
Abstract
Microvascular remodeling is a complex process that includes many cell types and molecular signals. Despite a continued growth in the understanding of signaling pathways involved in the formation and maturation of new blood vessels, approximately half of all compounds entering clinical trials will fail, resulting in the loss of much time, money, and resources. Most pro-angiogenic clinical trials to date have focused on increasing neovascularization via the delivery of a single growth factor or gene. Alternatively, a focus on the concerted regulation of whole networks of genes may lead to greater insight into the underlying physiology since the coordinated response is greater than the sum of its parts. Systems biology offers a comprehensive network view of the processes of angiogenesis and arteriogenesis that might enable the prediction of drug targets and whether or not activation of the targets elicits the desired outcome. Systems biology integrates complex biological data from a variety of experimental sources (-omics) and analyzes how the interactions of the system components can give rise to the function and behavior of that system. This review focuses on how systems biology approaches have been applied to microvascular growth and remodeling, and how network analysis tools can be utilized to aid novel pro-angiogenic drug discovery.
Introduction
A major hurdle in this area is the sheer complexity of microvascular growth and our incomplete understanding of the process. Angiogenesis, the process by which new capillaries sprout from preexisting vessels, involves the coordinated response of different cell types and molecular signals across various spatial and temporal scales. Early experimental progress in angiogenesis research highlighted the importance of vascular endothelial growth factor (VEGF) as a key angiogenic stimulus. 8 As a result of early success, most pro-angiogenic clinical trials to date have focused on increasing neovascularization via the delivery of a single growth factor or gene (most notably, VEGF). However, the disappointing clinical outcomes of single-factor therapies for promotion of angiogenesis serve to highlight the complexity of cell signaling networks that govern microvascular growth and remodeling. Even so, much attention is presently focused on preclinical studies that identify combinations of promising angiogenic agents, including the fibroblast growth factors (FGFs), platelet-derived growth factors (PDGFs), angiopoeitins (Ang-), and the transforming growth factors (TGFs).6,9,10 However, as screening libraries of potential drug candidates and drug combinations experimentally is prohibitively time consuming, expensive, and relatively limitless in scope, 11 many researchers have begun to turn to computational approaches. 12
Over the past 20 years or more, numerous mathematical and computational models of angiogenesis have been built to enhance our understanding of the process (for detailed reviews of the subject, see Refs.13,14). In 1998, a group of physiologists, biomedical engineers, and bioinformatics experts initiated the Microcirculation Physiome Project with the goal “to create a World Wide Web accessible database of the microcirculation.” The Microcirculation Physiome Project was created to integrate biological and functional data with computational models from the gene to the tissue level. 15 Although the vision of the founders has yet to fully materialize, significant progress has been made in the area of microvascular network modeling. Much of this work can be found at one online database, a resource for computational models on the microcirculatory system (www.physiology.arizona.edu/people/secomb/network.html) or via links from the Physiome Project Web page (www.physiome.org/Links/#group). 16
Systems biology can be defined as the integration and analysis of complex biological data from a variety of experimental sources, and how the interactions of the system components give rise to the function and behavior of that system. Scientists have worked from a reductionist perspective, which is by definition hypothesis-driven. In the past decade, however, systems biologists have challenged conventional experimentation and introduced new possibilities in testing scientific hypotheses. 17 These so-called holistic approaches aim to discover new emergent phenomena from combining the reductionist approach with network-level analysis of whole biological systems. In this manner, groups of data (-omics) have emerged, including genomics (gene sequencing), transcriptomics (RNA transcripts), and proteomics (protein expression and modification). Thus, the goal of systems biology is to develop a comprehensive integrated system in which all “omic” data can be incorporated and interpreted in a manner in which the whole is greater than the sum of the parts. 16 To this end, the use of network analysis tools has emerged to help explain experimental results and elucidate coordinated signaling pathways of entire networks of growth factors and genes that would otherwise not be possible through the examination of single-component regulation alone.
Transcriptomics and proteomics have recently contributed to unraveling the complexity of angiogenesis. During the complex process of neovascularization, the balance of pro- and antiangiogenic growth factors (i.e., statins and thrombospondin) controls the rate and extent of microvascular growth (Fig. 1). Matrix metalloproteinases (MMPs) degrade the extracellular matrix (ECM), allowing for endothelial cells (ECs) to form a sprout tip. ECM-bound factors that are released allow ECs to migrate and proliferate as a function of local growth factor gradients. As sprouts form, they can anastomose to adjacent vessels, retract in the absence of pro-angiogenic stimuli, or split or branch. The fate of several sprouts eventually result in a newly organized capillary network that, with the onset of flow, carries blood, oxygen, nutrients, and cells to surrounding tissues. Although the process of angiogenesis occurs under both physiological (i.e., exercise) and pathological (i.e., cancer) conditions, the signaling networks and governance of the process are tightly regulated. For example, studies using transcriptomics approaches systematically compared the gene expression profiles of ECs isolated from normal, tumor, and regenerating blood vessels. 18 Not surprisingly, ECs expressed different gene expression profiles depending on their pathological state.
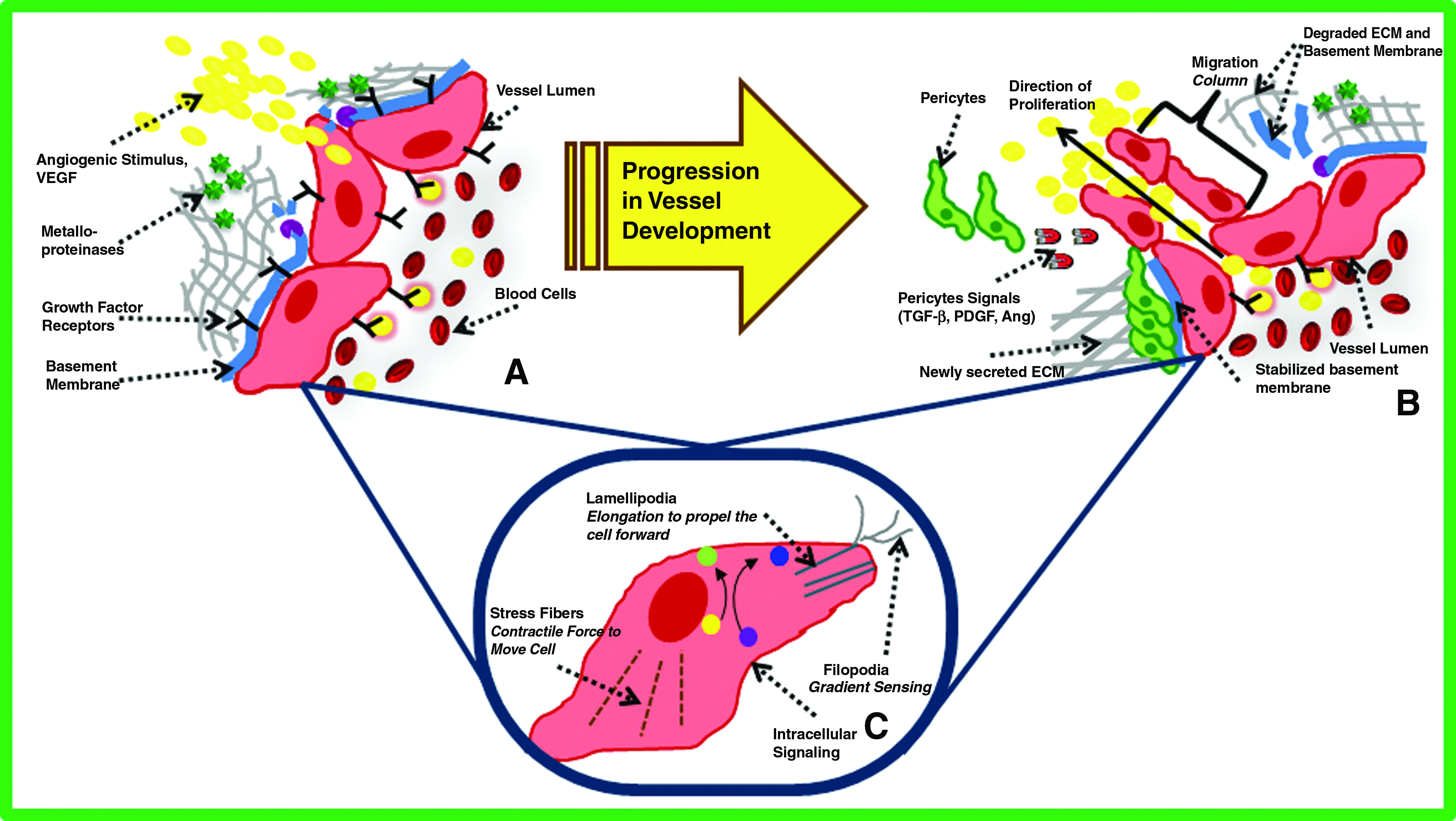
(
In this review, we will discuss both bottom-up and top-down methods that have been taken to elucidate pro-angiogenic targets. In addition, we will summarize the different types of systems biology approaches that have been employed to clarify our understanding of microvascular remodeling and identify potential therapeutic approaches. We will discuss several -omics approaches and how they have already been exploited to provide new insights to the fields of tissue engineering and vascular biology. Lastly, we will summarize the network analysis tools that can be utilized to elucidate signaling mechanisms of novel pro-angiogenic drug candidates.
Bottom-Up Modeling Approaches
In the bottom-up systems biology approach, information is initially gathered about all the components of a system and then constructed into a model that is subsequently used to predict systemic properties. For example, molecular signaling pathways may be combined to predict cellular functions, which can then affect tissue patterning or even organ development. Experimental validation of the models and appropriate refinement and iteration are critical. The ultimate goal of bottom-up systems biology is to combine the pathway models into a larger, unified model of the entire biological system of interest, that is, tissue or organ (Fig. 2).
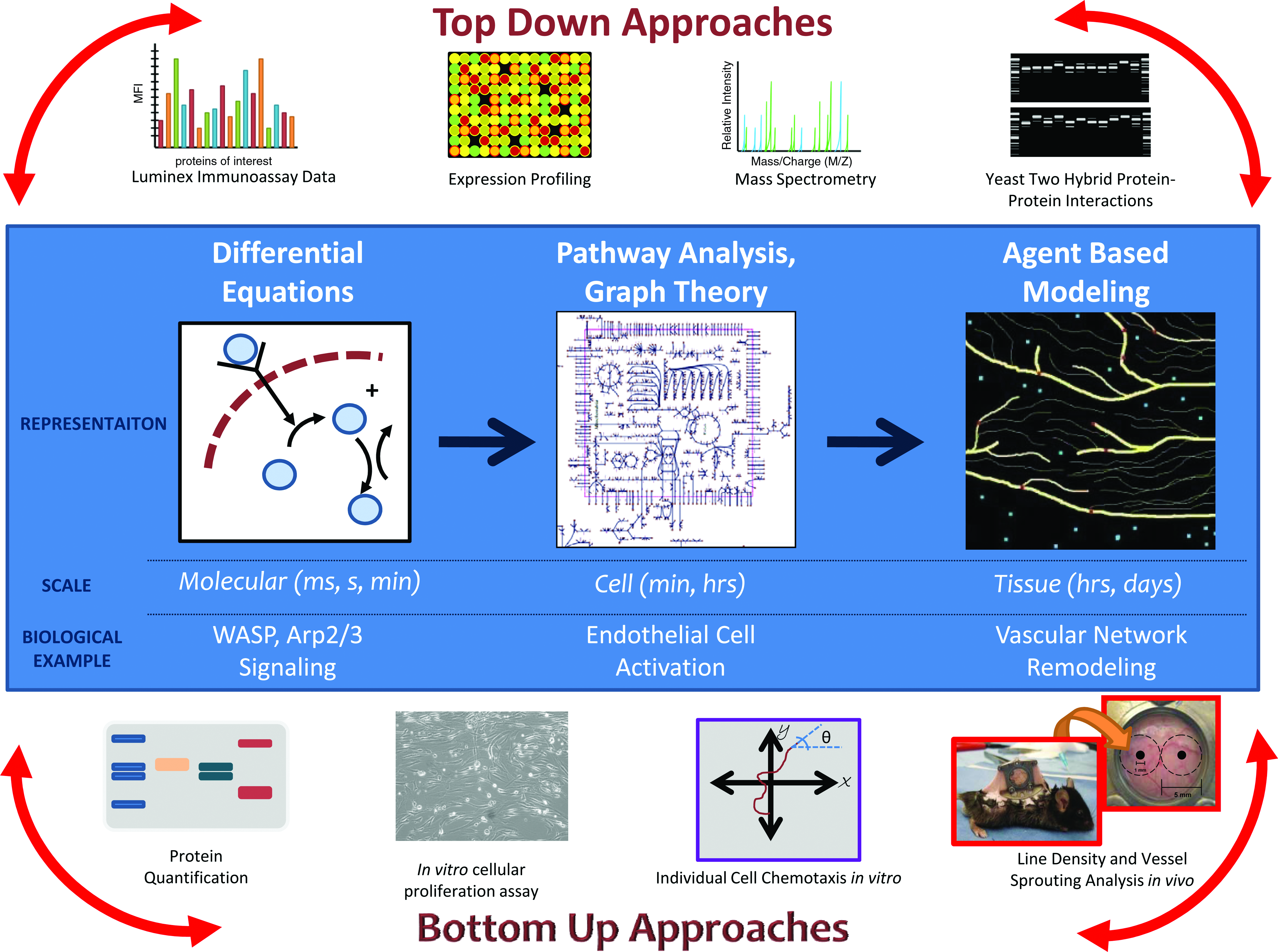
Systems biology approaches to engineer microvascular networks aid pro-angiogenic drug discovery. Bottom-up approaches focus on collecting and accumulating empirical data about the system and then incorporating these data into a larger, more unified model that explains emergent behaviors and properties. Top-down approaches are data-driven and focus on network level integration to identify novel interactions or mechanisms. Experimental validation of the models and appropriate refinement and iteration are critical. The ultimate goal is to combine top-down and bottom-up approaches to create a unified model of the entire biological system of interest spanning different spatial and temporal scales. Color images available online at www.liebertonline.com/ten.
Most angiogenesis models to date have taken the bottom-up approach. These modeling approaches can be classified into four subcategories: continuum models (processes are continuous in time and space), discrete models (system components are individual entities), stochastic models (probability dictates the occurrence of biological events), and deterministic models (system outcomes are determined by previous system inputs). 13 Although early models captured some of the key processes of angiogenesis (e.g., average capillary sprout density), they are limited because of their constraints in only one spatial dimension.19,20 Nearly two decades ago, Stokes and Lauffenburger developed a discrete, stochastic mathematical model of EC migration.21,22 The model predicted that the rate of vascular sprouting is primarily determined by EC migration rate in the direction of an angiogenic stimulus, namely, acidic FGF, but that a certain degree of randomness in migration direction is required for vessel anastomoses and capillary loop formation. Importantly, their model predictions were validated in vitro by measuring the displacement of ECs in the presence or absence of acidic FGF. Although in two space dimensions, the model did not account for interactions between the ECs and the ECM.
To this end, more recent models have combined the model strengths of their predecessors, including ECs, growth factors, and ECM molecules. Sun et al. elaborated on prior work by Anderson and Chaplain to create a model that accounts for the heterogeneity and anisotropy of the ECM.23,24 The continuous deterministic model included major mechanisms such as cell proliferation, branching, and anastomosis. Perturbations of the model's parameters revealed that capillary branching and extension is independent of starting geometry, that more tortuous networks with less branching occur with a highly anisotropic ECM (which slows down EC migration rate), and that there exists an optimal chemoattractant growth factor concentration that allows for proper branching and extension. 24
Cellular automata (CA) simulations, or agent-based models, have been used in the natural and social sciences to predict emergent patterns. This work has extended into the biological sciences to simulate vascular growth and predict blood vessel patterning across different spatial and temporal scales. In agent-based models, each agent behaves according to a given set of rules and can interact with and modify its surroundings. A decade ago, Markus et al. developed a CA model of vessel morphogenesis that describes leaf vein patterning based on rules for branching, chemotaxis, anastomosis, and tip death. 25 By adding an angiogenic gradient to their basic model, the authors showed a proof of concept that the CA could be applied to blood vessel growth; however, model revision and validation were not completed. Peirce et al. developed one of the first biological agent-based models to predict microvascular network patterning from a rule set based on epigenetic stimuli, molecular signals (PDGF-BB VEGF, TGF-β), and cellular behaviors (proliferation, migration, and differentiation). 26 The model predicted changes in microvascular patterning in response to hemodynamic forces and exogenous focal delivery of a growth factor (VEGF) over a 10- or 14-day period. Model predictions were validated with in vivo experimental results, demonstrating an increase in vascular length density in response to VEGF treatment or increased circumferential wall strain. Importantly, the model revealed changes in microvascular patterning in a multicell system (ECs, smooth muscle cells, perivascular cells, and interstitial precursor cells) and could prove to be a useful tool for the rationale design of therapeutic vascularization strategies. Most recently, Qutub and Popel reported on an agent-based model designed to predict tip cell activation, stalk cell development, and sprout formation as a function of local VEGF concentration. 27 Rules and parameters were based on literature values and in vitro experimental results, and included various growth factors (VEGF, hypoxia-inducible factor 1, alpha subunit [HIF1α]), and Notch ligand delta-like 4), MMPs, and cellular functions (migration, elongation, and proliferation). Another agent-based modeling study by Bentley, Gerhardt, and Bates explored the role of VEGF-A and delta-like 4 (D114)/notch signaling in tip cell formation. This group developed a hierarchical model focused on VEGF-A-stimulated tip formation and subsequent D114/notch VEGF receptor 2 (VEGFR-2) inhibition. They found that D114/notch inhibition in high VEGF-A environments increased aberrant vessel growth. Partial or total inhibition of D114/notch signaling normalized tip cell response in high VEGF-A environments. 28 To date, agent-based models have proven to be a useful tool for hypothesis testing and hypothesis generation by providing a platform for high-throughput and low-cost experiments.
Future iterations of CA models would benefit from adding detailed models of specific angiogenic signaling networks. In a true bottom-up approach, Mac Gabhann and Popel have created molecular-level models to investigate the role of VEGF receptors in angiogenesis. Because most of the clinical trials aimed at increasing functional vascularization in ischemic tissues have failed, computational and mathematical models that incorporate the molecular-level details of VEGF signaling that could then be utilized to predict effective therapies would be a major advancement. For example, Mac Gabhann et al. developed a three-dimensional model to approximate the concentrations of VEGF at the single-cell level in skeletal muscle during exercise.29,30 This type of information is currently unobtainable experimentally, but is critical to understanding cellular responses to local growth factor gradients. The model accounts for signaling network details of VEGF, as well as oxygen transport and blood flow. This type of model could be an extremely valuable tool for tissue engineers who seek to evaluate local, sustained VEGF administration in ischemic tissues. With the incorporation of other molecular-level details of growth factor signaling networks, the model could even prove more valuable.
In addition to molecular signals, mechanical stimuli also govern the structural remodeling response of the microcirculation. The role of hemodynamics in the microcirculation has been studied best by Pries and Secomb.31–34 The authors have investigated the structural adaptation of microvessels in response to mechanical forces, including blood flow (shear stress) and blood pressure (circumferential wall stress). Several mathematical models of angioadaptation (postnatal structural remodeling of blood vessels) have been developed to predict changes in vessel architecture (vessel number, diameter, wall thickness, and length) as a function of blood flow and pressure. 35 Although the mathematical equations governing blood flow are well documented and are not the focus of this review, their impact on angiogenesis and remodeling have inspired other tissue-engineering-focused models. Because of the mass transport limitations associated with tissues > 1 mm3, engineers have been trying to prevascularize tissues and engineered scaffolds to increase the rate of success of tissue-engineered biological implants. Janakiraman et al. developed a modeling approach for the rational design of blood vessel networks with mass transport characteristics that meet the metabolic demand of tissues. 36 A two-dimensional microscale model with a bifurcating network design was created and used to evaluate the effects of two different network geometries (rectangular vs. square ducts) on their mass and fluid transport characteristics. The model revealed that the rectangular ducts exhibited superior mass transport efficiency and were easier to fabricate than the square ducts. Additionally, mass transport efficiency decreases with an increase in network porosity, revealing a design criterion to minimize network bifurcations. Jabbarzadeh and Abrams developed a model to study growth factor (VEGF) diffusion in tissue-engineered constructs. 37 The authors varied the source of VEGF diffusion (line, line + point, and line + boundary release). Results revealed that a line source with release throughout the boundary of the construct led to a more uniform microvascular network distribution, with vascular coverage proportional to the amount of VEGF released. Model integration of Janakiraman and Jabbarzadeh would be useful to rationally design prevascularized tissue-engineered constructs with efficient mass transport characteristics and sustained growth factor release.
Many computational and mathematical angiogenesis models have been designed in the context of tumor vascularization because the field of therapeutic microvascular formation is still in its infancy. However, because the basic principles upon which tumor models have been built are similar to the processes governing therapeutic neovascularization, tumor models may prove useful for testing specific hypotheses, such as the identification of drug candidates. Arakelyan et al. developed a model to elucidate the role of specific growth factors on tumor angiogenesis (i.e., Ang-1, Ang-2, VEGF, and PDGF). 38 Importantly, the model predicted that only dual administration with anti-VEGF and anti-Ang-1 drugs resulted in significant decreases in tumor size, an outcome that was not observed with single administration of either factor alone. Perhaps most interestingly, the anti-VEGF drug was most effective when the percentage of immature vessels (vessels lacking pericytes coverage) was high (95%, relative to mature pericyte-containing vessels). This result may shed new light on therapeutic neovascularization approaches; perhaps a certain percentage of mural cells are needed to drive vascular growth and maturation.
Top-Down Modeling Approaches
Top-down systems biology approaches are data driven. In the advent of genomics data, these top-down approaches have emerged as new ways to study biological constituents and their interactions. The goal of top-down systems biology is to discover new molecular mechanisms through an iterative process between experimental data and hypothesis generation resulting from network level data integration. These new hypotheses can then be evaluated by experimental techniques and so forth. In this manner, previously unidentified interactions, mechanisms, and drug candidates can be identified. These -omics technologies have revolutionized biomedical research, providing new insights into mechanisms of disease and generating novel diagnostic and prognostic tools. 39 Example applications of these techniques and analyses are shown in Table 1.
VEGF, vascular endothelial growth factor; MAPK, mitogen-activated protein (MAP) kinase; VEGFR1/2, vascular endothelial growth factor receptor 1, or 2.
Although top-down, high-throughput approaches reveal novel facets of a particular system, they are not without limitations. This is the case for the VEGF signaling pathway, which has been the target of numerous system-wide studies. As such, Table 1 highlights high-throughput approaches in VEGF signaling, their strengths, limitations, and possible complementary approaches. In the end, the careful selection of additional assays that support conclusions drawn from systems analysis methods will be warranted. Table 1 is not an exhaustive list of VEGF systems biology approaches. (For a more thorough review of VEGF systems biology, we refer you to a review by Mac Gabhann and Popel. 40 )
Systems biologists use computational analysis tools to derive hypothesis from large data sets. Computational tools range in complexity and sophistication, and generate different hypotheses based on the nature of the analysis (Fig. 2). In the context of microarray data, a simple statistical analysis reveals which genes are significantly up-regulated or down-regulated compared to a predetermined threshold under a particular experimental condition. To better understand multiple gene interactions or patterns of activity, a more advanced clustering analysis may be required. These techniques are broadly applicable to any large data set and may be augmented depending on the nature of the system of interest. Quackenbush provides a more thorough introduction to the basic computational analyses available. 41 However, the field is constantly changing as new computational algorithms are developed to deal with complex data sets.
In recent years, genomic and proteomic approaches have elucidated many cellular and molecular mechanisms of angiogenesis and subsequently identified potential therapeutic targets for the manipulation of this process.42,43 Functional genomics approaches have been used to compare the gene expression profiles of ECs isolated from normal, tumor, or regenerating blood vessels.18,44–46 These studies have shown that ECs activate different signaling cascades depending on their location and physiological or pathological state. Such data can help guide therapies targeted to pro- or antiangiogenic drug delivery strategies.
In the context of tumor vascularization, top-down systems biology techniques have the capacity to unravel the complexity of angiogenesis. For example, a multiscale model depicting signaling dynamics between the pro- and antiangiogenic compounds within tumor cells qualitatively predicted the effect of endostatin gene therapy. The model determined a critical endostatin expression level required for therapeutic inhibition of angiogenesis. 47 Genomic analysis of annexin A1 knock out mice revealed that the protein is implicated in inflammatory and pro-angiogenic processes, making it an ideal candidate for cancer therapy. 48 Because tumor angiogenesis applications of systems biology are not the emphasis of this review, we direct you to Kreeger and Lauffenbuger, who provide a more thorough review of cancer systems biology techniques. 49
In the previous example of genomic annexin A1 analysis, the group also reported that annexin A1 overexpression may be a therapeutic means to increase wound healing. 48 Although the application of top-down approaches to therapeutic angiogenesis is limited, there are numerous ways to apply current approaches to developing functional microvasculature. Pathways analysis, for example, can be employed on genomic data from cDNA microarrays to discover the mechanisms of action of unknown drug candidates, to predict efficacy, or to identify new drug leads. 50
To provide an example, our group has been investigating the mechanism of action of the pro-angiogenic molecule phthalimide neovascular factor (PNF1), a synthetic small molecule that has demonstrated efficacy in enhancing EC proliferation and capillary network formation in vitro. 51 Additionally, local sustained release of PNF1 has led to increased neovascularization in the mouse dorsal skinfold window chamber model of microvascular growth and remodeling. 52 These results highlight PNF1 as a potential candidate for pro-angiogenic therapies. However, because its mechanism of action is unknown, transcriptional profiling was used to elucidate the mechanism underlying the biological activity of PNF1.53,54 cDNA microarray data containing ∼47,000 transcripts were generated for cultures of human microvascular ECs stimulated with PNF1 for various time points and compared to cultures treated with a vehicle control. Examination of the data using network analysis tools revealed a concerted regulation of many gene products that have known effects on vascular remodeling, providing a potential window for therapeutic manipulation with PNF1 delivery.
A novel compendium analysis was performed in the aforementioned interrogation of PNF1's mechanism. Traditionally, compendium analyses have been used to compare the genetic expression profiles of unknown drugs to signatures of known compounds, as drugs with similar genetic footprints are likely to share mechanistic targets. 55 Compendium analysis involves rating the similarity between a measured data set and the compendium, or compilation, of previously generated profiles. This technique is visually represented in Figure 3. Realistically, however, transcriptional profiles of similar drugs tested on the same cell type at similar concentrations often do not exist, making direct comparisons difficult and open to interpretation. Likewise, docking studies require an exhaustive screening process of all potential binding partners, which is both time consuming and prohibitively expensive. To this end, our novel compendium approach overlaid the transcriptional profile of a small molecule on a literature-derived pathway knowledge database (Ingenuity Pathways Knowledge Base [IPKB], www.ingenuity.com/products/pathways_knowledge.html). The IPKB is essentially a library of signaling interactions that may be used to elucidate associations between previously unrelated signaling network components. After identifying new network connections, Ingenuity Pathways Analysis iteratively compares and ranks these networks and markers relevant to the connections (Fig. 3). This process is similar to determining that two people are related in a family tree based on the acquisition of new knowledge that they share the same third cousin. Using Ingenuity Pathways Analysis to perform a novel compendium analysis, our group identified signaling networks that were most directly correlated with the gene expression data (for details, see Ref. 54 ). This methodology revealed that PNF1 predominately affects transcription of genes in the tumor necrosis factor-α (TNF-α) pathway at earlier time points (1- and 2-h poststimulation), whereas TGF-β-related signaling molecules are more influential at later time points (especially after 24 h). Both TNF-α and TGF-β have known activities in microvascular remodeling, and their identification as major players in PNF1-induced angiogenesis may now lead to drug optimization protocols and proper therapeutic utilization.
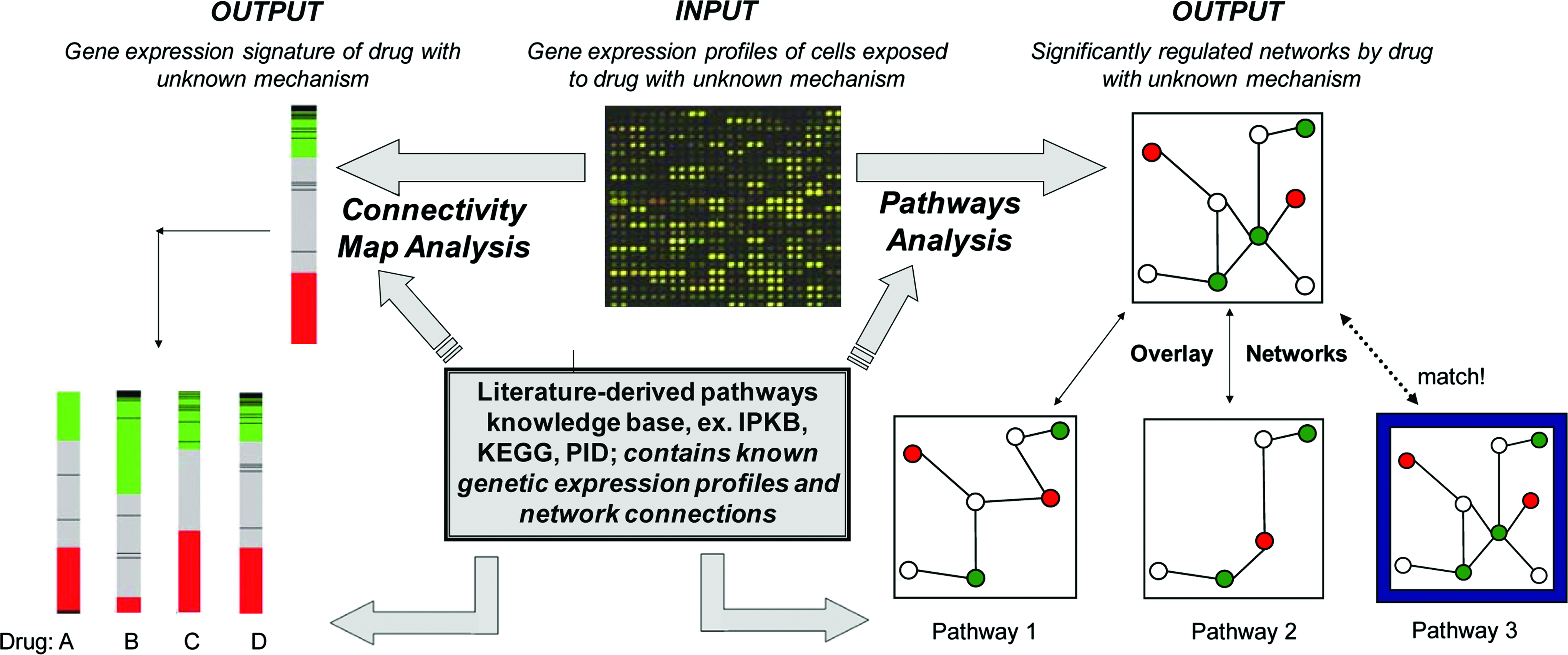
Network analysis tools. Microarray data from a drug of unknown mechanism can be analyzed using a compendium analysis approach (Pathways Analysis, shown on right) or a genomic-mapping approach (Connectivity Map Analysis, shown on left). Both top-down approaches rely on literature-derived knowledge bases that are manually curated and contain known genetic signatures and signaling networks. The unknown data are mapped against data from drugs with known mechanisms to identify areas of overlap, thereby indicating similarity in drug mechanism. Color images available online at www.liebertonline.com/ten.
Similar to using the IPKB to conduct compendium analyses, the Connectivity Map seeks to correlate expression data with a larger database (Fig. 3). This database contains catalogs of expression data from human cells exposed to various drug stimulations or disease states. 56 The Connectivity Map has evolved as a resource whereby a researcher studying a drug candidate, gene, or disease state, could compare its signature to a reference database to discover functional connections to known pathways. 56 This Web-based tool is publicly accessible at www.broad.mit.edu/cmap, allowing researchers to perform their own connectivity map analyses with user-defined signatures in real time. One large advantage over other compendia-based approaches is that the Connectivity Map enables the integration of internal reference data (genome-wide expression profiles) with external query data provided by users in the form of a genetic signature. The initial focus of the project is to include the genetic signatures of all the 1500 small-molecule drugs licensed by the Food and Drug Administration for human use; currently, the database contains gene expression profiles from 453 treatments of four cell lines with 164 bioactive molecules. 57 Ultimately, the goal of the Connectivity Map project is to connect human diseases with the genes that underlie them and the drugs that can treat them.
With emerging tools like the Connectivity Map, identifying genes responsible for the progression of cancer, for example, may become easier. The hallmark of cancer progression is the angiogenic switch, a shift of the angiogenic balance to a pro-angiogenic state. 58 Human tumors arise and can exist in the microscopic state avascularized for months or years. The switch to a pro-angiogenic phenotype results in rapid neovascularization, tumor growth, and subsequent metastasis. Although angiogenesis has been widely studied and several key components of the angiogenic switch have been identified, the molecular and genetic mechanisms mediating the switch are largely unknown. 59 Abdollahi and colleagues analyzed the human transcriptome by cDNA arrays using clustering techniques to observe how gene expression was altered after treatment with endostatin (antiangiogenic), VEGF alone, and VEGF plus basic FGF (bFGF) (pro-angiogenic). Although several well-known angiogenic genes were represented in the network (HIF1α and VEGFR-2), genes only recently reported as angiogenic were also highlighted by the clustering methodology. Further, genes not known to be connected to angiogenesis, such as peroxisome proliferator-activated receptor delta (PPARδ), interleukin-6 (IL6), MMP1, and others, were represented in high numbers, suggesting a broader participation in the angiogenic switch than previously hypothesized. To validate this prediction, the authors targeted the removal of the PPARδ hub node by using PPARδ−/− mice, and the results demonstrated a reduction in tumor growth and tumor microvascular density. This confirms a critical involvement of the signaling pathway in angiogenesis, and highlights the powerful utility of this top-down systems biology approach. 59 Interestingly, dual administration of VEGF and bFGF did not result in a significant upregulation of pro-angiogenic transcripts compared to either factor alone. One potential explanation is that VEGF and bFGF share a common pathway. The angiogenic phenotype observed in VEGF or bFGF stimulated tissues could be a result of activating similar downstream signaling cascades. There exist other known angiogenic stimuli, such as sphingosine 1-phosphate for example,60,61 that presumably have less crosstalk with VEGF signaling due to their different receptors (sphingosine 1-phosphate: G protein-coupled receptors, VEGF: receptor tyrosine kinase). It would be interesting to use systems biology tools to assess stimulation of two pathways, with minimal crosstalk, on angiogenesis. This method may identify novel targets of angiogenesis.
Future Directions
In silico strategies aimed at predicting clinical efficacy could have a major impact on the current pharmaceutical approach to drug discovery. The development of new drugs is a risky and costly process; approximately half of all compounds entering phase II clinical trials will fail, resulting in over $8 million per drug in amortized costs. 11 Thus, the application of systems biology approaches to drug discovery could streamline the entire process and save time, money, and resources. In this review, we have discussed both bottom-up and top-down systems biology approaches that have already been taken to identify pro-angiogenic drug candidates and engineer microvascular networks. Figure 2 visually summarizes how these different approaches probe the microvasculature at multiple scales. In isolation, each computational technique deepens the understanding of a particular facet of the microvascular system. However, bottom-up modeling and top-down modeling have yet to be combined in one comprehensive model.
To be ultimately successful, however, all of these systems methodologies depend on the quality and completeness of the data at hand. Several bioinformatics databases have evolved to integrate the various data forms and aid hypothesis generation.62–64 For example, the Pathway Interaction Database (http://pid.nci.nih.gov) is a highly curated, freely available collection of human signaling and regulatory pathways that was specifically designed to deal with incomplete knowledge, complex details, or generalizations. 63 Researchers can search for a single molecule to find its known mechanistic pathway or for a group of molecules to examine signaling interactions among them. The database is updated with new pathway information each month, including the National Cancer Institute (NCI)–Nature Curated collection, Reactome data, and the BioCarta collection (for a list of pathways, see: http://pid.nci.nih.gov/browse_pathways.shtml). Such breadth in imports will hopefully allow researchers to investigate novel networks and reveal parallel, yet alternative hypotheses, such as providing a mechanism by which a single pro-angiogenic compound is not effective and suggesting potential multiagent therapies. The future of systems biology will likely require curated databases and online workstations like the Physiome Project, Connectivity Map, and Pathway Interaction Database, among others. Although not without limitations, top-down and bottom-up systems biology approaches to drug discovery and microvascular network formation represent the future in bioinformatics strategies and have already led to important discoveries regarding the mechanisms of novel compounds and their interactions at the network scale.
Footnotes
Acknowledgments
Funding support was provided by NIAMS grant K01AR052352-01A1 to Dr. E.A. Botchwey and NSF CAREER award #0643548 to Dr. J.A. Papin.
Disclosure Statement
No competing financial interests exist.