Endothelial cells and mesenchymal stem cells were previously used in several studies as a combination with different biomaterials for tissue engineering in animal models.14,19,33,35,36 But to the best of our knowledge, this study describes the coimplantation of human ADSCs and HUVECs in the rat AVL model for the very first time. Previous studies have already demonstrated that the use of endothelial cells lead to an increase in vascularization.15,17,37 The ADSCs were convincing in contrast to MSCs used in other studies14,19,33,35 for various reasons. The ADSCs are easy to isolate from subcutaneous fat tissue, require undemanding cultivation, and have a great ex vivo expansion potential.38,39
To exploit the full osteogenic potential of ADSCs, osteogenic differentiation can easily be performed by varying the cell culture medium. In a previous study, we were able to prove that HUVECs stimulate osteogenic differentiation, as well as the production of angiogenic markers in cocultured ADSCs. Moreover, a ratio of 50% ADSCs and 50% HUVECs had the highest impact on osteogenic and angiogenic effects. 40 Therefore, this cell ratio was adopted to our in vivo AVL study.
To prevent a rejection reaction due to implanted human cells, T cell-deficient RNU rats were used. As previous studies have demonstrated,7,8 fibrin has a good biocompatibility. During the implantation period of 6 weeks, neither wound healing disorders nor disturbances in the general condition of the animals were observed, indicating a good biocompatibility of the implanted biomaterials. Regarding biodegradation, especially in the cell-containing groups, remodeling processes on the HAp granules were detected. With an average pore size of 0.78 ± 0.40 μm inside a single granule, it is important to remember that the granules have a packing density of ∼54% and provide therefore space for cell invasion and remodeling processes in between the granules. It was demonstrated 41 that an interconnected porosity, suitable for the cells, is necessary for nutrient supply and cell migration within the scaffold.
The experimental results showed that the remodeling processes were almost exclusively limited to the edge of the granules. To increase biodegradation and bone formation, smaller HAp granules with a more advantageous surface-to-volume ratio or a bimodal size distribution resulting in a higher packing density might be preferable for future studies.
Although the formation of a new vascular network was limited by implantation time, construct size, and space for the tissue growth, the vascular network was distributed over all areas of the constructs and there was no difference in the vessel count between the groups. According to our results, Verseijden et al. described in the mouse vasculogenesis model that not only the implantation of HUVECs but also the implantation of fibrin-embedded human ADSCs contributes to vascularization. 42 In addition to cell implantation, fibrin as a proangiogenic hydrogel also shows a positive effect on the formation of new vessels. 43 By linking the extrinsic and intrinsic vascularization mode, the titanium chamber allows continuous vascularization of the constructs. 14 This was demonstrated by the fact that the peripheral areas of the constructs were also permeated with vessels. The distances between the newly formed vessels and the AVL were only significantly different between groups B and D, which may be due to the different distribution of the bone tissue fragments, the position of the AVL or HAp granules.
By and large, the implantation of HUVECs did not lead to an increase in the vessel count, in contrast to the other experimental groups. Interestingly, the implantation of HUVECs led to bone formation, suggesting that implanted HUVECs stimulated bone formation originating from rat osteoprogenitor cells. This result may be caused by the fact that the extracellular matrix from HUVECs stimulate the osteogenic differentiation of osteoprogenitor cells. 44 The implantation of osteogenically differentiated ADSCs (group C) led to higher bone formation than the implantation of HUVECs (group B). But only the coimplantation of ADSCs and HUVECs (group D) led to a statistically significant larger bone area compared with the other cell-containing groups. This supports the results of earlier studies that endothelial cells stimulate proliferation, cell survival, and osteogenic differentiation of osteoprogenitor cells in vitro and bone formation in vivo.40,45–52
In relation to the newly formed tissue, a proportionate bone area of a minimum of 4% (group B) to a maximum of 22% (group D) was found without using any growth factors such as bone morphogenic protein 2. 14 Although coimplantation of ADSCs and HUVECs is a promising approach for bone tissue engineering, a number of factors, such as longer implantation time, the characteristics of the granules or the implantation of a higher cell count, 53 need to be modified in future studies to increase the de novo bone tissue.
Conclusion
For the first time, bone tissue using ADSCs and HUVECs could be successfully generated in the rat AVL model. Bone formation was confirmed in all cell-containing groups with the largest bone area in the HUVEC and ADSC coimplantation group. Moreover, all constructs demonstrated a complete vascular network enabling optimal conditions for bone formation by the implanted cells. Taken together, ADSCs and their osteogenic potential in combination with HUVECs represent a promising approach for bone tissue engineering applications in the AVL model.
Footnotes
Acknowledgment
The authors cordially thank Dr. Alexander Haydl for proof reading the article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by ELAN Fonds (17-10-18-1-Steiner). Furthermore, we acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—project number 326998133–TRR 225 (subprojects C03 and C04).