
Abstract
In membrane bioreactors the cells are isolated from the bulk medium through a semipermeable membrane. This concept, which is analogous to how the circulatory system supplies solid tissues with nutrients, allows the maintenance of cells at much higher densities than is possible in traditional cultures. The membrane-based microbioreactor described herein is easy to operate, requiring only a pipette to load and harvest cells. A 10 μL culture volume was isolated from 1 mL of bulk medium through a semipermeable membrane having a molecular weight cutoff of 10 kDa. Here we describe the benefits regarding the retention of both cells and their secretions within this small culture volume using the following two model systems: hematopoietic stem cell expansion and mesenchymal stem cell-derived cartilage matrix accumulation.
Introduction
Knazek et al. were the first to utilize hollow fibers, as an analogue to vascularization, to overcome mass transport limitations and achieve tissue density cell culture. 1 Hollow fiber-based bioreactor systems have since been used to culture mammalian cells at high density for the production of recombinant proteins, antibodies, tumor-infiltrating lymphocytes, and hepatocytes.2–7 Flat-plate membrane bioreactors, like hollow fiber-based systems, function to reduce mass transport constraints, and in doing so, they enable high cell density, but in a planar geometry. Flat-plate membrane bioreactors having single or double membranes to facilitate mass transfer have been widely used in the field of tissue engineering, especially in the generation and study of potential liver support systems.8–11
Optimization and use of membrane-based bioreactor systems can be compromised both by their inherent complexity as well as by their significant consumption of cell inoculum and culture medium. This consumption is generally a function of the large volumes required to supply the culture module as well as the external reservoirs, conditioning vessels, and circulation void volume. To identify potentially significant advantages associated with high-density cell culture from a scientific, therapeutic, and economic perspective, the aforementioned limitations of optimization within full-scale systems must be overcome. This can be achieved through the use of simpler, small-volume equivalents which enable rapid and inexpensive development of high-density cell culture protocols. The need for such micro-based culture systems has spawned the generation of a multitude of microbioreactor systems12–15 and journals dedicated to this development.
The membrane-based microbioreactor described in this article uses a parallel membrane geometry, and unlike existing membrane-based microbioreactors,1,11,13,16 this system requires no peripheral equipment beyond a pipette to load and harvest the cells. This level of operational simplicity enables the low-cost parallel study of multiple conditions with replicates and enables easy harvest of cells or intact solid tissues for analysis via conventional techniques such as flow cytometry or histology. As a proof of concept we report the use of a membrane-based microbioreactor to expand hematopoietic stem cells (HSCs) and enhance matrix accumulation in chondrogenically induced mesenchymal stem/stromal cells (MSCs).
HSC culture
HSC transplantation is a well-established therapy and can be curative for leukemic cancers following myeloablative therapy. However, the efficacy of this treatment is compromised by lack of suitable donors and delayed engraftment.17,18 The ability to manipulate the HSC populations ex vivo and achieve extensive HSC self-renewal has the potential to revolutionize this therapy and vastly improve clinical outcomes. Despite significant advances in our understanding and the ability to culture HSCs, attempts to achieve extensive self-renewal of a functional cell population in vitro have failed.19,20 It is now recognized that the HSC niche is instrumental in this process and that recapitulation of this microenvironment may be an essential step in the development of HSC-based cellular therapies. We propose that the use of bioreactors, which enable control over the microenvironment, will play a significant role in the optimization of in vitro niche recapitulation.
HSCs are traditionally cultured statically at densities up to approximately 106 cells/mL or approximately 2 orders of magnitude less than the cell density in the bone marrow. 21 This significant disparity may be one of the variables limiting the successful self-renewal of HSCs in vitro, which can be evaluated using culture devices that enable the efficient delivery of metabolites even at high cell densities. Theoretical application of high-density culture may reach beyond the generation of tissue mimics, playing a role in cellular therapies. The production of a unit of red blood cells (2 × 1012 cells 22 ) at traditional static densities (106 cells/mL) requires a 2000 L culture volume. Assuming only a 100-fold improvement (108 cells/mL), high-density cultures would reduce the working volume to a more manageable 20 L. The purpose of the HSC experiments conducted here is to identify if a higher cell density could be achieved using the membrane bioreactor and what implications this has on HSC expansion.
MSC to Chondrocyte Culture
Articular cartilage is composed primarily of extracellular matrix and a single cell type, the chondrocyte. Once damaged, articular cartilage lacks the ability to properly repair and regenerate damaged tissue.23,24 Strategies that enable the repair of cartilage defects will thus have significant value to both the public and the emerging field of tissue engineering. Autologous chondrocyte implantation25–28 is perhaps the newest and most promising tissue engineering cartilage repair strategy. The autologous chondrocyte implantation process involves in vitro chondrocyte expansion followed by in vivo cartilage regeneration. Strategies used to regenerate cartilage in vitro will have to address cell expansion as well as cartilage matrix production and retention. The primary matrix components of cartilage are collagen II and aggrecan which are largely responsible for the mechanical strength and compressive characteristics of the tissue. 29 We routinely observed only 40–50% retention of the total glycosaminoglycans (GAGs) produced during bone marrow-derived MSC (BM-MSC) chondrogenesis, with the remaining GAG lost to the bulk medium. This results in a matrix composition with much lower fractions of these important components than are present in native cartilage. Both collagen II and aggrecan have molecular weights > 10 kDa. 29 By isolating the developing cartilage tissue from the bulk medium using a dialysis membrane with a 10 kDa molecular weight cutoff (MWCO) it may be possible to retain both expensive growth factors and valuable matrix secretions on the cartilage side of the membrane, thus resulting in more physiologically relevant in vitro cartilage generation. This concept is evaluated in the second set of experiments described in this article.
Materials and Methods
Microbioreactor design and fabrication
A schematic of the membrane microbioreactor used in this study is shown in Figure 1A. The dual membrane design decouples the supply of oxygen from other metabolites, which was seen as a necessary first step to adequately supply each nutrient to the high-density cell mass in the absence of convection. The cells access medium metabolites in the upper reservoir through a cellulose membrane having a specific MWCO and gas exchange is achieved through the bottom membrane fabricated from polydimethylsiloxane (PDMS, Sylgard 184; Dow Corning, Midland, MI). The selection of a specific MWCO enables the tailored retention of proteins within the culture volume. In this proof-of-concept study, a 500 μm deep by 6 mm diameter culture chamber utilized Cuprophan™ (Membrana GmbH, Wuppertal, Germany) with a 10 kDa MWCO and a 300-μm-thick PDMS base. The unique feature, which makes this bioreactor particularly easy to use, is that the upper and lower sections bind each other reversibly through electrostatic interactions, thereby producing a seal which retains the cell culture inoculum within the microwell and enables easy access to the culture for harvest and analysis. Further, this design enables the loading and harvest of cell inoculum using only a pipette.
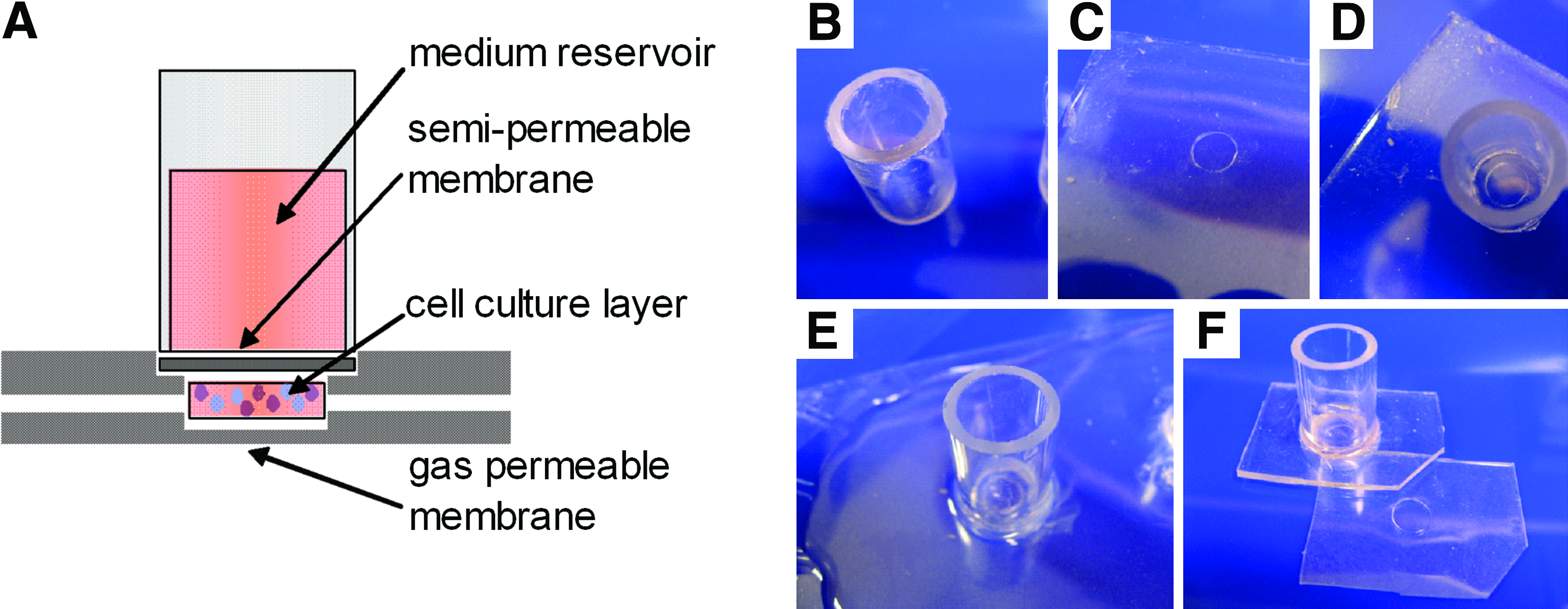
(
The sequential fabrication of the bioreactor (Fig. 1) will be described as steps 1B–F. In step 1B the Cuprophan membrane is fixed to one end of a glass cylinder (10 mm ×17.5 mm) using silicone sealant (Selleys, Auckland, NZ) to make the upper medium reservoir and semipermeable membrane. This section is then bonded to a PDMS membrane which enables the reversible electrostatic sealing of this upper bioreactor portion with the lower portion. This 300-μm-thick PDMS membrane is formed by spin-coating PDMS on to an overhead transparency at 500 rpm for 60 s, curing at 80°C for 10 min, and then repeating. Then (as shown in step 1C) a 6 mm hole is punched out of the PDMS membrane. A thin layer of silicone sealant is placed on the PDMS film around the 6-mm hole with care to avoid placing sealant in the hole. The upper reservoir (step 1D), with Cuprophan membrane facing down, is centered over the hole and then brought into contact with the membrane at which point it is anchored in place by the thin layer of silicone sealant. In Figure 1E PDMS is cast around the glass medium reservoir, further anchoring the reservoir to the PDMS film as well as increasing the membrane thickness to approximately 2 mm resulting in a more rigid structure which is easier to handle. The lower section of the bioreactor is a single 500-μm-thick PDMS membrane (step 1F) fabricated by spin coating PDMS onto a silica wafer having an SU-8 (SU-8 100; Microchem, Newton, MA) microwell feature (6 mm in diameter and 200 μm high), using well-established procedures. 30 As shown in Figure 1F the upper and lower PDMS membranes are cut such that when the edges align the microwell in the bottom membrane and the hole punched in the upper section which exposes the Cuprophan membrane are also aligned.
Microbioreactor operation
The microbioreactors are steam sterilized at 120°C for 20 min. Sterility is maintained by enclosing the microbioreactors within a modified Petri dish with 6-mm holes over which the microwells are located to allow unhindered gas transport. Figure 2 shows the correct loading of cells and bulk medium into the microbioreactor system. Cells can be harvested by peeling the upper PDMS seal from the bottom membrane and recovering cells with a pipette. A critical operational consideration is selection of the inoculum volume to be used in the microbioreactor. The actual microwell volume (6 mm diameter × 500 μm depth) is ∼14 μL. However, we utilize an inoculation volume of 10 μL as we have found that this eliminates the risk of accidentally displacing any portion of the inoculum volume outside the microwell when sealing the upper section with the lower section. Because of the hydrophobic nature of PDMS, and the hydrophilic nature of the Cuprophan membrane, the inoculation fluid forms an instant connection with the upper membrane which is maintained throughout the culture duration. The small void volume that remains within the microwell does not compromise cultures and only functions to facilitate gas mass transport through the culture well.

Cell culture in microbioreactors. (
Model of metabolite supply
The assumption that it is possible to maintain HSCs within the microbioreactor up to densities of 108 cells/mL was evaluated by mathematically modeling both the oxygen and glucose profiles through the cell mass at this density. The model assumes that the cells are evenly distributed throughout the microwell and that oxygen is supplied exclusively through the PDMS base while glucose is supplied through the upper membrane. Our calculations showed that at maximal oxygen consumption by the cell mass within the microwell, less than 4% of oxygen flux occurs via the medium reservoir, and thus this component of the flux was excluded from the model. Table 1 provides the measured values utilized to resolve the model (similar models are outlined in detail by Muschler et al.
31
) and Figure 3 shows both the predicted oxygen and glucose profiles through the cell mass. Equation 1 was generated by performing a shell balance and was used to predict the metabolite profile through the microwell.
Glucose and oxygen profile through cell mass within microbioreactor. The middle layer contains the evenly distributed cell mass. Glucose is supplied from the medium reservoir through the Cuprophan (green) membrane. Oxygen is assumed to be exclusively supplied through the bottom PDMS membrane (blue). Supply of both metabolites is sufficient to maintain HSC cell densities of 108 cells/mL. HSC, haematopoietic stem cell. Color images available online at
HSC, haematopoietic stem cell; PDMS, polydimethylsiloxane.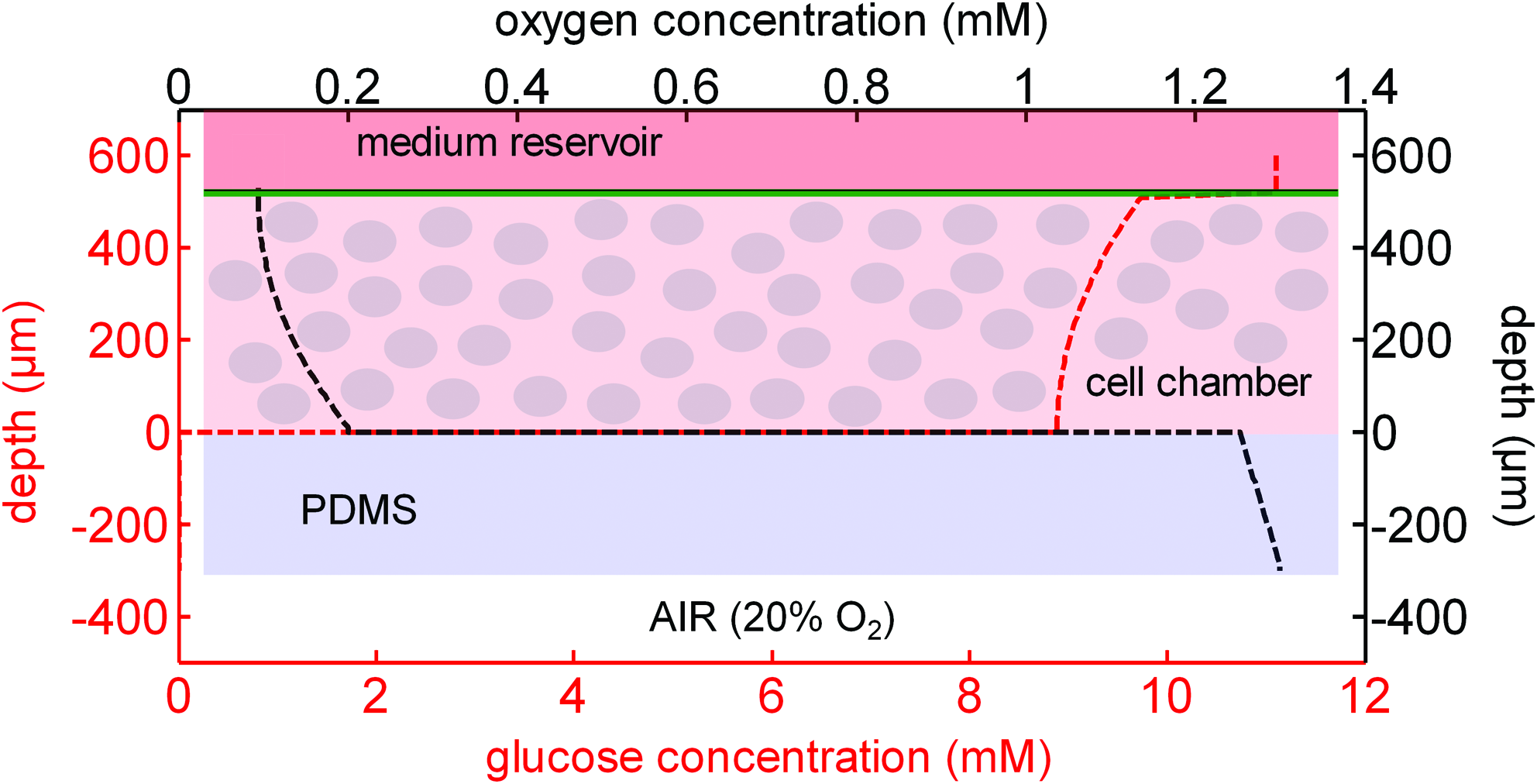
where C is the concentration, U is the specific cellular uptake rate, D is the metabolite diffusion coefficient, z is the vertical position in the microwell, Z is the total microwell height, and Co is the metabolite concentration at the interface with the supplying membrane.
The model demonstrated that at the proposed cell densities and bioreactor dimensions, the oxygen concentration will not drop below that equivalent to equilibrium with a 5% oxygen atmosphere and the glucose concentration will not drop to less than 5 mM (lower physiological blood glucose concentration) even if a third of the glucose in the medium (Rosswell Park Memorial Institute, (RPMI)) is depleted.
HSC cell culture
Umbilical cord blood from full-term deliveries was obtained with informed consent, from both the Royal Brisbane and Women's Hospital Human Research Ethics Committee and the University of Queensland Medical Research Ethics Committee. Mononuclear cells were isolated by centrifugation on Ficoll-Paque (Amersham Biosciences, Uppsala, Sweden) and enriched for CD34+ cells using Mini-MACS columns (Miltenyi Biotech, Bergisch Gladbach, Germany). We found the cell populations to be 95% CD34+ after two passes through the MACS column. Two HSC expansion experiments are described in this article which demonstrates that HSC expansion in the microbioreactor is equivalent or superior to that observed in traditional static well-plate cultures.
High-density HSC expansion
In the first HSC experiment we were interested in evaluating high-density HSC culture methodologies. Bioreactor cultures were contrasted against traditional well plate cultures. Thus seeding densities of 106 cells/mL were utilized. Cultures were initiated with 10 μL inoculation volumes containing 10,000 CD34+ selected cells suspended in Stemline II medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10,000 ng/mL each of stem cell factor (Stemgen; Amgen, Sydney, Australia), thrombopoietin peptide 37 (AusPep, Parkville, Australia), and granulocyte colony-stimulating factor (GCSF, Neupogen; Amgen), 1000 ng/mL Flt3-ligand (PeproTech, Rocky Hill, NJ), and 1 μL of Insulin-Transferrin-Selenium-X (Gibco-Invitrogen, Carlsbad, CA). These 10 μL inoculation volumes were seeded either into the bioreactor microwell or into 1 mL of Stemline II medium in a single well of a 24-well plate (Becton Dickinson, Franklin Lakes, NJ). The bulk medium used in the microbioreactor reservoirs was 1 mL of Stemline II. Cultures were maintained for 5 days in humidified incubators at 37°C that were set at either 2% or 20% oxygen with 5% carbon dioxide atmospheres.
Low-density HSC expansion
In the second HSC experiment we were interested in evaluating how the use of a membrane bioreactor system to isolate cells and their secretions might rescue cell expansion when the cells were seeded at densities known to compromise or delay cell expansion. Bioreactor cultures were contrasted against traditional well plate cultures. The inoculation was prepared in a similar way; however, the inoculation contained only 3000 CD34+ cells and excluded GCSF. This represents a cell density of 3000 cell/mL in each system; however, the cells in the bioreactor are localized within the microwell resulting in a local density of 300,000 cells/mL. Cultures were maintained in a 2% low oxygen or a 20% normoxic environment for 7 days. Protein-free RPMI alone was used as bulk medium to progress toward a more economical protein-free expansion protocol.
HSC cell analysis
Cell phenotype and number were determined using flow cytometry. The cells were labeled with monoclonal mouse anti-human CD34-FITC (Miltenyi Biotech) and monoclonal mouse anti-human CD133/2 (AC141)-PE (Miltenyi Biotech) as per manufacturer's instructions. The cell number was determined by spiking culture samples with a known number of counting beads (Beckman Coulter, Fullerton, CA) and calculating the relative ratio of cells to beads.
MSC to chondrocyte experiment
In this experiment, bioreactors with or without the semipermeable membrane were utilized to assess this membrane's impact on the chondrogenic induction of MSCs, specifically through the evaluation of sulfated GAG production.
MSC isolation
Fully informed patient consent was obtained in all cases. Approximately 10 mL bone marrow was taken from the iliac crest of healthy donors. The sample was diluted 1:1 with phosphate-buffered saline (PBS) and underlayed with 12 mL Ficoll-Paque Plus (GE Healthcare, Buckinghamshire, United Kingdom). The tubes were spun at 535 g for 20 min (no brake). The interface cells were washed and resuspended in tissue culture media consisting of low-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco-Invitrogen), with 20% fetal calf serum (Gibco-Invitrogen) and 50 μg/mL gentamicin (Pharmacia, Stockholm, Sweden), and placed in a 75-cm2 tissue culture flask (Gibco-Invitrogen). After 48 h, nonadherent cells were removed by washing with PBS and remaining adherent cells further cultured in tissue culture media. The media was changed every 3–4 days thereafter. The cells were generally approaching confluence after 14–20 days. At this stage, they were passaged and further expanded. After the second passage the cells were immunophenotyped by flow cytometry (all mAb were from Pharmingen) and were functionally assessed for differentiation potential. Excess cells at passage 3 were stored for subsequent experiments. The cells were deemed MSC if they were CD45−, CD73+, CD90+, and CD105+ and showed adipo, osteo, and chondro differentiation potential as described previously by Brooke et al. 38
MSC chondrogenesis and GAG accumulation
Human BM-MSCs were expanded in low-glucose DMEM supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (1% penicillin streptomycin (PS); Gibco-Invitrogen). For chondrogenic differentiation studies, BM-MSCs were grown as pellet cultures for 3 days in 15-mL polypropylene tubes and then transferred to membrane bioreactors or bioreactors assembled without the membrane and maintained up to day 14. Briefly, monolayer-cultured BM-MSCs were grown to near confluence and then detached using recombinant trypsin replacement (TrypLE Select; Gibco-Invitrogen) and placed in a serum-free chondrogenic induction medium consisting of high-glucose DMEM (Gibco-Invitrogen) containing 10 ng/mL transforming growth factor-β1 (TGF-β1) (PeproTech), 10−7 M dexamethasone (Sigma-Aldrich), 0.2 mM ascorbic acid 2-phosphate (Sigma-Aldrich), 100 μg/mL sodium pyruvate (Sigma-Aldrich), 40 μg/mL proline (Sigma-Aldrich), 1 × Insulin-Transferrin-Selenium-X (Gibco-Invitrogen), and 1% PS. Pellet cultures were formed by centrifuging 2 × 105 cells/mL at 500 g in chondrogenic induction medium and then culturing in a 2% oxygen atmosphere as we have repeatedly observed that this results in higher GAG production than differentiation under 20% oxygen atmosphere. At day 3, the pellets were transferred to membrane bioreactors which did or did not contain the upper Cuprophan membrane. The pellets were supplemented with 10 μL of medium composed of DMEM containing 1000 ng/mL TGF-β1 and 1 μL Insulin-Transferrin-Selenium-X. One milliliter of bulk medium containing the same concentrations of dexamethasone, ascorbic acid 2-phosphate, sodium pyruvate, proline, and PS as described earlier was added to the medium reservoir. These medium supplements are below the membrane MWCO and thus pass freely into the microwell. The bulk reservoir medium was collected and stored at −80°C at each medium replacement, every 3 to 4 days. Change of medium in bioreactors with membranes consisted only of this bulk medium, whereas those without membranes were also supplemented with 10 ng/mL TGF-β1 at each change as is typical of normal pellet culture. At the end of 14 days, the pellets were rinsed with PBS and digested with 1.6 U/mL papain (Sigma-Aldrich) at 60°C overnight. The sulfated GAG content and DNA were quantified with 1,9-dimethymethylene blue (Sigma-Aldrich) and Hoechst 33342 (Gibco-Invitrogen). Shark chondroitin sulfate (Sigma-Aldrich) and calf thymus DNA (Sigma-Aldrich) were used as the respective standards. Additionally, the 1,9-dimethymethylene blue assay was used to quantify sulfated GAGs released into the medium that was collected at days 3, 6, 10, and 14.
Results and Discussion
High-density HSC expansion
We first investigated HSC expansion under standard 20% oxygen culture conditions with medium containing stem cell factor, GCSF, thrombopoietin peptide, and Flt3-ligand which is suitable for the expansion of cells down the myeloid lineage.39,40 Cells that are dual for both the CD34 and CD133 antigens have been termed stem cell candidates, acknowledging that likely only a fraction of this population will truly be such. 41 We observed that the expansion of both the total population and stem cell candidates in the membrane-based microbioreactor system was significantly greater than in a traditional 24-well plate at day 5 (Fig. 4). The final cell density in the microbioreactor system was 4.5 × 107 cells/mL or a 45-fold expansion, while the final cell density in well plates was 3 × 105 cells/mL or a 30-fold expansion. Some cultures were maintained for an additional 48 h and cell expansion reached an excess of 150-fold within the microbioreactor system, generating viable cell densities > 1.5 × 108 cells/mL (data not shown). Figure 5 shows the density of HSCs contained in a microbioreactor at day 5 of culture. HSCs are traditionally expanded in static culture, where cultures at 106 cells/mL are generally considered confluent. A bioprocess that produces units of red blood cells must produce 2 × 1012 cells/unit, which at a maximal cell density of 106 cells/mL is equal to 2000 L/unit. The use of a membrane-based bioreactor system would be expected to allow a cell density increase of 10–100-fold, and therefore, a 10–100-fold reduction in working culture volume over static systems. From an economic and feasibility perspective, such a reduction may be paramount. The use of membranes having specific MWCOs allows for the retention of expensive soluble proteins within the reduced culture volume and thus potentially more efficient use of these factors. Small volume tools that enable the study and optimization of high-density cell culture protocols will facilitate the evolution of high-density cell bioreactor systems such as those which might be utilized in future blood pharms.
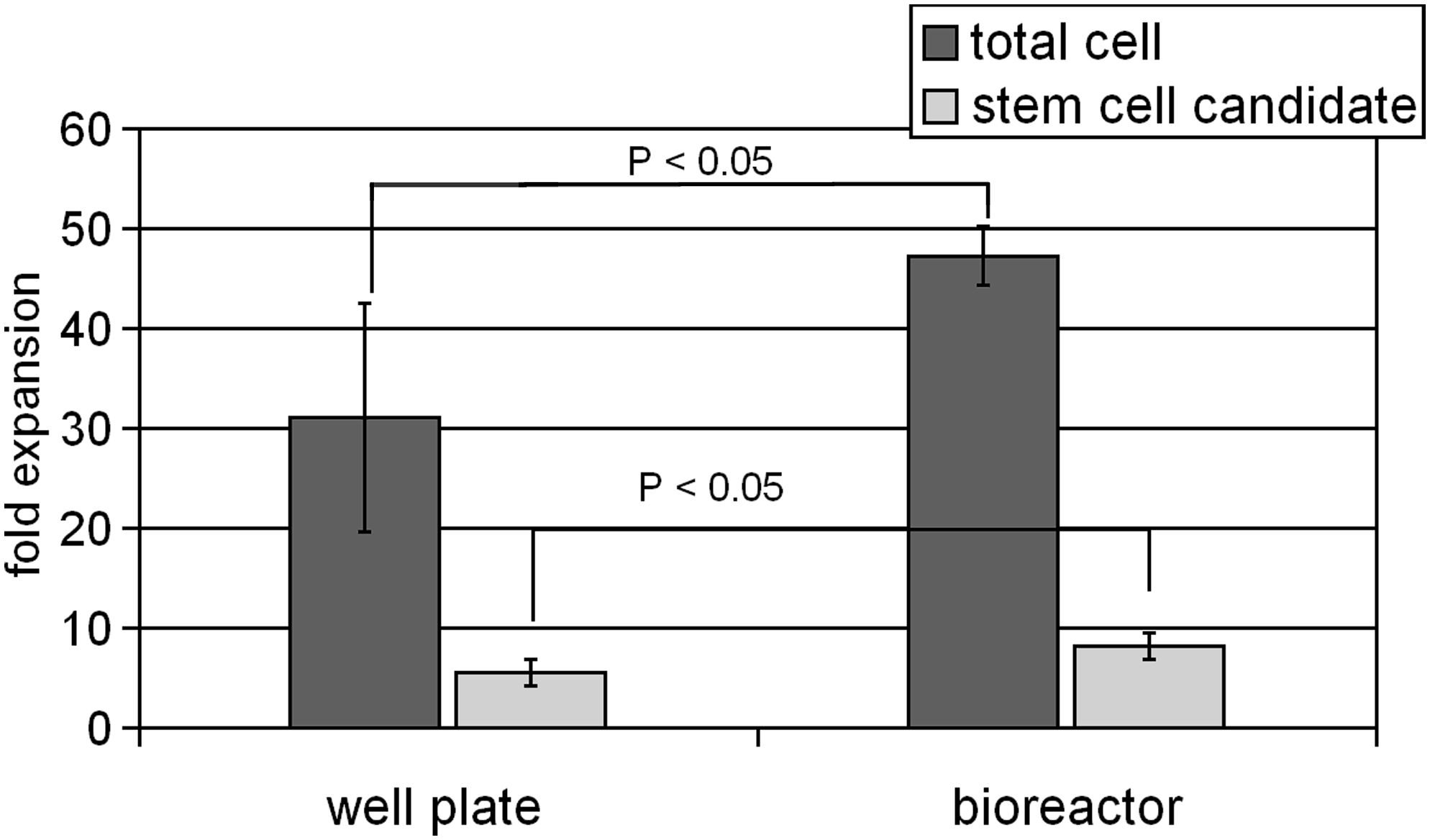
Day 5 HSC expansion at 20% oxygen within the bioreactor, in which the medium contained stem cell factor, granulocyte colony-stimulating factor, thrombopoietin peptide, and Flt3-ligand, is significantly (p < 0.05, Student's t-test) greater than that observed in traditional static culture (n = 6; error bars reflect 1 standard deviation).
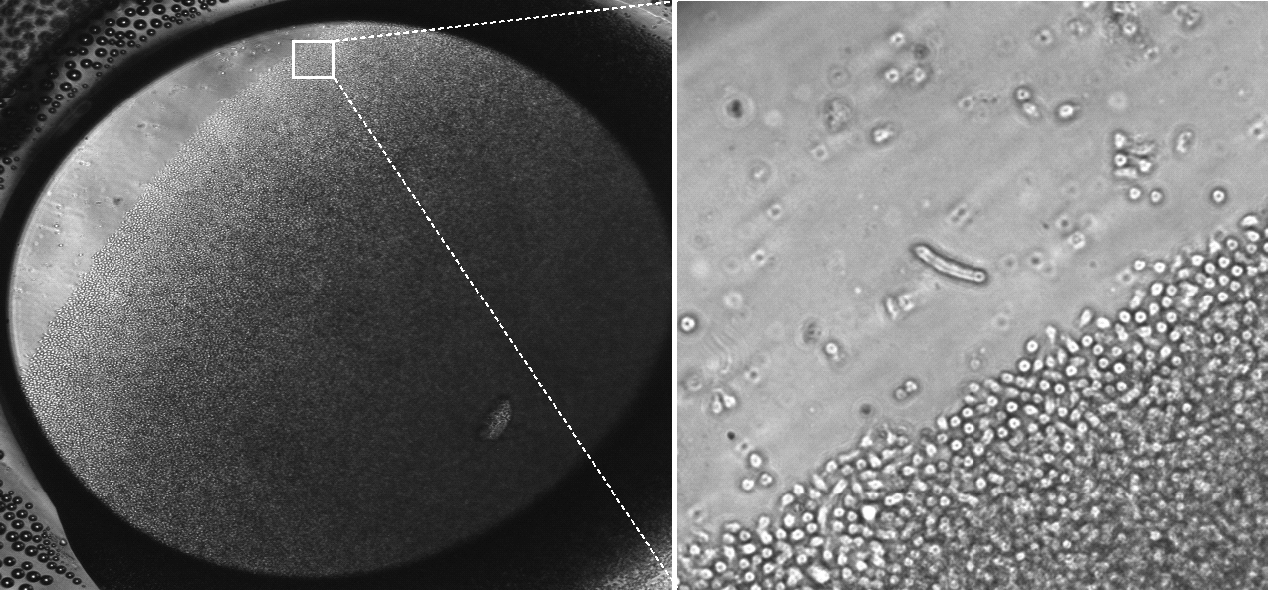
Day 5 of HSC culture in the microbioreactor (4 × magnification). The bioreactor has been tilted to reveal the depth of the cell layer.
Low-density HSC expansion
Although the total cell culture volume in both the microbioreactor (including the reservoir) and well plate is the same, the actual cell culture volume in the bioreactor is 10 μL or 1/100th of that in the well plate. This feature generates a high local cell density which, when coupled with the semipermeable membrane designed to retain cellular secretions greater than 10 kDa, effectively reduces the necessary cell numbers required to condition the medium and support cell expansion. This was confirmed by reducing the number of cells used to initiate cultures to 3000 CD34+ cells. In our laboratory we have observed that initiating traditional well plate HSC cultures at densities less than 10,000 cells/mL compromises cell expansion; therefore, reducing the inoculation number to 3000 cells/mL total volume provides an assessment of any benefits obtained by increasing the local cell density in the microbioreactor. Cell expansion, as shown in Figure 6, demonstrates that increasing the local density through utilization of the membrane bioreactor system rescues the expansion potential of the HSC culture. The ability to initiate cultures with a reduced cell number provides the following two significant advantages: (1) the more efficient use of rare cell subsets and (2) the ability to achieve greater total expansion in a single culture system.
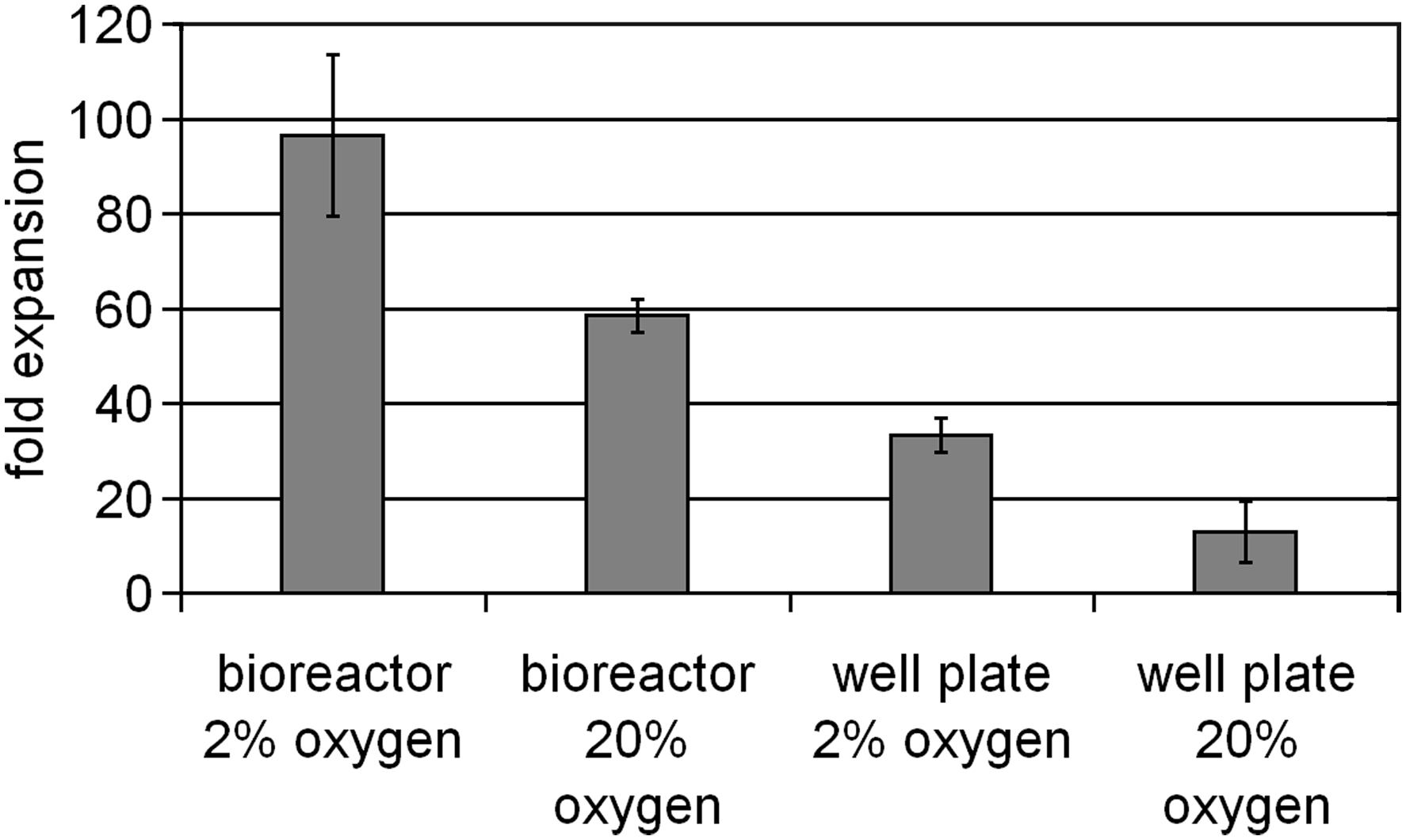
Day 7 HSC expansion initiated from 3000 CD34 +cells. Bioreactor cell expansion is significantly (p < 0.01, Student's t-test) greater than equivalent well plate cultures maintained in either 2% or 20% oxygen atmosphere (n = 6; error bars reflect 1 standard deviation).
Chondrocyte differentiation
Using an established method for in vitro chondrogenic induction of MSCs we evaluated whether the design of the membrane bioreactor would allow for similar levels of matrix production. Typically, chondrogenic medium is changed every 2–4 days, with new TGF-β added with each medium replacement. Because our membrane bioreactor design does not allow this critical growth factor to pass freely through the membrane, it is not refreshed during medium replacement. Thus we investigated whether this would have a negative impact on the cartilage-like matrix formation by quantifying the amount of sulfated GAGs that were produced over 2 weeks compared with pellets in bioreactors without membranes. The MWCO of the membrane ensures that any GAGs produced will be retained within the microwell but it was unknown whether this process would proceed as usual in this setup. At day 14 of culture, or after 11 days in the bioreactors, the pellets were harvested and the GAG composition relative to DNA was quantified. The total GAGs produced by pellets in the bioreactor with membrane was similar to the total GAGs produced in the bioreactor without membrane (Fig. 7). However, the fraction that was maintained within the matrix of the cell mass was approximately 50% in bioreactors without membranes. The remaining 50% was found from the sum of the medium supernatant at days 6, 10, and 14. In contrast, there were no detectable GAGs in the bulk medium supernatant at any of days 6, 10, or 14 as expected when the medium was recovered from bioreactors having membranes. The control of oxygen and the provision of mechanical stimulation have been previously acknowledged as significant design features to be addressed in the development of cartilage bioreactor systems, but the loss of cartilage matrix material to the bulk medium has not been addressed.42,43 Here we showed that MSCs in bioreactors were able to produce GAGs to at least the same level as MSCs in a more typical chondrogenic system. Although more specific evaluations of the membrane bioreactor-produced tissue need to be performed to characterize it as MSC-derived cartilage, we have demonstrated that a semipermeable membrane can be utilized to reduce matrix component loss to the bulk medium without diminishing the amount produced per cell and without the need for replenishing growth factors. With further optimization this technology has promise for enhancing the quality of in vitro cartilage production.
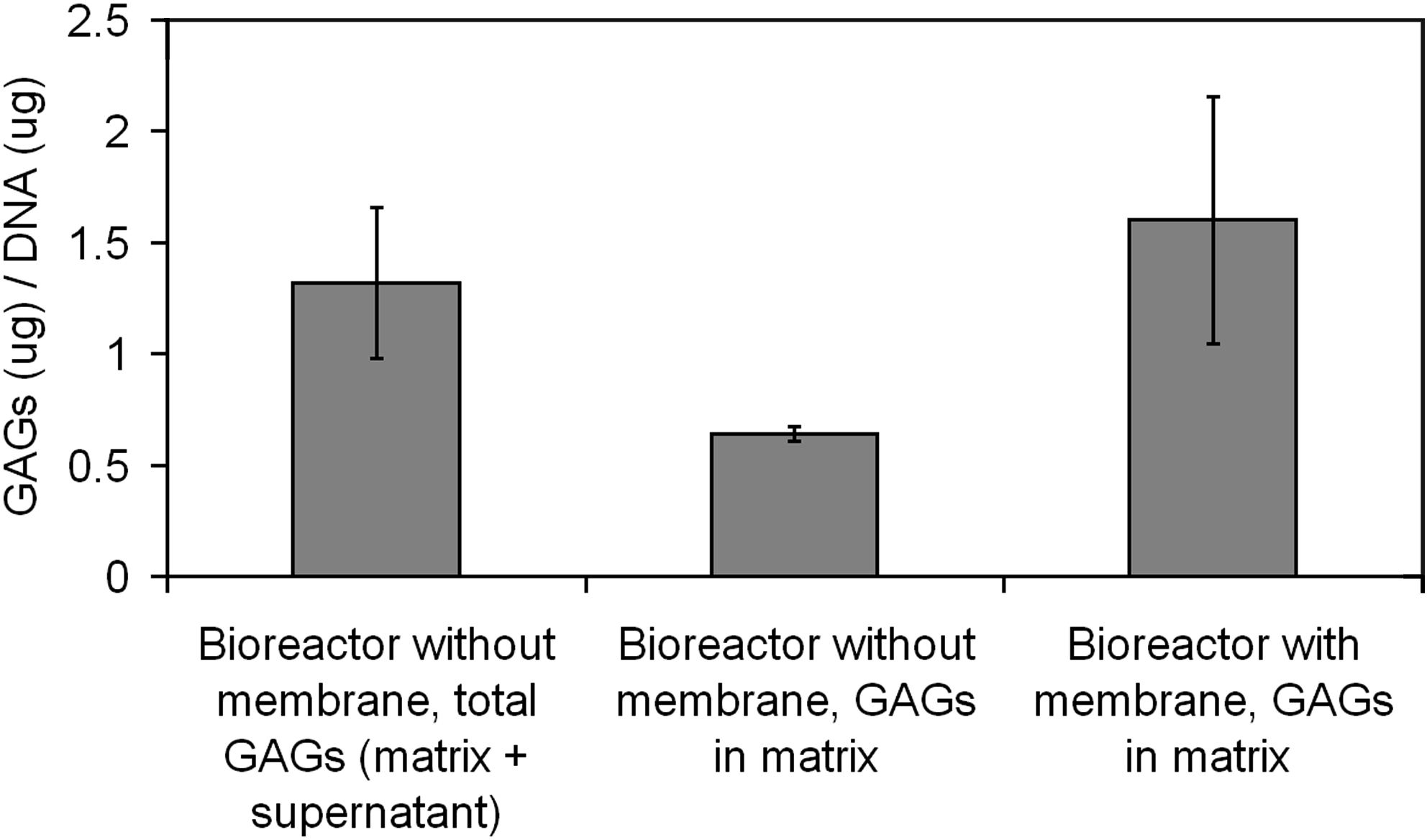
Chondrogenic cultures in membrane bioreactors were able to produce similar levels of total GAGs over 14 days as cultures without membranes. For the membrane bioreactors, no GAGs were detectable in the supernatant collected from the bulk medium at days 3, 6, 10, and 14. For bioreactors without membrane, the total amount of GAGs is the cumulative amount from supernatant measured at days 3, 6, 10, and 14 in addition to that within the matrix. Values are mean GAGs/DNA (μg/μg) ± 1 standard deviation from n = 4 bioreactors with membranes and n = 4 without. GAGs, glycosaminoglycans.
Conclusion
In this article we have outlined the design and fabrication of an easy-to-use, small volume, membrane bioreactor. We believe that such devices will find utility in the development of high-density cell culture for either cellular therapies or the generation of tissue mimics. Further, the use of membrane bioreactors enables the isolation and retention of molecules having molecular weights greater than the selected cutoff of the semipermeable membrane. The retention of high-molecular-weight factors within the small volume culture may offer significant cost savings, especially for those cultures that require frequent medium exchange. The semipermeable membrane can also be utilized to retain cellular secretions which may either condition the microenvironment or contribute to the composition of valuable extracellular matrix deposits. Specifically, we have shown (1) that it is possible to culture HSCs up to static densities of 4.5 × 107 cells/mL, (2) by isolating sensitive HSCs from the bulk medium though a semipermeable membrane it is possible for them to more rapidly condition their environment and thus rescue cell expansion from very low seeding densities, and (3) a semipermeable membrane can be used to prevent cartilage matrix (GAG) loss to the bulk medium and thus improve the matrix composition of developing cartilage pellets. We hope that the bioreactor concept presented here and the demonstrated applications will contribute to the evolution of membrane-based bioreactor systems and the tissue engineering field.
Footnotes
Acknowledgments
The authors thank the University of Queensland Early Career Grant Scheme and the Australian Research Council for their support.
Disclosure Statement
No competing financial interests exist.