
Abstract
Millions of people worldwide suffer from trauma- or age-related orthopedic diseases such as osteoarthritis, osteoporosis, or cancer. Tissue Engineering (TE) and Regenerative Medicine are multidisciplinary fields focusing on the development of artificial organs, biomimetic engineered tissues, and cells to restore or maintain tissue and organ function. While allogenic and future autologous transplantations are nowadays the gold standards for both cartilage and bone defect repair, they are both subject to important limitations such as availability of healthy tissue, donor site morbidity, and graft rejection. Tissue engineered bone and cartilage products represent a promising and alternative approach with the potential to overcome these limitations. Since the development of Advanced Therapy Medicinal Products (ATMPs) such as TE products requires the knowledge of diverse regulation and an extensive communication with the national/international authorities, the aim of this review is therefore to summarize the state of the art on the clinical applications of human bone marrow-derived stromal cells for cartilage and bone TE. In addition, this review provides an overview of the European legislation to facilitate the development and commercialization of new ATMPs.
Introduction
T
However, while autologous transplantation can lead to chronic pain in the explant region and is limited by material availability, allogenic transplantation potentially faces complications such as graft rejection and disease transfer. Moreover, synthetic substitutes and prosthesis alone often lack adequate mechanical or biological properties for large defect regeneration,1–4 and their architecture doesn't often allow repopulation of the bone marrow cavity—hosting cells of the hematopoietic but also mesenchymal and endothelial lineages important for bone regeneration.
For these reasons, TE bone and cartilage products represent a promising and alternative approach 5 with the potential to overcome the limitations of the clinically established therapies.
Advanced Therapy Medicinal Products (ATMPs) started acquiring a proper legislative identity and the due attention from the market with the institution of the European regulatory framework concerning gene therapy medicinal products and somatic cell-therapy medicinal products (European Directive 2001/83/EC). Consequently, a definition of principles and guidelines for the manufacturing of products intended for human use and investigational medicine and complying with international standards was finally needed.
Since the development of TE products for human applications requires an extensive communication with the national and international authorities, the aim of this review is to provide the reader with the state of the art on the applications of human bone marrow-derived stromal cells for cartilage and bone TE in the clinical scenario and to provide the required legislative background for the development of new ATMPs in Europe able to push forward the actual clinical state of the art.
ATMPs and Good Manufacturing Process: The European Legislative Framework
The directive 2003/94/EC 6 lays down the guidelines for good manufacturing process (GMP) definition, establishing three important pillars for a patient- and product-centered standardization of the medicinal product: (1) the institution of quality control procedures allowing standardization of the manufacturing procedures and traceability of materials and final products; (2) the integration of procedures for complaint reception, product recall, and emergency unblinding of the studies; (3) and the establishment of self-inspection analysis to monitor and eventually correct deviations from the internally determined quality standards.
These principles have subsequently become integral part of the two major legislative redlines concerning TE products, which are the establishment of quality and safety criteria for the donation, procurement, testing, processing, preservation, storage, and distribution of human tissues and cells, regulated by the directive 2004/23/EC 7 later implemented by the directive 2006/17/EC 8 ; and the regulation of ATMPs, introduced by the regulation 1394/20079 and the directive 2009/120/EC 10 amending the directive 2001/83/EC. 11 According to these regulations, the main requirement for the production of ATMPs for human use is the accreditation, designation, authorization, or licensing of tissue establishments and tissue and cell preparation processes (Art. 6-7, 2004/23/EC 7 ) that include established standard operating procedures (SOPs), guidelines, training and reference manuals, reporting forms, donor records, and information on the final destination of tissues or cells (Art. 16) in their quality management systems (QMS).
Manufacturers should ensure confidentiality of any health-related information and traceability (Art. 8) of all the employed cells and materials from the product to the donor for a minimum of 30 years after clinical use and submit an annual report to the authorities (Art. 10) on their activities.
As such, the European regulation attempts to restrict the ATMP manufacturing processes to structures able to provide a transparent reporting of their activities, standardize their production, and minimize the risk for the patient, while leaving relative freedom on the scientific and medical aspects of the product design. Annex I of 2006/17/EC 8 defines in fact both deceased and living donors as possible sources of cells and tissues, and together with Annex II identifies a series of exclusion criteria and biological tests to characterize healthy donors or donors with a clinical history not resulting detrimental for the quality of the tissues or cells to be retrieved.
The definition of ATMPs was set with the regulation No. 1394/2007, 9 and more specific requirements were defined in 2009/120/EC. 10 In this regulation, the definition of “Tissue Engineering products” and of “combined-ATMPs” incorporating medical devices or implantable medical devices (directive 90/385/EEC 12 and 93/42/EEC 13 ) is added to what was already defined in 2001/83/EC. 11 With the 1394/2007, 9 a Committee for Advanced Therapies (CAT) was instituted as official consulter responsible for expressing an opinion on the quality, safety, and efficacy of each ATMP for final approval. TE products in particular are defined as a subclass of “somatic cell therapy medicinal products” and must contain human or animal engineered cells or tissues.
Engineered cells/tissues are by definition subject to substantial manipulation to be used for a different function in the recipient as in the donor. Cutting, grinding, shaping, centrifugation, soaking in antibiotic/antimicrobial solutions, sterilization, irradiation, cell separation/concentration/purification, filtering, lyophilization, freezing, cryopreservation, and vitrification are not considered substantial manipulations according to current regulation (Art. 2.1.c 1394/20079), and therefore, many of the standardly used procedures for cell isolation and culture are not significantly affected by it. Xenogeneic cell-based products may be used upon submission of information regarding the source of animals, specific acceptance criteria, measures to prevent and monitor infections in the source animals, testing for infectious agents, and evidence of the suitability of the animal facilities (Art. 3.3.2.1 2009/120/EC 10 ).
The testing of any additional substance like scaffold, matrices, devices, biomaterials, biomolecules, or other components combined with the engineered cells should be described. Furthermore, the manufacturing process should be validated to ensure batch/process consistency and functional integrity from the manufacturing to the moment of application (Art. 3.3.2.2). This includes characterization of the cells used in terms of identity, purity, viability, potency, karyology, tumorigenicity, genetic stability, suitability for the intended use, and compatibility of additional substances with the used cell types.
Finally, the primary pharmacological studies should be adequate to demonstrate the proof of concept (Art. 4.3), and the therapeutic amount of product should be indicated; while conventional pharmacokinetic studies are not required, parameters such as viability, longevity, distribution, growth, differentiation, migration, toxicity, carcinogenicity and genotoxicity, immunogenic and immunotoxic effects, and zoonosis transmission risk should be investigated. A summary of a typical pipeline for ATMP development and approval is shown in Figure 1, while a timeline of the evolution of the European legislation is shown in Figure 2.
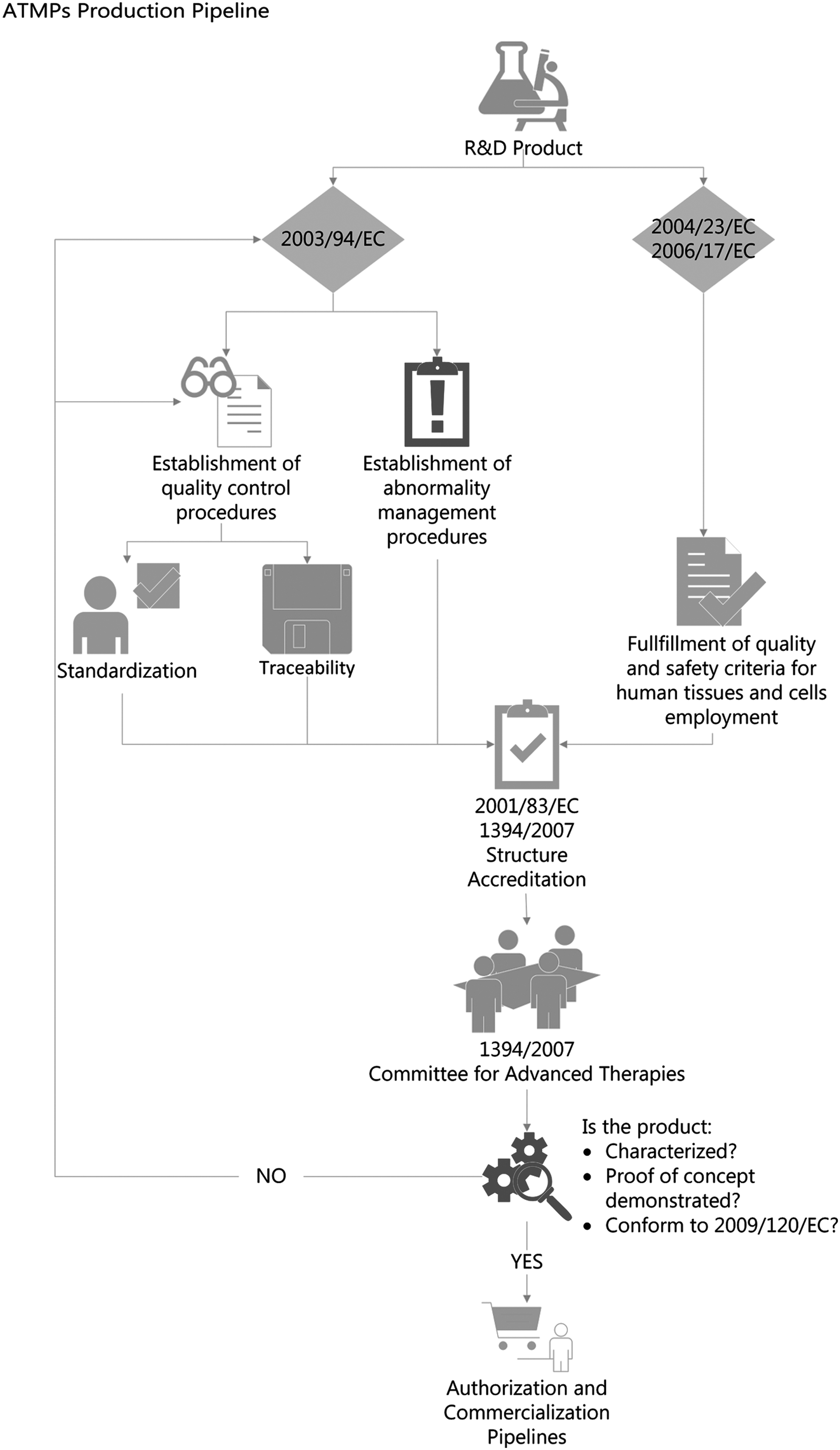
Pipeline for the production of an ATMP. After the R&D phase, quality control procedures ensuring standardization and traceability of the product, alongside procedures for the management of abnormalities, have to be established according to 2003/94/EC. Moreover, criteria for quality and safety of human tissues and cell employment need to be conformed to 2004/23/EC and 2006/17/EC. After accreditation of the structure according to 2001/83/EC and 1394/2007, the Committee for Advanced Therapies is responsible for the evaluation of the documentation regarding the product characterization, proof of concept, and conformity to 2009/120/EC. Upon authorization from the Committee, the product can enter the EU-authorization and commercialization pipelines. ATMPs, Advanced Therapy Medicinal Products.
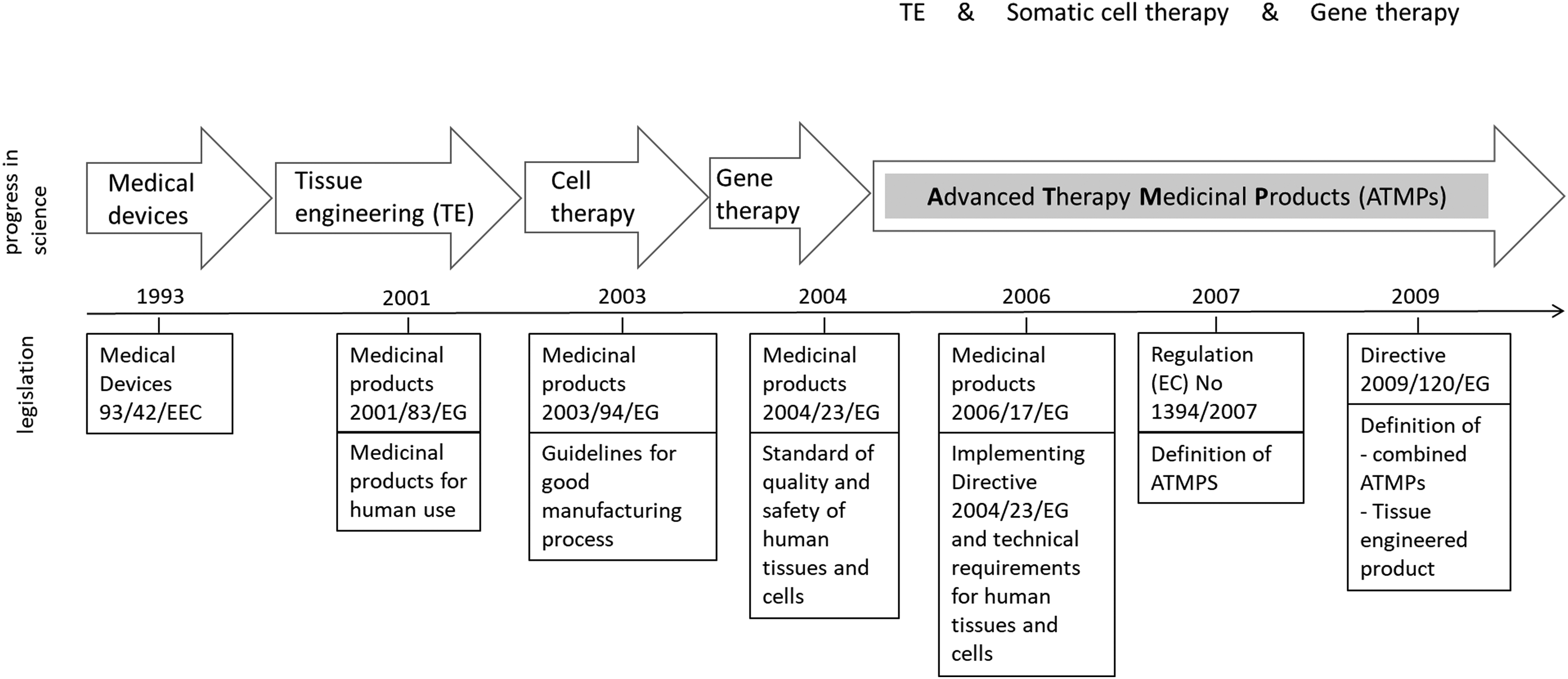
Timeline of the evolution of the European legislation regarding ATMPs in relation to progress in science addressing tissue engineering methods.
Characteristics of Mesenchymal Stromal Cells
Mesenchymal stromal cells (MSCs) constitute a heterogeneous population of spindle-shaped cells that can be isolated from many tissues, including adipose tissue, 14 bone marrow, 15 umbilical cord blood,14,15 skeletal muscle, 16 deciduous dental pulp, 17 synovial, 18 Wharton's jelly, 19 and skin. 20 According to the International Society for Cellular Therapy, human MSCs have to address at least the three following minimal criteria. First, adherence to plastic surfaces. Second, presence of the surface antigens CD73 (Sh3/4), CD90 (Thy1), and CD105 (SH2) and absence of CD14 or CD11b, CD19 or CD79a, CD34, CD45, and HLA-DR. And third, the cells must have the potential to differentiate into adipogenic, chondrogenic, and osteogenic lineages in vitro. 21
miRNA and mRNA analysis of differentiation markers of MSCs extracted from different sources revealed an “environmental-niche memory” for these cells, resulting in a better differentiation in the cellular types characteristic of the original host tissue. 22 In this context, the influence of interacting neighboring cell populations, the exposure to proteases, and the oxygen pressure in the surrounding environment can contribute to the functional development of a specific MSC population in a particular tissue, resulting in differentially regulated intracellular responses that can finally direct MSC function and fate. 23 Studies from Jeon et al. 24 and from Heo et al. 25 recently revealed that MSCs exhibit different differentiation potentials depending on their source despite having similar morphology and surface antigen expression, presenting 90 differentially expressed proteins when comparing the differentiation potential of human bone marrow-, placenta-, and adipose tissue-derived MSCs. For this reason, the choice of the source of MSCs plays an important role in the final outcome, and bone marrow-derived MSCs constitute the best candidate for bone and cartilage TE.
MSCs are characterized by self-renewal capacity, minimal to no immunogenic 26 and tumorigenic tendencies, 27 and potential to be expanded through many passages in vitro maintaining their pheno- and genotype. 28 Among their clinical application, MSCs hold the potential to reduce graft versus host disease, 29 increase cell engraftment, 30 migrate to inflamed tissue where they modulate inflammation, and recruit host cells leading to tissue repair.31,32 Taken together, these characteristics make these cells extremely appealing for TE and RegMed purposes, and their clinical application promises to overcome the issue of limited availability of healthy primary differentiated cells from patients.
Clinical Application of MSC in Bone and Cartilage Treatments
Two major MSC-based approaches are clinically used for knee reconstruction of osteoarthritic patients: either injection of autologous bone marrow-derived MSCs into the knee33–35 or seeding of MSCs on natural derived scaffolds serving as biological transplant/implant.36–38 A selection of published clinical studies is summarized in Table 1 for cartilage reconstruction and in Table 2 for bone applications.
β-TCP, β-tricalcium phosphate; HA, hydroxyapatite.
A comparative study in which cartilage defects were either treated with autologous chondrocytes or MSCs showed the potential of MSCs as promising alternative to autologous chondrocyte implantation. Both groups resulted in a similar clinical outcome in cartilage repair tissue but, in contrast to autologous chondrocyte implantation, MSCs did not show any age-related decrease in performance. 34 Similarly, intra-articular injection of MSCs in patients improved cartilage quality 35 and reduced articular pain.39,40 When MSCs were embedded in a hydrogel formulation such as platelet-rich fibrin glue 36 or acid soluble collagen I gel37,38 and then implanted in the defect site, this resulted in the improvement of clinical knee scores, with partially to completely repaired tissue at 12 months follow-up.36–38
Despite the consensus on strategies for cartilage defect repair, the determination and standardization of the clinically relevant cell dose for such treatments are still sources of debate. First results published by Jo et al. suggest a dose-dependent healing capacity of adipose-derived MSCs in osteoarthritic knees, 41 with significant reduction in the size of the cartilaginous defect and an increase in the volume of regenerated cartilage in a high-dose group (1·108 MSCs) compared to lower dose (≤5·107 MSCs).
Similarly to what is reported for cartilage, approaches for the use of MSCs for bone repair are either based on injection of the cells alone or on the implantation of cells in combination with scaffolds. In the clinical practice, the main applications of MSCs are in maxillofacial reconstruction, in which expanded MSCs 42 or bone marrow concentrates 43 are co-implanted with β-tricalcium phosphate (β-TCP) or hydroxyapatite synthetic scaffolds, 44 decellularized bone powders or granules, 43 or embedded in platelet-rich plasma derivatives. 45
While applications for MSCs in maxillofacial reconstruction are already routinely used in the clinic, applications for critical size long bone defect reconstruction are still mainly lacking or under experimentation in preclinical models. In this context, Rastogi et al. reported a preliminary study on the use of autologous MSCs injected at the site of lesion for the management of osteonecrosis of femur. 46 Nonetheless, risk assessment and long follow-up times of the studies prevent from drawing conclusive statements on the success of these approaches.
Sensebe et al. summarized more detailed information on potential risks of MSCs, concluding that they do not pose significant risks. 31 There are no reports of immunological related side effects of MSCs alone 41 and MSC-seeded TE constructs for (osteo-)chondral applications in humans, highlighting the particular suitability of MSCs for clinical use. 23 Although there are not enough data available addressing the immunogenicity of osteogenic or chondrogenic differentiated MSCs after implantation, several reports point out that differentiated cells may retain low immunogenicity and a certain degree of immunomodulation in vitro.47–49
In Vitro Differentiation of MSCs into Chondrogenic and Osteogenic Lineages
The production of ATMPs for bone and cartilage regeneration requires the proper combination of stimuli able to trigger cell differentiation and matrix deposition. Among the different environmental cues able to drive cell proliferation or differentiation into different lineages, the most used laboratory approaches are based on biochemical stimuli and culturing substrate-derived stimuli, of which the scaffold material itself acts as a combination of the previous two. Although many of the growth factors and scaffolds used in the manufacturing process do not meet the GMP requirements for human application, the actual ATMP regulation does not limit a priori their use for ATMP manufacturing.
Therefore, the employment of GMP-compliant equivalents or the implementation of GMP-compliant manufacturing processes from a very early phase of the product research and development pipeline can speed up the transition of TE products to their clinical application. The effect of biochemical and scaffold-derived stimuli on MSC differentiation will be reviewed in the following paragraphs.
Biochemical parameters: stimulation with media supplements, co-culturing and oxygen concentration
Chondrogenic differentiation in vitro requires high cell density in a three-dimensional (3D) setup under chemically defined culture conditions. 50 Reported biochemical stimuli able to drive chondrogenic differentiation mostly include members of the transforming growth factor-beta (TGF-β) family, 51 although other growth factors like bone morphogenetic protein (BMP) or insulin-like growth factor (IGF) may have chondrogenic potential when in combination with TGF-β. 52 Despite contrasting results, the supplementation of TGF-β and dexamethasone53–55 in combination with ascorbic acid-2-phosphate, L-proline, insulin, transferrin, selenium, bovine serum albumin, and linoleic acid appears to hold the highest chondrogenic differentiation potential in vitro.
Osteogenic differentiation can be stimulated with chemicals such as β-glycerophosphate, ascorbic acid-2-phosphate, and dexamethasone, 56 but important growth factors for differentiation are also 1,25-dihydroxyvitamin D3, 57 prostaglandin E258,59 as a regulator of BMPs, growth hormone, 60 interleukin-6, 61 BMP-2, 62 BMP-4, 63 BMP-6, 64 BMP-7, 65 IGF-1, 66 parathyroid hormone receptor, 67 TGF-β, 68 fibroblast growth factor, 69 and some components of the platelet-rich plasma. 70
For bone repair strategies, coculture of MSCs with other cell types sharing the same niche in the bone marrow, and most likely producing many of the previously reported cytokines and growth factors, has been explored. Among them CD169+ macrophages, 71 hematopoietic stem cells, 72 and endothelial cells 73 are the most likely candidates contributing to the bone marrow niche. The development of such a co-culture system may in the future reveal to be of crucial importance for the improvement of cell engraftment, scaffold vascularization, and reconstitution of the bone marrow cavity and microenvironment, 74 especially when dealing with critical bone defects in long bones and load bearing regions. 75 Depending on the specific requirements of the relevant cell types, more complex culture devices might therefore be needed.
Of particular importance during fracture or wound repair is the regulation of oxygen tension in the tissue. While hypoxic conditions characterize the bone marrow environment, low oxygen tension differentially regulates the differentiation into the osteogenic or chondrogenic lineages.
In fact, severe hypoxia lower than 1% O2 for up to 48 h induces downregulation of osteogenic differentiation markers such as cbfa-1/Runt-related transcription factor 2 (Runx2)—an important early osteoblastic transcription factor, osteocalcin—a late osteogenic marker—and type I collagen—the main component of bone matrix, while having no effect on MSC survival. 76 Conversely, an oxygen tension of 20% has been reported to be optimal for promoting osteogenic differentiation. 77 In contrast, oxygen tensions less than 1% on the joint surface are typical of the cartilage tissue. 78 In vitro pellet cultures of MSCs under this condition promote chondrogenic differentiation—increasing proteoglycan synthesis.79,80 Moreover, hypoxic conditions can suppress apoptosis during chondrogenesis and thus prevent MSCs from undergoing the process of endochondral ossification. 80
Biomaterials used in bone and cartilage TE
Since Badylak et al. 81 highlighted the potential of biomaterials mimicking native extracellular matrix (ECM) in structure and composition, naturally derived zonal structured scaffolds have become of great interest for cartilage TE.82–86 Materials used for this application vary from natural polymers, including agarose, 86 alginate,87,88 collagen,89–92 fibrin glue,93,94 and silk,95–98 to synthetic polymers like poly-lactid and poly-glycolides and their copolymers, poly-lactic glycolic acid, 99 polycaprolactone,100,101 and polyethyleneglycol.102,103 Since hyaline cartilage contains up to 80% of total weight by water, hydrogels represent the ideal scaffold formulation for cartilage regeneration,84,104 inducing chondrogenic differentiation, allowing uniform cell distribution, and providing easy handling, injectability, and printability.104–107 Although protein based hydrogels (collagen and fibrin) present poor mechanical stability and often suffer from shrinking during chondrogenesis, 108 combination with glycosaminoglycans like chondroitin sulfate or hyaluronic acid improves their mechanical properties and enhances chondrogenic differentiation.91,109
For clinical application collagen I hydrogels are widely used, as they can be applied with minimally invasive procedures, for example, through injection into defect. 110 This approach owns his appeal to a simpler execution in the operation theater, but fails to reproduce the particularly structured architecture of the articular cartilage, underlying the need for the development of layered scaffolds for cartilage repair mimicking natural tissue characteristics in structure and composition, hence functionality. 84 Porosity, pore distribution, and structure of the scaffold are, for example, important factors that influence cell–cell interaction in high density cell cultures, MSC condensation, and cartilage formation.104,111 Macroscopic pores (400–500 μm) have been reported to be beneficial for ECM synthesis,112,113 whereas micropores (100–250 μm) appear to promote cell migration, proliferation, and mass transport.104,112,113 Thus, a combination of micro- and macropores on the same scaffold results to be beneficial for cartilage TE.
In the context of bone TE, different materials have been used to mimic either the organic phase114–116 or the mineral phase of the bone matrix. These materials traditionally aim to maintain physical stability and stiffness while having enough porosity to allow cell infiltration, proliferation, and vascularization. 117 Scaffold porosity has been proven to be of crucial importance in directing bone formation, which has been reported to occur at the lining surface of the pores, being either porous macrostructures 118 or well-defined concavities. Different kinds of ceramics, 119 such as β-TCP, 120 calcium-deficient hydroxyapatite, 121 and β-CaSiO3, 122 and bioactive glasses 123 can be manufactured to host pores with inner diameter between 100 and 500 μm and porosity up to 90%.
Alternatively to ceramics, several approaches attempted to use synthetic materials in different combination with hydroxyapatite124–128 and/or β-TCP 129 to find a compromise between the request for initial stability and stiffness and the ability to support further mineral deposition. Different approaches with demineralized 130 or remineralized 131 bone matrix have finally provided promising results in terms of MSC differentiation and mineral deposition. Moreover, other scaffold formulations are aimed at the development of a vascularized network and at achievement of a more natural spatial cell distribution into the matrix. This includes cellulose sponges, 116 collagen microcarriers,114,132 electrospun fibers, 125 or multilayered scaffolds for vascularization.133,134
The proper proportion between organic and mineral phase, together with scaffold architecture, contributes in providing the cells with the needed signals for osteogenic differentiation. The cytoskeleton of adhering cells is normally maintained in a state of tension (tensegrity), pulling on the cell membrane through focal adhesions and resulting in degradation of the ECM. ECM degradation causes cell spreading and development of tensile forces, which in turn initiate proliferation and tissue folding. Application of tension enhances osteoblastic gene expression, including the calcitonin receptor, β-catenin, Runx2, and Wnt-8.135,136 MSCs grown on stiff substrates exhibit extensive production of stress fibers, while cells on collagen gels show less spreading and fewer stress fibers, but more filopodia. 137
Bioreactors for Bone and Cartilage Tissue Formation
Generation of bone or cartilage tissue based on human MSC usually involves two steps: (1) expansion of human mesenchymal stromal cells (hMSCs) to obtain the needed amount of cells for the specific defect dimension and (2) their subsequent differentiation into osteoblasts or chondrocytes. For both processing steps, the use of bioreactors offers advantages over two-dimensional static culturing conditions as described in more detail in this paragraph.
In terms of hMSC expansion, main benefits of bioreactor usage are better process monitoring and control by integrating in-line sensors for temperature, pH, oxygen tension, and metabolites. Furthermore, bioreactors are used for better space-time yield, for example, by improving nutrient exchange and increasing growth surface and system automation. Available bioreactors and culture concepts for hMSC expansion, especially regarding GMP and Process Analytical Technology requirements for ATMP manufacturing, were summarized by Cierpka et al. 138
For hMSC differentiation, bioreactors are similarly used because of the possibility to improve process control thanks to the integration of sensors and automation steps. Furthermore, maintenance of cell viability and homogenous cell distribution in 3D TE products requires extensive mass transfer to overcome the diffusion limit of 100–200 μm in depth139,140 and provide the cells with the appropriate nutrient supply. Different bioreactor systems able to overcome such a limit have therefore been developed. 141 However, in contrast to the expansion systems, bioreactors used for chondrogenic and osteogenic differentiation processes should include stimuli triggering cell differentiation as well. In this regard, integration of mechanical stimulation devices is a central issue in bioreactor development for culturing bone and cartilage tissue engineered products.
As chondrocytes are mainly exposed to low oxygen tension and dynamic compressions in vivo, a bioreactor system should combine different mechanical stimuli and oxygen control to induce MSCs to undergo chondrogenesis, increase matrix synthesis, 142 and collagen fiber alignment. While hydrostatic pressure alone stabilizes cartilage phenotype, 143 several hybrid bioreactors attempt to combine two or more physical stimuli such as shear stress and dynamic compression144,145 or mechanical and electromagnetic stimulation 146 on cell-seeded scaffolds simultaneously or in sequence.
Dynamic deformation and compression can in fact induce MSC chondrogenic differentiation and increase matrix synthesis.147–149 Moreover, the effect of mechanical stimulation on chondrogenic differentiation is increased by addition of TGF-β. 150 Conversely, it has been shown that dynamic compression starting from the first day of culture can inhibit chondrogenic differentiation of MSCs. 151 Mouw et al. indicated that dynamic compression applied on MSCs predifferentiated with TGF-β1 upregulated Smad2/3 phosphorylation, 152 while other reports show the potential of preventing terminal differentiation of MSCs toward hypertrophic chondrocytes by dynamic compression and hydrostatic pressure.143,153,154
As far as bone TE is concerned, bioreactors should allow both significant mass transfer inside the porous scaffold and mechanically stimulate the construct. For this reason, commercially available bioreactors often comprise perfusion systems, generally used in conjunction with chambers—columns or cartridges to hold the cell-seeded scaffold or construct155,156 and spinner flask bioreactors. 157
MSC osteogenic differentiation has been reported to be positively regulated by tension158,159 —upregulating BMP2 expression and matrix stiffness 137 —resulting in activation of Ras homolog gene family, member A (RhoA), and Rho associated coiled-coil containing protein kinase 2 (ROCKII). Similarly, fluid flow and shear strain154,160 promote osteogenic differentiation by regulating canonical and noncanonical Wnt pathways. Finally, cyclic strain 161 can lead to increased calcification depending on the matrix and start of loading. Periodic vibration, 162 perfusion, and magneto-mechanical stimuli 163 can also be used to prime osteogenic differentiation.
To summarize, bioreactors offer the possibility of automating the culturing process, saving time and allowing to develop products in closed systems more suited for clinical uses. Moreover, specifically defined culture conditions like pH value, oxygen, CO2, and glucose concentration, and in-line monitoring of administered soluble factors, allow standardization and reproducibility of the in vitro culturing environment. 164
Although a universal consensus on the optimal bioreactor solution for bone and cartilage TE is yet to be reached, such advantages allow the manufacturing of products that are compliant with the ATMP regulation already from an early R&D phase, and we therefore believe that a broader employment of the bioreactor technology in the RegMed research community will greatly accelerate the transition of new products to the clinical practice.
And Now, Where Do I Start from? Thinking the “GMP Way”
Many scientists in the TE and RegMed field think about the GMP regulations as a chimera made of many “don'ts” and few “cans”. All the opposite, regulation 1394/2007 came to shed light on the real identity of ATMPs for the legislators. Not surprisingly, scientists already knew that tissue engineered products contain for definition “engineered cells or tissues” (Art 2.1.b 1394/2007) that are by definition “subject to substantial manipulation” (Art 2.1.c 1394/2007). And now that the concept is also known to the authorities, there's no need to worry about it.
Producing GMP-compliant products is about certifying manufacturing processes to ensure the delivery of safe and reliable products for human use. This means that few limitations apply to the product and many to the process. All manipulations listed in Annex I 1394/2007 and described in the beginning of this article are considered as nonsubstantial manipulations, while all others, including seeding of the cells on a scaffold, are potentially classified as substantial manipulations. The evaluation of the substantial manipulations allowed for a particular product is done on a case-to-case base and follows the criteria of risk vs. benefit. This means that all products should be tested to provide preliminary safety data to the local authorities and to the CAT. In addition, methods evaluating safety and functionality can be agreed and validated according to authorities' indications. In case of doubt, both authorities and CAT will be able to advice on the possibility to introduce a specific manipulation in the manufacturing process at an early stage.
First step for the manufacturing of a GMP-compliant product is the establishment and accreditation of a QMS conforming to international standards, such as the ISO 9000 and ISO 9001. The QMS should describe all the SOPs related to the manufacturing of the product and in general all the procedures in the company or institute where it is produced, including relationship with the suppliers, management of deviations from the SOPs and risk management, reviewing of customer satisfaction and complaints, traceability of the products and all its components, data storage, training of personnel—all part of the so-called “quality assurance”—and testing or “quality control.” The ISO 9000 and 9001 standards are valid for a wide range of companies, and they are not specific for ATMPs and pharmaceutical products. It is the responsibility of the manufacturer to make sure that all contractors have a QMS in place and, in the case of suppliers, can provide GMP-compliance certifications for their products to be used as raw material. Integral part of the development of the QMS is the certification of an own/contracting of a third-party GMP-manufacturing facility being conformed to GMP guidelines as described in EudraLex—Volume 4 165 where the product will be manufactured.
Once technical documentation and risk analysis has been performed, an audit from the local authorities will verify conformity to regulation and eventually set contacts with the European authorities. While the local authorities will be responsible for issuing a manufacturing authorization, a GMP certificate and a country-specific approval, approval from European Medicines Agency (EMA) after consultation of the CAT and the CHMP (Committee for Medicinal Products for Human Use), will be required for marketing authorization at the European level. Marketing authorization for countries outside the European Union requires conformity with the local regulations.
While adoption of the ISO 9000 or 9001 will ensure a good adaptability of the processes to extra-European regulation, the International Conference for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) provides optimal indications for the harmonization of GMP guidelines worldwide. Once the GMP certificate and the manufacturing authorization have been issued, the product is ready to enter the clinical trials and once all safety data are collected, can enter the path to marketing authorization.
To sum up, the whole process of a product's approval for human use can be simplified in three steps: (1) Development and testing of a product according to state-of-the-art scientific technology, (2) Development of a process for its manufacturing, including adoption of a QMS to ensure standardization, traceability, and safety for the patient, and (3) Early-as-possible start of relationships with local and/or European authorities to certify the process and product conformity to regulation, perform clinical trials, and eventually acquire marketing authorizations.
Challenges and Perspectives
MSCs play an important role in TE and RegMed holding the potential to replace adult and terminally differentiated cells often limited both in availability from the native tissue and in in vitro expansion potential in their final application. However, up to now no MSC-based ATMP for bone or cartilage regeneration is available on the European market, and only few companies own marketing authorizations for cryopreserved concentrated bone marrow-derived MSCs.
The development of the ATMP-fabrication strategy depends in fact on a variety of different factors, among which the careful combination of scaffold materials and biochemical and mechanical stimuli used to direct tissue formation plays a crucial role. The development of bioreactor systems specifically tailored to simulate the in vivo situation during product manufacturing should take into account the normal response of the native organism to external materials and tissues and should therefore consider the possibility to establish cocultures with other cell types, with particular attention to the immune response of the host. Finally, the whole production process should be carefully designed to match the regulatory requirements in terms of standardization, reproducibility, performance, and safety and to match the needs of the final users of the product, be them the surgeon or the patient.
For this reason, an effective communication with the responsible authorities and the establishment of surrogate internal parameters to assess product functionality and quality during manufacturing are pivotal for a successful transition of TE products to the market.
With this review, we attempt to give a comprehensive understanding of the state of the art of bone marrow-derived MSC employment in bone and cartilage TE clinical application, and we hope to provide important insights on the legislative requirements of ATMPs able to promote and stimulate the early transition of new TE products from the research and development phase to their clinical application.
Footnotes
Acknowledgments
This work was supported by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 309962 (HydroZONES) and no. 607051 (BIO-INSPIRE).
Author Contributions
D.C. and A.S. contributed equally to this work, they wrote and revised the article; and H.W. and F.E. reviewed and revised the article.
Disclosure Statement
No competing financial interests exist.