
Abstract
Background:
Previous studies showed a close association between several types of human cancers and somatic mutations of thyroid hormone receptor β (TRβ) and reduced expression of TRβ due to epigenetic inactivation and/or deletion of the THRB gene. These observations suggest that TRβ could act as a tumor suppressor in carcinogenesis. However, the mechanisms by which TRβ could function to inhibit tumorigenesis are less well understood.
Methods:
We used the human follicular thyroid cancer cell lines (FTC-133 and FTC-236 cells) to elucidate how functional expression of the THRB gene could affect tumorigenesis. We stably expressed the THRB gene in FTC cells and evaluated the effects of the expressed TRβ on cancer cell proliferation, migration, and tumor growth in cell-based studies and xenograft models.
Results:
Expression of TRβ in FTC-133 cells, as compared with control FTC cells without TRβ, reduced cancer cell proliferation and impeded migration of tumor cells through inhibition of the AKT-mTOR-p70 S6K pathway. TRβ expression in FTC-133 and FTC-236 led to less tumor growth in xenograft models. Importantly, new vessel formation was significantly suppressed in tumors induced by FTC cells expressing TRβ compared with control FTC cells without TRβ. The decrease in vessel formation was mediated by the downregulation of vascular endothelial growth factor in FTC cells expressing TRβ.
Conclusions:
These findings indicate that TRβ acts as a tumor suppressor through downregulation of the AKT-mTOR-p70 S6K pathway and decreased vascular endothelial growth factor expression in FTC cells. The present results raise the possibility that TRβ could be considered as a potential therapeutic target for thyroid cancer.
Introduction
T
To test the hypothesis that the loss of normal functions of TRβ contributes to thyroid cancer development and progression, several genetic engineered mice have been developed (10). The ThrbPV/PV mouse, harboring a knock-in dominant negative mutation, known as PV, in the Thrb gene locus, spontaneously develops metastatic follicular thyroid carcinoma (FTC) similar to human FTC (17). As ThrbPV/PV mice age, pathological changes in thyroid glands progress from hyperplasia to capsular invasion, vascular invasion, anaplasia, and distant metastasis (18). Moreover, another mouse model with the loss of total functional TRs (Thra1−/− Thrb−/− mice) also leads to spontaneously developed FTC with a similar pathological progression (19). These findings indicate that the loss of normal functional TRβ by deletion or mutation contributes to thyroid carcinogenesis in mice. However, the functional consequences and the molecular actions of the re-expression of the silenced THRB gene in human thyroid cancer cells have not been well studied.
In the present study, we adopted the gain-of-function approach by expression of the THRB gene in human FTC cells, FTC-133 and FTC-236. These two cell lines were derived from the same patient with FTC, but were from the primary thyroid lesion and a neck lymph node metastasis, respectively (20). We stably expressed the THRB gene in these two cell lines and evaluated the effects of the expression of TRβ on cell proliferation and migration and tumor development in xenograft models. We found inhibition of cancer cell proliferation and impediment of migration by the expression of the THRB gene in FTC cells. These inhibitory responses were mediated by downregulation of the AKT-mTOR-p70 S6K signaling pathway due to the expression of TRβ. Moreover, in xenograft models, the expression of TRβ in FTC-133 and FTC-236 significantly reduced tumor growth and inhibited generation of new blood vessels in tumors. Further studies indicated that the decreased angiogenesis was mediated by downregulation of vascular endothelial growth factor (VEGF) by the expression TRβ in FTC cells. Thus, TRβ could act as a tumor suppressor in thyroid carcinomas by modulation of cancer angiogenesis.
Materials and Methods
Cell lines
FTC-133 and FTC-236 cells (provided by Orlo H. Clark, MD, San Francisco) were obtained from a primary human FTC and a metastatic lymph node, respectively. These cells were maintained in Dulbecco's modified Eagle's medium/Ham's F12 (1:1) medium (Invitrogen) supplemented with 10% fetal bovine serum, 10 μg/mL bovine insulin (Sigma Aldrich), 1 mIU/mL bovine thyrotropin (Sigma Aldrich), 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen), as previously described (21). The human THRB cDNA encoding TRβ1 subtype was cloned into pFH-IRESneo plasmid (a generous gift from R.G. Roeder, Rockefeller University, New York, NY) (22) to obtain Flag-hemagglutinin-tagged (FH)-TRβ1 cells. The preparation of FTC cells stably expressing FH-TRβ1 and control cells harboring the empty vector was similar to steps described for HeLa cells (23). The FTC-133 and FTC-236 cells expressing FH-TRβ1 were designated FTC-TRβ and the control cells expressing the Neo only were designated FTC-Neo cells. Cells were selected with 200 μg/mL G418 (Invitrogen) after transfection with expression plasmids encoding TRβ1 or the Neo. G418-resistant colonies expressing FH-tagged proteins were expanded for subsequent experiments. The expression of TRβ1 protein was verified by Western blot analysis using anti-TRβ1 C4 antibodies (24).
Cell proliferation assay and wound healing assay
Cell proliferation assay was done as previously described (21). Briefly, 5×104 cells were cultured for 24 hours in a 6-well plate, and nonattached cells were removed by gently washing twice with 2 mL of phosphate-buffered saline. The number of attached cells was counted every 24 hours by using a cell and particle counter (Beckmann Coulter). To evaluate the effect of T3 on cell proliferation, cells were cultured in the medium containing 10% thyroid hormone–deficient bovine serum (Td medium) for 24 hours. Cells were re-seeded in 6-well plates at a density of 5×104 cells/well in Td medium (day 0). Cells were further cultured in the medium with T3 (100 nM) or without T3, and cells were counted each day for 6 days.
Wound healing assay was carried out as previously described with some modifications (21). The wound was applied with a pipette tip on the confluent cells, and nonattached cells were removed by washing with phosphate-buffered saline followed by addition of a complete culture medium. We visualized cell migration with an inverted microscope at ×100 magnification at baseline, 8 hours, and 24 hours. The cell migration rate was quantified by measuring the distance between the edges of the wound, and the percentage of migration was determined as the ratio between migrated distance and initial distance of the wound.
To evaluate the T3 effect, a similar protocol was used, except that cells were seeded at 1,000,000 per well of a 6-well plate in Td medium for 24 hours. Cells were further cultured in the medium with T3 (100 nM) or without T3, and the wound healing assay was performed as described above.
Western blot analysis
Preparation of whole-cell lysates from FTC-133 and FTC-236 cells was carried out as previously described (25). The protein samples (30 μg of lysates from FTC-133 and FTC-236 cells) were loaded and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. After electrophoresis, the protein was electrotransferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore). Anti-TRβ C4 antibodies were used for detection of TRβ expression. The antibodies against phosphorylated (p-)retinoblastoma (Rb) (1:500 dilution), total Rb (1:250 dilution; C-15, Cat# SC-50; Santa Cruz), p-AKT (1:1000 dilution), total AKT (1:1000 dilution), p-p70 S6K (1:1000 dilution), total p70 S6K (1:1000 dilution), p-eIF4B (1:1000 dilution), total eIF4B (1:1000 dilution), p-GSK3β (1:1000 dilution), total GSK3β (1:1000 dilution), p-4EBP1 (1:1000 dilution), total 4EBP1 (1:1000 dilution), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1000 dilution) were purchased from Cell Signaling Technology. Antibodies against cyclin B1 (1:200 dilution), cyclin D1 (1:300 dilution), cyclin E (1:200 dilution), and VEGF (1:200 dilution) were purchased from Santa Cruz Biotechnology. The blots were stripped with Re-Blot Plus (Chemicon) and re-probed with antibodies against GAPDH and poly(ADP-ribose) polymerase. Band intensities were quantified by using NIH Image software (ImageJ 1.44o; Wayne Rasband, NIH, Bethesda, MD).
In vivo mouse xenograft study
The National Cancer Institute Animal Care and Use Committee approved the protocols for animal care and handling in this study. Four-week-old male athymic NCr-nu/nu mice were obtained from the NCI-Frederick animal facility. FTC-133 cells or FTC-236 cells (5×106 cells) in 200 μL of suspension mixture with Matrigel (BD Biosciences) were subcutaneously inoculated into the right flank of mice, similar to our previous report (21). Mice were randomly divided into each group (six mice per group). Tumor size was measured with calipers, and tumor volume (V) was calculated as (length×width2)/2. Tumor weight was determined after dissection of tumor tissues from euthanized mice at the study endpoint.
Histopathological analysis
Tissues from xenografted tumor were fixed in 10% neutral buffered formalin (Sigma-Aldrich) and subsequently embedded in paraffin. Five-micrometer-thick sections were prepared and stained with hematoxylin and eosin. Immunohistochemistry was performed as previously described with some modification using Vectastain ABC kit (Vector Laboratories) (26). A rabbit anti-CD31 antibody (1:50 dilution; Abcam) was used as primary antibody.
Statistical analysis
Data are presented as mean±SE or as median/range. One-way analysis of variance with Tukey's post-hoc test was used to compare continuous variables between groups. Two-way analysis of variance with Bonferroni's post-hoc test was used to compare cell proliferation and cell migration according to the time and groups. p-Values were two-sided throughout, and p<0.05 was considered statistically significant. Data were analyzed using GraphPad Prism 4.0a (GraphPad Software).
Results
TRβ expression in FTC-133 cells inhibits cancer cell proliferation and migration
To elucidate the functional consequences of re-expression of TRβ in thyroid cancer cells, we generated FTC-133 cells stably expressing TRβ (TRβ8 and TRβ13) or only the neomycin gene as controls (Neo3 and Neo5). The protein abundance of TRβ in FTC-133-TRβ8 and FTC-133-TRβ13 was confirmed by Western blot analysis (Fig. 1A), whereas no TRβ was detected in control Neo cells (Fig. 1A). When culturing in the medium containing 10% regular fetal bovine serum, cancer cell proliferation was clearly less in FTC-133 cells expressing TRβ (TRβ8 and TRβ13) than in control FTC-133 cells (p<0.001 and p<0.001, respectively; Neo3 and Neo5; Fig. 1B-i). No significant differences in proliferation rates between two TRβ clones (TRβ8 and TRβ13) were observed (Fig. 1B-i). We further evaluated the effect of T3 on the cell proliferation of TRβ8 cells and Neo cells by first culturing cells in the medium containing Td serum followed by culturing the cells in the Td medium with or without T3. As shown in Figure 1B-ii, no effect of T3 was observed on the proliferation of Neo cells that did not express TRβ. In contrast, T3 further potentiated the inhibitory effect on the proliferation of FTC133 cells expressing TRβ.
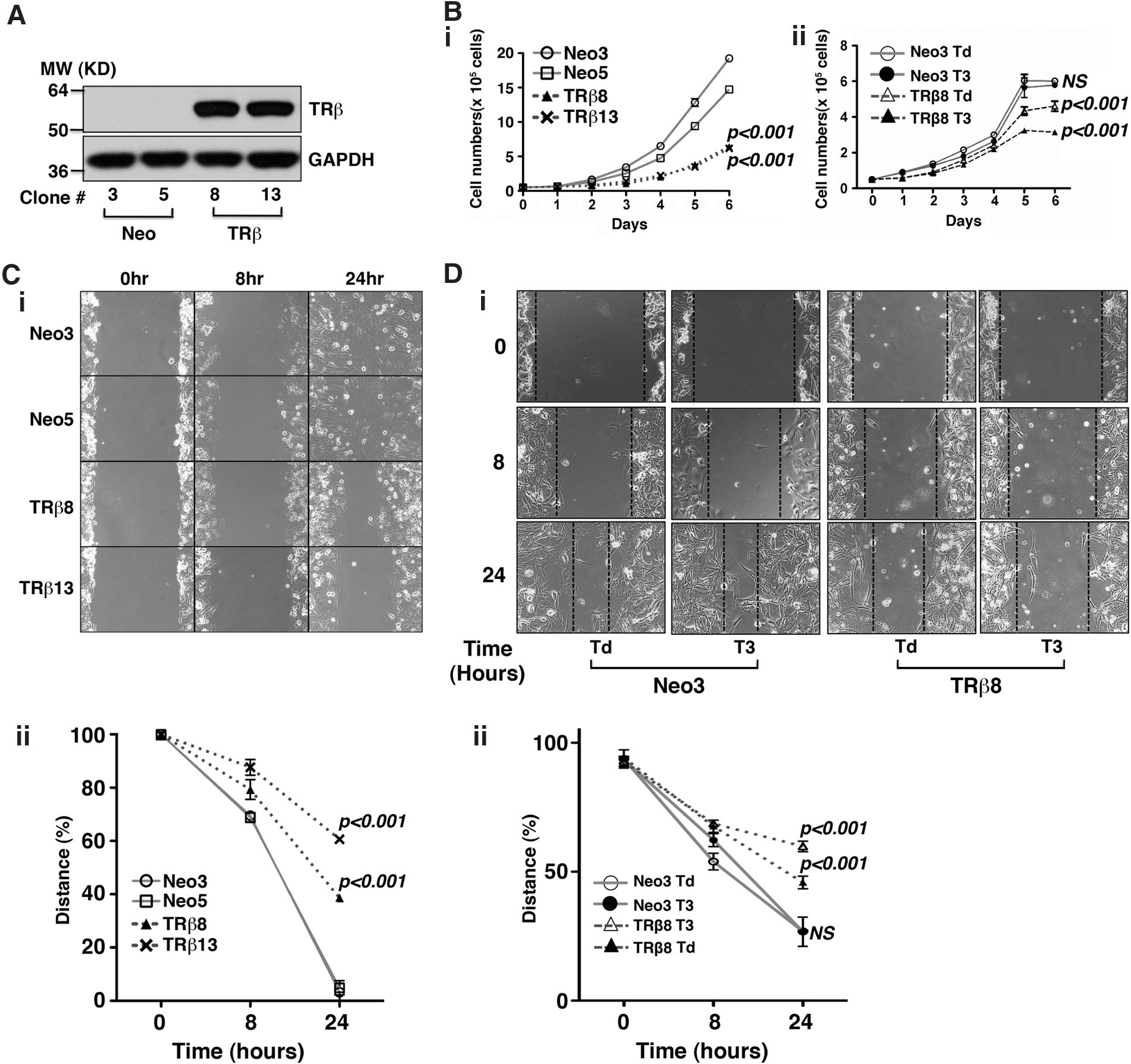
Stable expression of TRβ in FTC-133 thyroid cancer cells reduces cell proliferation and migration.
The effect of TRβ expression in FTC-133 cells on cancer cell motility was evaluated by wound healing assays. When the wound healing assays were conducted in cells cultured in the medium containing 10% regular fetal bovine serum, the cell migration was markedly impeded in FTC-133 cells expressing TRβ (TRβ8 and TRβ13 clones) compared with the control Neo FTC-133 cells (Fig. 1C-i). Quantitative analysis showed that the migration was significantly inhibited (p<0.001 and p<0.001, respectively (Fig. 1C-ii). We further evaluated the effect of T3 on migration of TRβ8 cells and Neo cells by first culturing cells in the medium containing Td serum followed by culturing the cells in the Td medium with or without T3. As shown in Figure 1D-i, no significant effect of T3 was observed on the proliferation of Neo cells. In contrast, T3 further impeded the migration of FTC133 cells expressing TRβ (Fig. 1D-i). Quantitative analysis showed that the migration was further impeded by T3 in FTC133 cells expressing TRβ (p<0.001 and p<0.001, respectively (Fig. 1D-ii).
We further examined the abundance and activity of key regulators involved in cell cycle progression in thyroid cancer cells. The level of p-Rb represents the G1-S cell cycle progression because unphosphorylated Rb acts as a negative regulator of the G1-S phase. Western blot analysis shows that the protein abundance of p-Rb (S780) was clearly less in FTC-133 cells expressing TRβ (TRβ8 and TRβ13) than in control FTC-133 cells (Fig. 2A-i) without apparent changes in the protein abundance of total Rb (Fig. 2A-ii). Figure 2A also shows that the protein levels of cyclin E, cyclin B1, and cyclin D1 were lower in FTC-133 cells expressing TRβ than in control FTC-133 cells (Fig. 2A-iii–v). The band intensities were quantified, and the results are shown in Figure 2B, indicating the ∼50% decrease in p-Rb/total Rb ratios (data group 1) and 75%, 80%, and 25% reduction in the protein abundance of cyclin E (data group 2), cyclin B1 (data group 3), and cyclin D1 (data group 4), respectively. These findings suggested that TRβ expression could suppress thyroid cancer cell proliferation by delaying cell cycle progression.
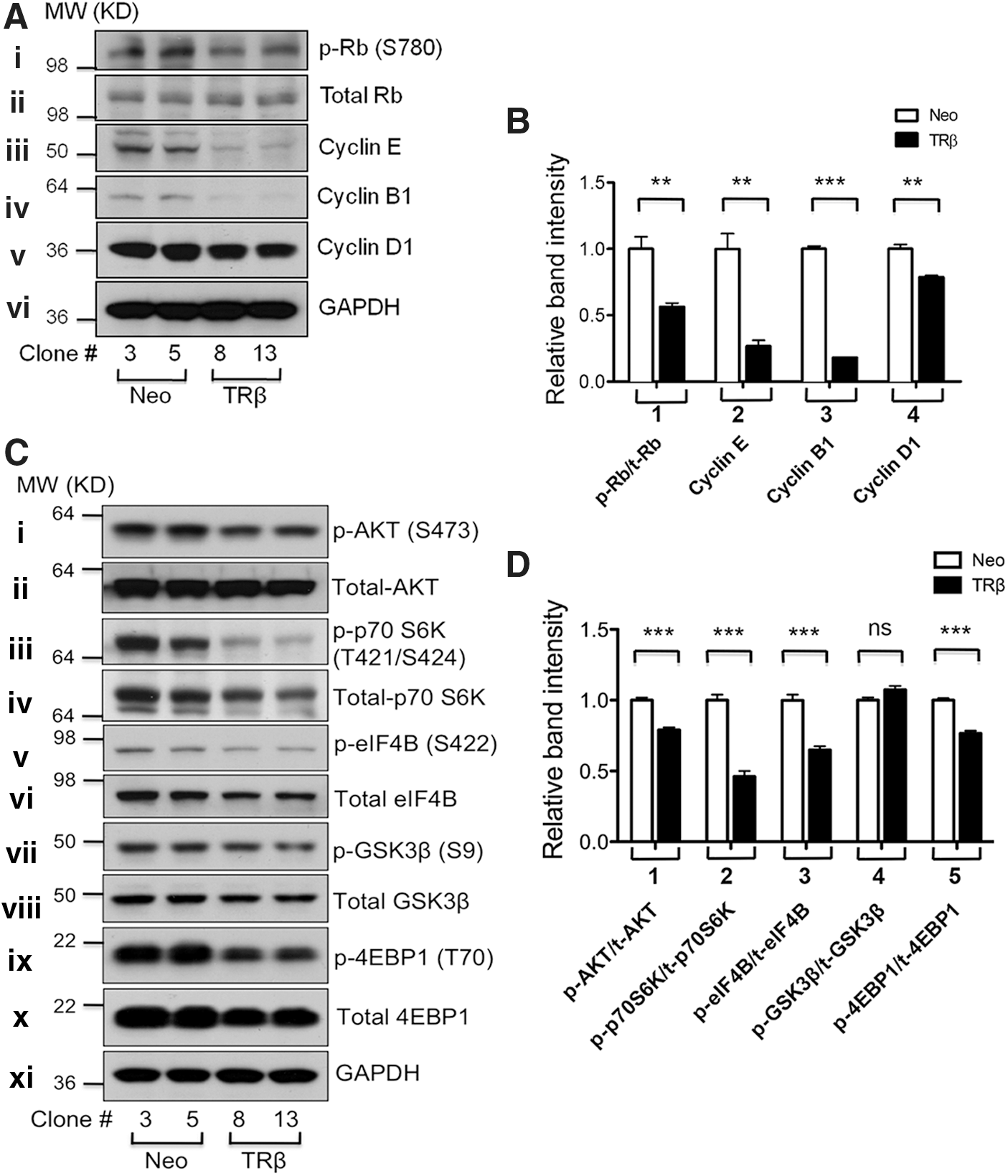
Stable expression of TRβ in FTC-133 thyroid cancer cells attenuates the phosphoinositide 3 kinase (PI3K)-AKT signaling pathway.
AKT is a central player in the signal transduction pathways activated in response to many growth factors, hormones, cytokines, and nutrients. Via several downstream phosphorylation cascades, it controls a myriad of cellular functions, including proliferation and survival, metabolism, angiogenesis, and motility. Studies indicate that aberrant AKT activation is common in a variety of human cancers, including thyroid cancer (27,28). Our previous studies revealed that increased AKT activities contribute to thyroid carcinogenesis in the ThrbPV/PV thyroid cancer mouse model (29). Therefore, we evaluated whether AKT activity was altered via TRβ re-expression by focusing on AKT-mTOR- p70 S6K signaling in FTC-133 thyroid cancer cells. As shown in Figure 2C-i, the protein level of p-AKT was decreased in TRβ-expressing FTC-133 cells (TRβ8 and TRβ13) compared with control FTC-133 cells (Fig. 2C-i). No apparent changes in the total AKT level were observed (Fig. 2C-ii). The level of p-p70 S6K (T421/S424), the downstream target of AKT-mTOR signaling, was lower in FTC-133 cells expressing TRβ than in control FTC-133 cells (Fig. 2C-iii). Consistent with these findings, the protein abundance of p-eIF4B (S422), the downstream target of p70 S6K, was also lower in TRβ-expressing FTC-133 cells than in control FTC-133 cells (Fig. 2C-v). Even though there was no significant change in the protein abundance of p-GSK3β and total GSK3β (Fig. 2C-vii,viii), the protein abundance of p-4EBP1 (T70) was significantly decreased in FTC-133 cells expressing TRβ without changes in total 4EBP1 protein levels (Fig. 2C-ix,x). Figure 2D shows the relative ratios of p-AKT/total AKT, p-p70 S6K/total p70 S6K, p-eIF4B/total eIF4B, p-GSK3β/total GSK3β, and p-4EBP1/total 4EBP1. These ratios were obtained after normalization of the band intensities against GAPDH signals shown in Figure 2C. These ratios indicated significant attenuation of the AKT-p70 S6K-p-4EBP1 pathway by re-expression of TRβ in FTC-133 cells.
TRβ expression inhibits growth of tumors induced by FTC-133 and FTC-236 cells in mouse xenograft models
To further validate the findings of the cell-based studies described above, we evaluated the effects of the expression of TRβ on tumorigenesis using in vivo mouse xenograft models. FTC-133 cells expressing TRβ (TRβ8 and TRβ13) and control cells (Neo3 and Neo5) were subcutaneously inoculated into the right flanks of athymic nude mice. As shown in Figure 3A, the volume of tumors induced by FTC-133-TRβ8 and FTC-133-TRβ13 was significantly less than that of tumors induced by control FTC-133 (Neo3 and Neo5) cells (p<0.001 and p<0.001, respectively). The weights of FTC-133-induced tumors were measured at the endpoint (4 weeks after cancer cell inoculation). The weight of tumors induced by FTC-133-TRβ8 and FTC-133-TRβ13 was also significantly less than that of control tumors (p<0.001 and p<0.001, respectively; Fig. 3B).
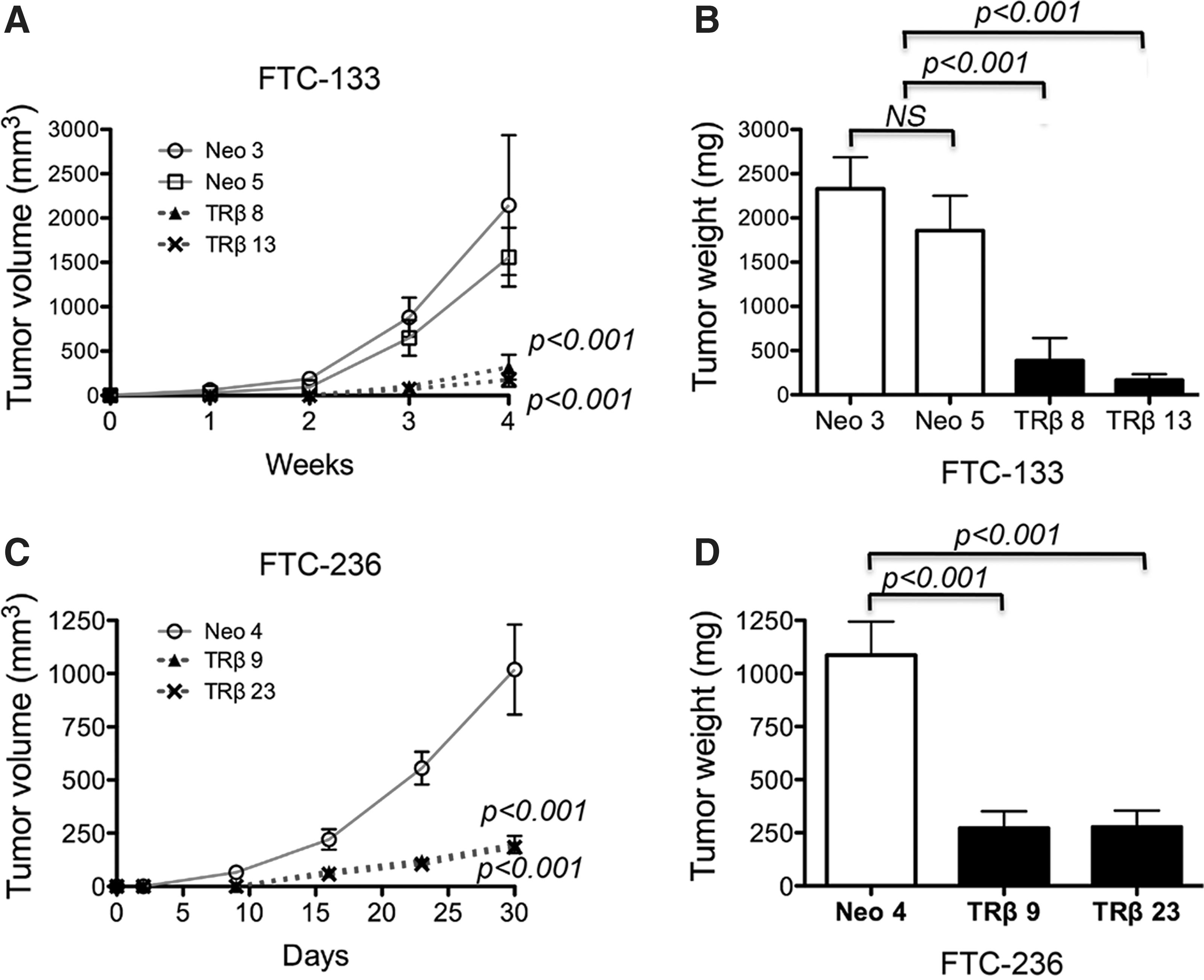
TRβ expression inhibits growth of tumors induced by FTC-133 cells and FTC-236 cells in mouse xenograft models.
We also inoculated FTC-236 cells in athymic nude mice and evaluated the effects of the expression of TRβ tumor growth. FTC-236 cells were derived from neck lymph node metastases of the same patient from whom FTC-133 cells were derived using the primary tumor (20). Previous studies showed that FTC-236 cells had a more invasive phenotype and were less responsive to thyrotropin than FTC-133 cells (20,30). In our previous study, we showed that the THRB gene was inactivated by promoter hypermethylation in FTC-236 cells, and re-activation of the THRB gene led to a less aggressive phenotype in an in vitro study (21). We, therefore, examined the in vivo effect of TRβ expression in FTC-236 cells to confirm the effect of TRβ expression and to ascertain whether common signaling pathways inhibited tumor growth. Figure 3C shows that, indeed, the volume of tumors induced by FTC-236 cells expressing TRβ (FTC-236-TRβ9 and FTC-236-TRβ23) was also significantly lower than that of control FTC-236-Neo4-induced tumors (p<0.001 and p<0.001, respectively). At the study endpoint, the weight of tumors induced by FTC-236 cells expressing in TRβ was significantly lower than that of FTC-236-Neo4 (p<0.001 and p<0.001, respectively; Fig. 3D). These findings confirmed the tumor-suppressing effect of TRβ in vivo, not only in FTC-133, but also in FTC-236 thyroid cancer cells.
TRβ expression reduces tumor growth of thyroid cancer cells by inhibiting new vessel formation
To understand how TRβ expression inhibits in vivo tumor growth, we evaluated the pathohistological features of tumors induced by FTC-133 cells without the expression of TRβ (Fig. 4A-i) or with it (Fig. 4A-ii). Hematoxylin and eosin–stained cells revealed the presence of undifferentiated cancer cells with brisk mitotic activity and with minimal apoptosis (Fig. 4A-i, ii). There was no major apparent histological difference in tumor cells induced by FTC-133 cells with or without TRβ. However, it is important to note that the number of vascular structures clearly visible in tumors induced by control FTC-133 cells (indicated by arrows in Fig. 4A-i) was decreased in tumors induced by TRβ-expressing FTC-133 cells (Fig. 4A-ii).
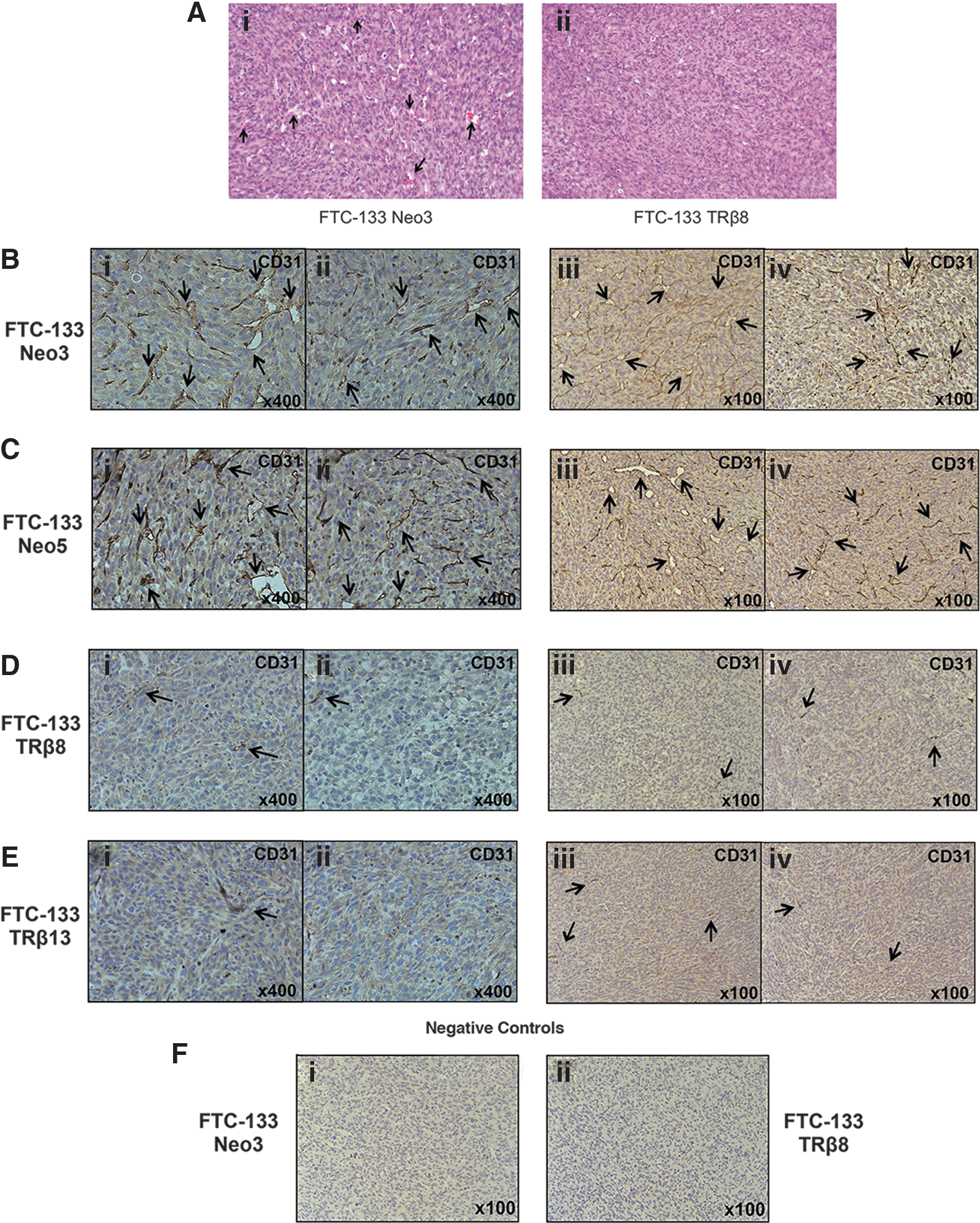
TRβ expression inhibits neovascularization of tumors induced by FTC-133 cells in a mouse xenograft model.
New vessel formation is known to be a critical factor for tumor growth, and an antiangiogenesis approach has been proposed as an important cancer therapy (31). Therefore, we evaluated the extent of new vessel formation in FTC-133 cell-induced tumors by immunostaining the endothelial marker, CD31. The expression of CD31 in TRβ-expressing FTC-133 cell-induced tumors (indicated by arrows in Fig. 4D-i, ii and Fig. 4E-i, ii [high magnification] and in Fig. 4D-iii, iv and Fig. 4E-iii, iv [low magnification]) was clearly less than with control FTC-133 tumors (indicated by arrows in Fig. 4B-i, ii and Fig. 4C-i, ii [high magnification] and in Fig. 4B-iii, iv and Fig. 4C-iii, iv [low magnification]). The negative controls are shown in Figure 4F for the control Neo cells and FTC-133 cells expressing TRβ8 (Fig. 4F-i and F-ii, respectively). These findings suggested that TRβ expression reduces tumor growth in FTC-133 thyroid cancer cells by inhibiting new vessel formation.
TRβ expression inhibits expression of VEGF in FTC-133 and FTC-236 cells
VEGF is a main regulator of tumor-induced angiogenesis and acts on vascular endothelial cells to induce vascular permeability and to reprogram gene expression, leading to endothelial cell proliferation and migration in vitro, and to generation of new blood vessels in vivo (31 –33). Therefore, we examined the level of VEGF in TRβ-expressing cells and control cells. As shown in Figure 5A, the protein abundance of VEGF was lower in FTC-133-TRβ8 and FTC-133-TRβ13 cells than in control FTC-133 cells (Neo3 and Neo5). The quantification of the band intensities shows that the protein abundance of VEGF was decreased by about 50% in TRβ-expressing FTC-133 cells (Fig. 5B). Consistently, we also found that the protein abundance of VEGF was lower in FTC-236-TRβ9 and FTC-236-TRβ23 cells than in control FTC-236 cells (Neo1 and Neo4; Fig. 5C). The quantification of the band intensities is shown in Figure 5D. These findings revealed the downregulation of VEGF expression by TRβ to be an important mechanism by which TRβ expression in thyroid cancer cells inhibits new vessel formation.
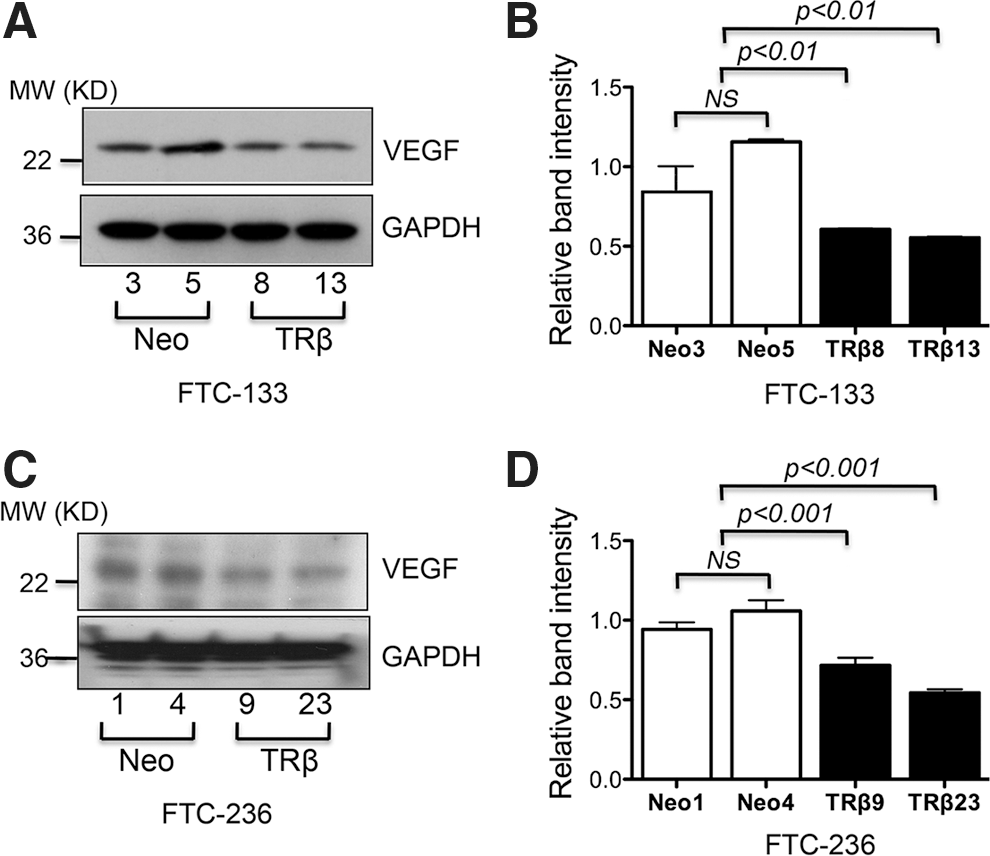
TRβ expression decreases protein abundance of VEGF in FTC-133 and FTC-236 thyroid cancer cells.
Discussion
Previous reports indicating a close association of reduced expression and somatic mutations of the THRB gene with several human cancers support the hypothesis that TRβ could act as a tumor suppressor (3 –10). In line with these studies, we have recently shown a significant correlation of reduced THRB gene expression with more advanced cancer stage in human differentiated thyroid cancers (21). Direct in vivo evidence to support the tumor suppressor role of TRβ in thyroid cancer came from the observations that mice deficient of all functional TRs (Thra1−/−Thrb−/− mice) or with a targeted homozygous mutation of the Thrb gene (ThrbPV/PV mice) spontaneously develop metastatic thyroid cancer similar to human cancer (17,19).
In the present study, we adopted the gain-of-function approach by expression of TRβ in human thyroid cancer cells. The expression of TRβ in FTC cells led to less tumor cell proliferation and impeded the migration of cancer cells. Moreover, in addition to these cell-based results, we showed that the growth of tumor induced by TRβ-expressing FTC cells was inhibited in in vivo xenograft models. Importantly, we found that new vessel formation, critical for carcinogenesis and cancer progression, was suppressed in tumors induced by TRβ-expressing FTC cells. The expression of a critical growth factor that stimulates angiogenesis, VEGF, was suppressed in TRβ-expressing FTC cells. Therefore, the present study provides additional evidence to show that TRβ acts as a tumor suppressor in thyroid carcinogenesis.
Tumor angiogenesis is an essential process for blood supply to support tumor growth and metastasis (34). To obtain this blood supply, tumor cells can tilt the balance toward stimulatory angiogenic factors to drive vascular growth by attracting and activating cells from within the microenvironment of the tumor (34). VEGF is one of the essential angiogenic factors needed to induce activation of endothelial cells, and its expression is commonly elevated in most types of human cancer cells (35). Recently, many kinds of antiangiogenic agents targeting VEGF signaling pathways have been developed and are in clinical use. In the present study, we found that tumor growth induced by TRβ-expressing FTC cells was decreased through inhibition of tumor angiogenesis. The expression of endothelial marker CD31 was clearly reduced in tumors induced by TRβ-expressing FTC cells (Fig. 4D, E). Currently, the molecular mechanisms by which TRs act to affect tumor angiogenesis are not known. In models of thyroid hormone-induced cardiac hypertrophy, thyroid hormone could enhance angiogenesis by engaging in crosstalk with the VEGF signaling, and a derivative of thyroxine, tetraiodothyroacetic acid (tetrac), was shown to inhibit vascular budding in response to VEGF (35). The present findings that the expression of VEGF was decreased by expression of TRβ in FTC cells are consistent with the observations of reduced new vessel formation in xenografted tumors. Clearly, further investigation is needed to elucidate the role of TRβ in tumor angiogenesis.
It was reported that FTC-133 cells have hemizygous PTEN allele deletion and a splice variant IVS4-19G→A in the remaining allele (36). The decreased PTEN mRNA and protein levels result in the overactivation of the AKT-mTOR-p70S6K signaling pathway in FTC-133 cells. The present studies showed that the overactivated AKT-mTOR-p70S6K pathway was attenuated by TRβ expressed in FTC-133 cells (Fig. 2). Aberrant activation of the AKT-mTOR-p70S6K pathways, as well as activated VEGF/receptors signaling, has been described in both thyroid cancer mouse models of Thra1−/−Thrb−/− mice and ThrbPV/PV mice (19,29). Our previous studies also showed that treatment of ThrbPV/PV mice with a PI3K inhibitor, LY294002, effectively delays thyroid tumor progression and inhibits distant metastasis (37). We also reported that another mouse model of follicular thyroid cancer, ThrbPV/PVPten+/− mice, displayed overactivation of the AKT-mTOR-p70S6K signaling pathway (38). Treatment of ThrbPV/PVPten+/− mice with a specific mTOR inhibitor (RAD001) reduced thyroid tumor growth (39). The findings from these mouse models showed that the loss of normal functions of wild-type TRβ due to gene knockout (Thra1−/−Thrb−/− mice) or mutations (ThrbPV/PV mice and its derived strain ThrbPV/PVPten+/− mice) promotes thyroid carcinogenesis. In line with the findings from animal models, the present study shows that re-gaining of TRβ functions inhibits tumor development in xenograft models, at least in part, via attenuation of the AKT-mTOR-p70S6K signaling pathway. Interestingly, TRβ, when overexpressed in FTC-236 cells, acts to attenuate β-catenin signaling to decrease cell proliferation (21). However, in contrast to FTC-236 cells, we did not find that the re-expressed TRβ in FTC-133 cells affected the β-catenin signaling (data not shown). These observations suggested that the tumor suppressor actions of TRβ differ in FTC-133 and FTC-236 cells. FTC-133 cells were derived from the primary lesions, whereas FTC-236 cells were derived form a lymph node metastasis from the same patient (20). Thus, these two cells are known to have distinct phenotypes (30,40). It is reasonable to expect that the signaling pathways affected by TRβ would be different due to different genetic alterations in these two thyroid cancer cell lines.
In summary, TRβ expression in FTC cells decreased cancer cell proliferation, impeded tumor cell migration, and inhibited tumor growth in vivo through downregulation of the AKT-mTOR-p70 S6P signaling pathway and suppression of VEGF expression. These findings indicate that TRβ could act as a tumor suppressor in thyroid cancer by modulation of cancer angiogenesis.
Footnotes
Acknowledgment
The present research was supported by the Intramural Research Program at the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Author Disclosure Statement
The authors have nothing to disclose.