
Abstract
Background:
The presence of single nucleotide polymorphisms (SNPs) in the REarranged during Transfection (RET) gene has been investigated with regard to their potential role in the development or progression of medullary thyroid cancer or pheochromocytomas (PHEO) in patients with the multiple endocrine neoplasia type 2 (MEN2) syndrome. The aim of this study was to evaluate the spectrum of RET variants in France between 2003 and 2013, and to evaluate the impact of SNPs on the MEN2 A phenotype.
Methods:
In this retrospective cohort study, RET variants were screened in 5109 index cases, and RET pathogenic variants were screened in 2214 relatives. Exons 5, 8, 10, 11, 13, 14, 15, and 16 were characterized by Sanger sequencing. RET pathogenic variants, RET variants with unknown functional significance (VUS), and four RET SNP variants—G691S (rs1799939), L769L (rs1800861), S836S (rs1800862), and S904S (rs1800863)—were characterized and are reported in index cases. In silico analysis and classification following the recommendation of the American College of Medical Genetics and Genomics was performed for RET VUS. Each patient's age at the time of diagnosis, sex, and the endocrine neoplasias present at molecular diagnosis were recorded.
Results:
Twenty-six single VUS in RET without any well-defined risk profiles were found in 33 patients. Nine of these were considered probably pathogenic, 11 of uncertain significance, and six as probably benign. Three double pathogenic variants found in three patients were classified as pathogenic. A study of the entire cohort showed that patients carrying pathogenic variants or VUS in RET together with PHEO were diagnosed earlier than the others. The presence of the G691S SNP, or a combination of SNPs, increased the risk of developing PHEO but did not modify the date of the diagnosis. No association was found between SNPs and medullary thyroid cancer or hyperparathyroidism.
Conclusions:
The findings propose a classification of 15 of the 26 VUS in RET without any well-defined risk profiles and suggest that the G691S SNP, or a combination of SNPs, may be associated with the development of PHEO.
Introduction
T
Germline gain-of-function RET variants mostly occur in exons 5, 8, 10, 11, 13, 14, 15, and 16. Various consortia, as well as the American Thyroid Association (ATA), have defined the risk profiles of each variant for the risk and aggressivity of MTC, giving estimations of the penetrance of MEN, recommending the optimal age for prophylactic thyroidectomy, and suggesting that asymptomatic patients carrying a germline disease-causing RET variant should be screened for PHEO and HPTH (1,2). Some RET variants have not been categorized because of insufficient data. More information about these cases will be needed to provide adequate genetic counseling to patients and their families. In 2010, an extensive study of RET variants in exon 10 evaluated the risk profiles of these patients and estimated the penetrance of MEN2 (3). The results showed that 50% penetrance was achieved by 36 years of age for MTC and by 68 years of age for PHEO. Another study analyzed clinical risk profiles and outcomes for RET variants at codon 634. The outcomes varied according to the amino acid substitution, and C634R seemed to be associated with a more aggressive MEN2A phenotype (4). However, some of the conclusions concerning RET variants are controversial. A recent study focusing on the RET Y791F variant, classified as moderate risk by the ATA, reported no association between this variant and susceptibility to MTC (5). The authors found a frequency of 0.008 in the tumor-free cohorts originating in Central Europe. They, and other authors, also reported subjects harboring RETY791F without the MEN2 syndrome (5,6). It is therefore important to collect more data about patients in order to update the classification of RET variants in MTC.
The presence of single nucleotide polymorphisms (SNPs) in the RET gene has been investigated with regard to their potential role in the development or progression of MTC or PHEO in patients with the MEN2 syndrome. Four SNPs— G691S (rs1799939), L769L (rs1800861), S836S (rs1800862), and S904S (rs1800863)—have been regularly studied in the context of MEN2. For each SNP, the findings were contradictory. Some clinical studies found an increased prevalence of RET SNPs in MTC (7 –11), whereas others did not (12 –14). A meta-analytic study on G691S, combining results from 968 cases of MTC and 2115 controls, found that female patients were at a higher risk of the disease than males were (15). Other studies suggest a potential additive effect of these SNPs on the susceptibility to MTC or PHEO (8,11). Taken together, these findings suggest that the consideration of the possible modifying role of these SNPs may contribute to better clinical and follow-up management.
The present study describes the spectrum of RET variants observed in the French population over the last 10 years collected from a nationwide database. A classification of 32 genotypes that have not been classified by the ATA is proposed (16). Finally, the effects of multiple RET SNPs on the MEN2A phenotype are explored.
Patients and Methods
Organization of RET gene analysis in France
In France, genetic counseling and RET molecular analysis are recommended in the presence of MTC, isolated or in combination with PHEO and HPTH. It is provided free of charge to patients and their next of kin by the French National Cancer Institute (INCa). This institute, created under the French Public Health Act of 2004, plays a pivotal role in the national coordination of actions aimed at fighting cancer. Through its coordinated programs, INCa has funded a total of 11 laboratories across the country to perform RET molecular analyses, constituting the nationwide TenGen network.
Database
TenGen, which is affiliated with the French Endocrine Society, has set up an exhaustive national database to collect clinical and genetic information about these patients. The database has been declared to the French National Commission for Computerized Data and Individual Freedom (CNIL) and is registered under no. 91513. The TenGen network has verified all the data included in the RET database from 2003 to 2013. This study is nationwide, and all centers performing RET gene analysis have contributed to the RET database.
Patients
Over the 10-year period from 2003 to 2013, 5109 index cases and 2214 relatives underwent RET proto-oncogene analysis. Index cases were diagnosed with various combinations of endocrine neoplasias, that is, MTC associated or not with PHEO or HPTH. Each index case belonged to apparently unrelated families according to the genetic counseling and the family name. Each laboratory was invited to anonymize and complete a form for each index case. The information collected included each patient's age at the time of diagnosis, sex, the endocrine neoplasias present, and the RET genetic variants. For screened relatives only, the presence or absence of the RET pathogenic variant was available.
Molecular characterization
The germline RET variants, including SNP, in exons 5, 8, 10, 11, 13, 14, and 16 were analyzed (Genbank reference NM_020975.4). Genomic DNA was obtained from peripheral blood leucocytes, and the genotyping was done using direct sequencing.
This study uses “variant” as a neutral term, “pathogenic variant” for variants affecting RET function, and “polymorphism” or “SNP” for a variant found at a frequency ≥1% in the general population. The RET SNPs studied included G691S (rs1799939), L769L (rs1800861), S836S (rs1800862), and S904S (rs1800863). For variants for which a functional effect was not well-defined and that were not listed in the ATA MTC guidelines (1,2,17), the term “variant of unknown significance” (VUS) was used. The variants were also classified following the recommendation of the American College of Medical Genetics (ACMG) (16). In silico analysis was performed by three different in silico bioinformatics algorithms, that is, the scale-invariant feature transform (SIFT), polymorphism phenotyping (PolyPhen-2), and the MutationTaster.
Ethics
Informed consent for genetic analysis and the use of data was obtained from all patients or their legal guardians according to the French governmental regulations.
Statistical analysis
Ordinal or nominal variables were summarized in terms of the frequency of occurrence and percentage, or the mean ± standard deviation (SD) for continuous variables. Baseline characteristics were compared using either the chi-square test for qualitative variables or Student's t-test for quantitative variables. Kaplan–Meier curves were used for estimating the age-related penetrance of MEN2A according to the presence of PHEO, and comparisons between curves were made with a log-rank test. Statistical analyses were performed using SPSS v15 (SPSS Inc., Chicago, IL).
Results
Pathogenic variants and VUS in RET in France
Over the 10-year period from 2003 to 2013, 7323 tests were carried out, of which 5109 concerned index cases, and 2214 were used for family studies (Table 1). The frequency of pathogenic variants and VUS in RET was 10.1% in the index cases, that is, 514/5109 patients tested, and 39.1% in the next of kin, that is, 867 carriers of RET pathogenic variants found in 2214 patients studied. Genetic data were collected in 480/514 (94.2%) index cases in which the eight RET exons had been analyzed during the period 2003–2013. The mean age of the patients was 47.3 ± 17.7 years, and 59.9% of the patients were women. The clinical feature at the time of diagnosis was MEN2A in 448 (93.3%) patients and MEN2B in 32 (6.7%) patients.
Report completed by the INCa each year for index cases and screened relatives.
Bold: patients with RET variants.
RET, REarranged during Transfection gene; VUS, variants with unknown functional significance; MEN2, multiple endocrine neoplasia type 2.
Among the 480 index cases, 477 patients had missense single variants (Fig. 1). Of the single variants found, 51.6% are located in the extra-cellular domain of the receptor, affecting the cysteine codons. The most frequent variants were V804M (17.2%), C634R (11.0%), C634Y (9.5%), L790F (8.9%), and M918T (6.0%), whereas the others represented <5% (Supplementary Table S1; Supplementary Data are available online at
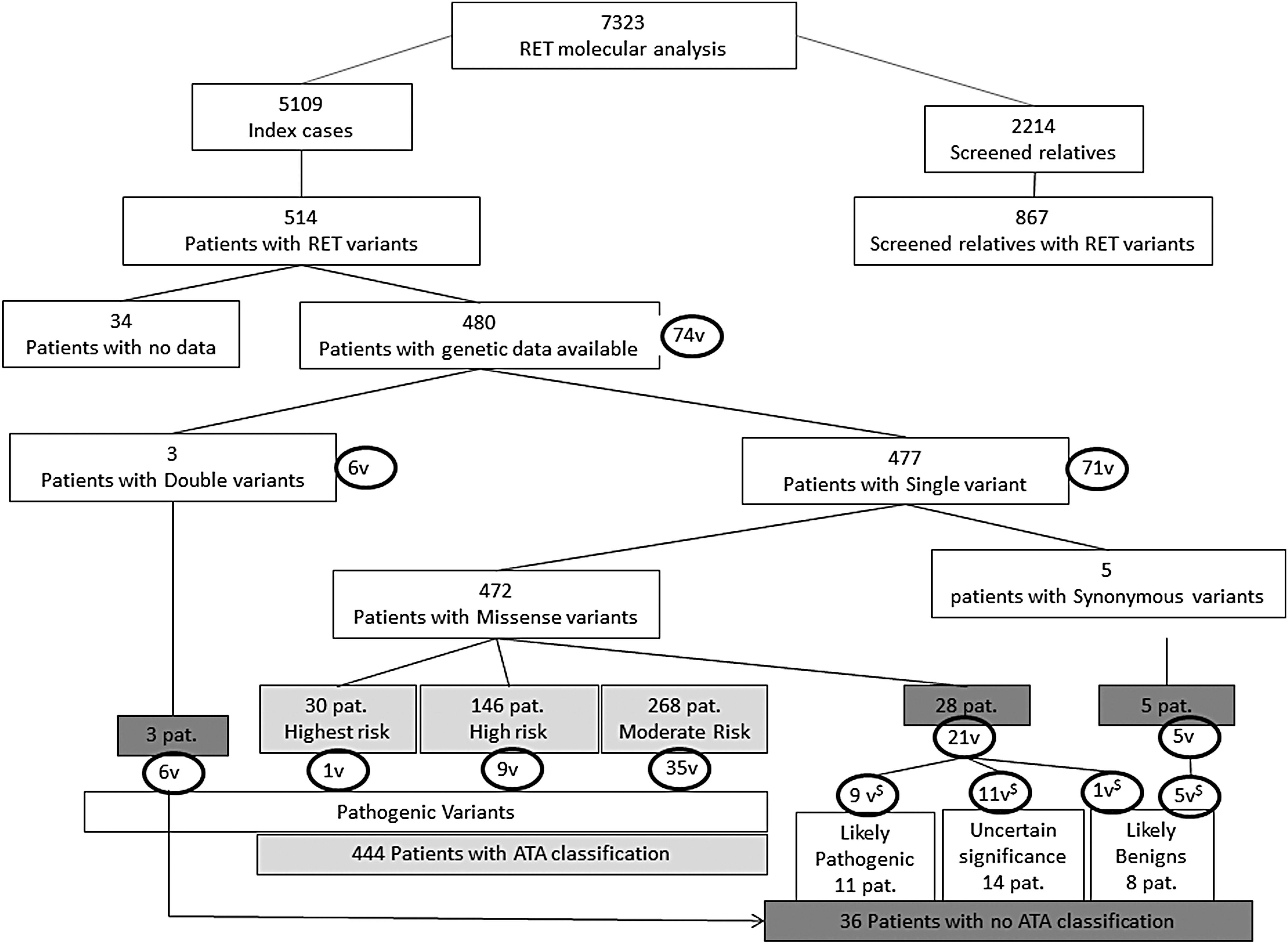
Pathogenic variants or variants with unknown functional significance (VUS) in REarranged during Transfection (RET) for index cases and screened relatives from 2003 to 2013. Classification by risk groups following the 2015 American Thyroid Association (ATA) clinical guidelines for medullary thyroid cancer and proposition of classifying variants not found in the guidelines following the consensus recommendation of the American College of Medical Genetics. $Classification following the recommendation of the ACMG. Light gray variants classified in ATA; dark gray variants not classified in ATA.V, pathogenic variants or VUS in RET; Pat., patient.
Three of the patients had three different double variants: (V804M; R844L), (K603Q; L790F), and (D631Y; Y791F). Each of the variants observed was classified as pathogenic by the ATA. However, the double variants were not classified.
Clinical and oncological features of patients carrying ATA classified variants
Following the 2015 ATA Clinical Guidelines for MTC (1), the variants of 444 (92.3%) patients were listed in the guidelines, and these patients had a well-defined risk profile. Among these, 268 (55.7%) patients could be qualified as carrying a “moderate risk” (corresponding to a combination of the former class A and class B pathogenic variants included in the 2009 ATA clinical guidelines) (2), 146 (30.4%) patients as being at “high risk” (former class C pathogenic variants, which includes patients with the RET codon C634 and A883F pathogenic variants), and 30 (6.2%) patients at the “highest risk” (former class D pathogenic variants, which includes MEN2B and RET codon M918T; Fig. 1).
Clinical and oncological features of patients carrying non-ATA-classified variants
Among the index cases, 36 (7.7%) patients had variants not classified by the ATA, including 28 patients with 21 different single missense variants, five patients with five different synonymous RET variants, and three patients with double missense variants. Thirty patients were diagnosed with MTC and/or PHEO after 40 years of age, whereas six patients were diagnosed before 40 years of age (Table 2).
ACMG, American College of Medical Genetics; MTC, medullary thyroid cancer; PHEO, pheochromocytoma; T, tolerated; D, deleterious; P, possibly damaging; ND, not done; CCH, C cell hyperplasia.
Two of the five synonymous RET variants—C620C (c.1860C>T) and S699S (c.2097C>T)—have never been reported either in the NCBI dbSNP or in the Exome Aggregation Consortium (ExAC) databank. All patients with these variants were diagnosed after 40 years of age.
Of the 21 RET single variants not listed in the ATA classification (Table 2), five have never been reported either in the NCBI dbSNP or in the ExAC databank. Three of the latter—C515F (c.1544G>T), C541R (c.1621T>C), and N783S (c.2348A>G)—were qualified as “damaging” by three different in silico bioinformatics algorithms. The patient who carried the C541R mutation was diagnosed before one year of age with a suspicion of MEN2B and Hirschsprung's disease. The parents were asymptomatic, and no genetic analysis was performed for them. Unfortunately, access was not available to additional clinical data on the patient to confirm MEN2B or Hirschprung's disease. The two other variants—V704F (c.2110G>T) and F644V (c.1930T>G)—were qualified as “benign” or “damaging,” depending on the bioinformatics tool used. The patient carrying the F644V substitution was diagnosed with MTC at 35 years of age, and the patient carrying the V704F variant was diagnosed with HPTH at 49 years of age.
Of the 16 single variants not classified by the ATA listed in the NCBI dbSNP or the ExAC databank, according to the three algorithms used, four were qualified as “benign” (one of the patients was diagnosed at 21 years of age), five were qualified as “damaging” or “possibly damaging,” and seven RET variants were qualified as being either “benign” or “damaging” according to the algorithm used (two of these patients were diagnosed before 40 years of age; Table 2).
The criteria for classifying these 21 single RET variants were used with the consensus recommendation of the ACMG. Nine variants for 11 patients could be classified as probably pathogenic, six as probably benign, and the others as VUS.
Clinical and oncological features of patients carrying RET double pathogenic variants
Each single variant found in these cases is already listed in the ATA MTC guidelines with a moderate risk of aggressive MTC, but the risks for double mutations have not been reported. One patient with a concomitant V804M and R844L substitution was diagnosed with MTC at 51 years of age with a metastatic node. Her twin sister was diagnosed with MTC at the same age but without any metastatic nodes. The two pathogenic variants were on the same allele. The patient with K603Q and L790F had MTC alone at 60 years of age. No family study was available for this patient. A 31-year-old male patient, diagnosed with PHEO, harbored D631Y and Y791F. The two variants were in distinct alleles. His father, carrying D631Y, was diagnosed with metastatic bilateral PHEO at 65 years of age. No information was available for his mother.
The variant spectrum of the RET codon in MEN2A is associated with PHEO and HPTH
Complete clinical information was collected at the time of molecular analysis for 366/448 MEN2A patients. Among these patients, 77.3% had MTC alone, 17.5% had MTC and PHEO, 3.3% had MTC and HPTH, and 1.9% had MTC, PHEO, and HPTH.
The frequency of patients for whom PHEO was the first appearing lesion was 19.4% (71/366 MEN2A patients). All the variants were pathogenic and already listed in the ATA MTC guidelines, except for G548S and K666T. Sixty-one of the 71 patients with PHEO had predominantly variants in exons 10 or 11, and these mainly concerned cysteine residues for these exons, except for the K666T variant. The mean age at the time of molecular diagnosis of MEN2A was significantly different between patients with or without PHEO: 41.8 ± 3.0 and 48.7 ± 16.3 years, respectively (p = 0.001, Student's t-test, and p < 0.0001, log-rank test; Fig. 2).
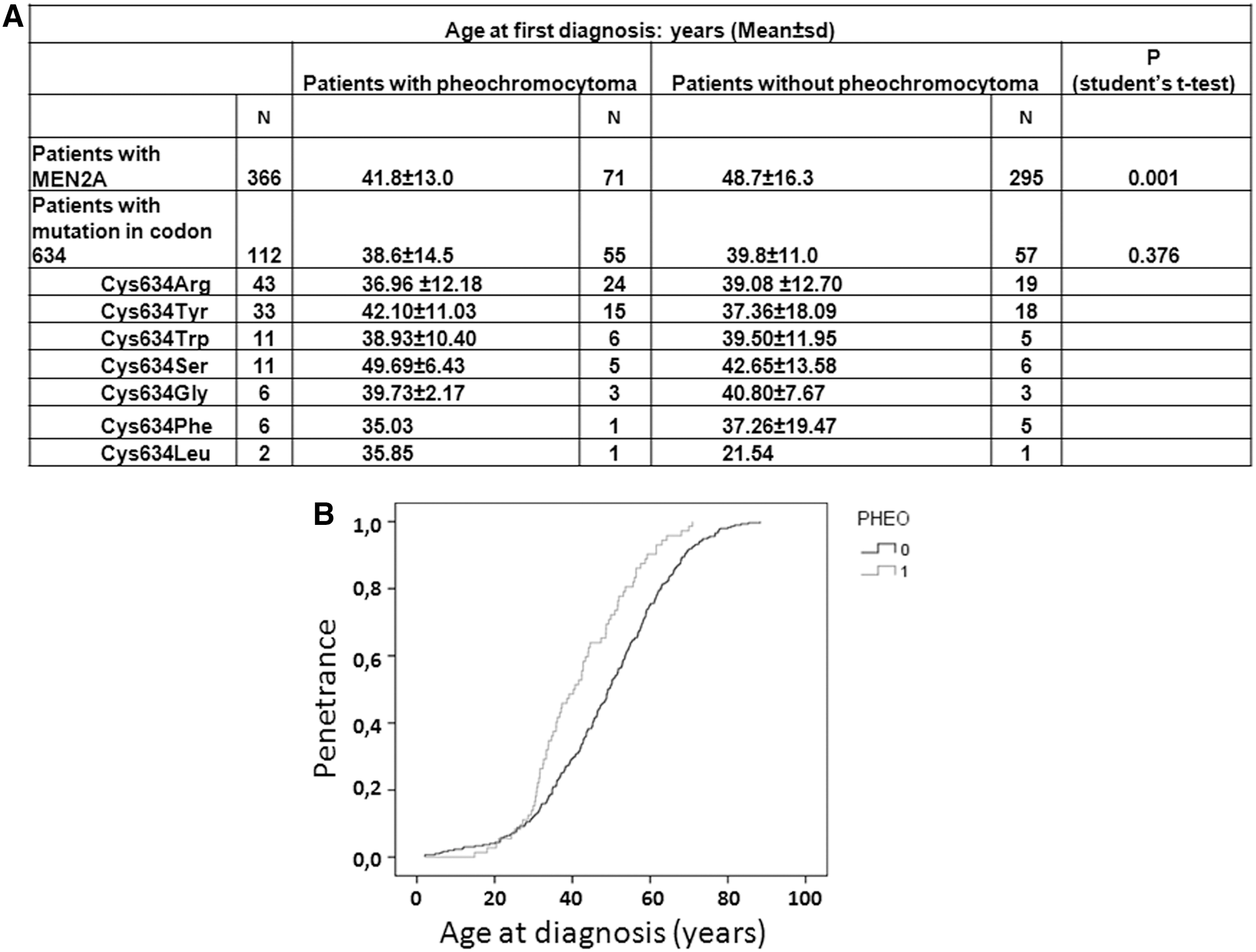
Patients with multiple endocrine neoplasia type 2A (MEN2A) carrying RET germline pathogenic variants or VUS (N = 366 index cases). (
Codon 634, which was most frequently mutated, was found to be affected in 112 patients, among which 55 (49.1%) had PHEO. In this group of 112 patients, 43 had C634R (38.4%), 33 had C634Y (29.5%), 11 had C634W (9.8%), 11 had C634S (9.8%), six had C634G (5.4%), six had C634F (5.4%), and two had C634L (1.8%). The risk of PHEO was about 50% for each variant except for C634F where it was lower at 17%. The mean age at the time of diagnosis did not significantly differ between patients, with or without PHEO, harboring a codon 634 pathogenic variant.
HPTH was found in 19/366 (5.2%) MEN2A patients. All the mutations were pathogenic variants already listed in the ATA MTC guidelines, except for V704F. Of these 19 patients with HPTH, 13 (68%) harbored pathogenic variants affecting cysteines in exons 10 or 11. The most frequent was C634R, found in 7/19 patients. The other variants were found in the transmembrane domain (S649L in one patient) or the intracellular domain (V704F in one patient, and V804M in four patients).
RET polymorphic alleles
Clinical and molecular data were analyzed according to four SNPs—G691S (rs1799939), L769L (rs1800861), S836S (rs1800862), and S904S (rs1800863)—in 244/366 MEN2A patients with clinical information. The frequency of RET SNPs was similar to that reported in the literature, that is, G691S 24.3%, L769L 22.1%, S836S 8.4%, and S904S 19.9%.
The presence of the G691S SNP was associated with a higher risk of developing PHEO (p = 0.036; Fig. 3A). The same tendency was observed when in analyses of the two groups of the ATA classification “high-risk” group (patient carrier of variant in 634 codon) and “moderate-risk” group (patient carrier of variant other than 634), without reaching the significance threshold for both group (p = 0.07). However, for the patient with PHEO, statistical testing was not able to detect a difference in age at the time of molecular analysis in groups with or without G691S (46.7 ± 1.5 years with G691S and 44.3 ± 1.0 years without; p = 0.8). The study examined whether the G691S SNP would change the age at which PHEO might have been diagnosed according to the three ATA categories of pathogenic variants. No difference between the different risk levels was found.
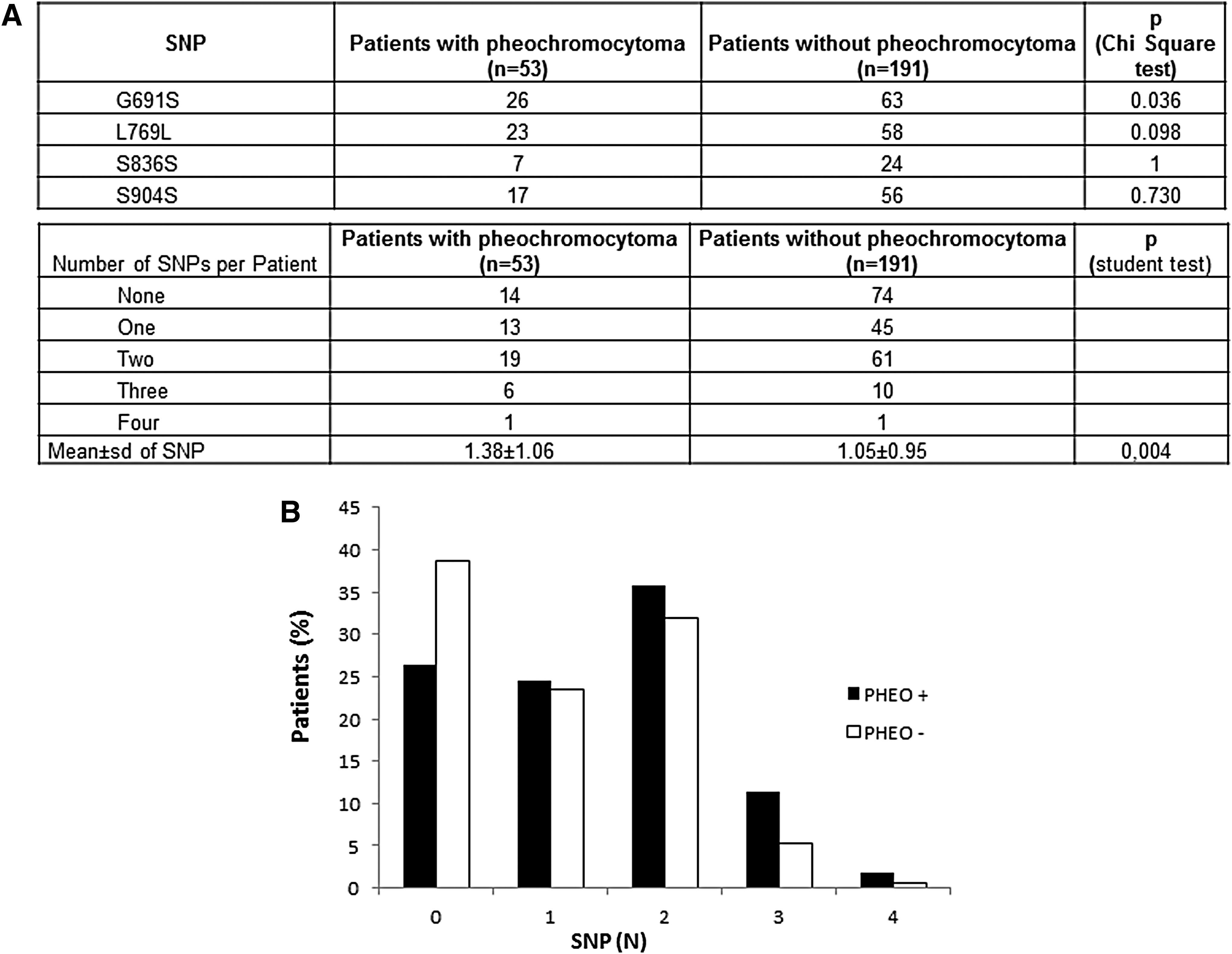
Additive effect of RET polymorphisms on PHEO in patients with MEN2A (N = 244 index cases). (
The study also examined whether the presence of one, two, three, or four RET SNPs might increase the risk of PHEO. The distribution of the number of polymorphisms was significantly different between the four groups of patients (p = 0.04). Patients with PHEO were found to have a higher SNP number (1.38 ± 1.06) than patients without PHEO (1.05 ± 0.95; Fig. 3).
Discussion
This study reports the rate of new cases carrying pathogenic variants or VUS in RET per year and their distribution in the French population studied between 2003 and 2013. As recommended by INCa and based on the literature, the exons bearing activating mutations (exons 5, 8, 10, 11, 13, 14, 15, and 16) were analyzed in all patients. In the 480 index cases, 74 different pathogenic variants or VUS were found. Codon 634 was the most frequently altered codon, as already described (18,19). However, the frequency spectrum of these variants is not universal, and there are differences among ethnic groups, presumably caused by founder effects. For instance, in Greece, the G533C variant, with a prevalence of 36.2%, appears to be characteristic of a specific ethnic group (20). This variant has rarely been found in other series (18,19,21,22), and in this study, the frequency amounts to only 0.6% of patients. It was found that 8.9% of the patients were carriers of L790F, a pathogenic variant that seems to be present more often in Germany and Slovenia but rarely in other European countries (Supplementary Table S2) (22,23). The spectrum of RET variants is similar to that found in Germany, but quite different from that in Italy, Spain, or Greece (18 –21).
The accumulation of genotype–phenotype data over the last decade has made it possible to define risk profiles, to estimate the penetrance of MEN for index cases, and to recommend the optimal age for a prophylactic thyroidectomy or PHEO and HPTH screening in asymptomatic carriers of RET pathogenic variants (1,2,24).
In the present study, 92.3% of the patients carrying pathogenic RET variants received such recommendations. However, for 36 patients carrying RET variants not classified by the ATA, the data were not sufficient to support a recommendation. The majority of these patients had been diagnosed after 30 years of age, but two patients had been diagnosed at the age of one year and 21 years. The patient diagnosed at one year of age had a C541R variant associated with a MEN2B-like phenotype and Hirschprung's disease. Unfortunately, there are no current available clinical data to confirm the clinical diagnosis further. This variant concerns one of the extracellular cysteine residues. The C541R variant is predicted to be deleterious by the three bioinformatics algorithms used. Based on the consensus recommendation of the ACMG for the characterization of variants, the substitution is considered as probably pathogenic. A nonsense change in codon 541 in patients with Hirschsprung's disease has been described in the literature, but there is no precedence of a codon 541 variant with an activating mutation (25). The functional impact of C541R awaits confirmation by further studies. The patient diagnosed at 21 years of age had a V648I variant. With functional tests, this variant is qualified as having a low oncogenic potential (26), and the three bioinformatics algorithms have predicted it to be a tolerable substitution. This variant has been reported in trans with C634R for a patient with MTC and bilateral PHEO, but his two children with the V648I variant had no evidence of the presence of tumors (27). Considering all these arguments, this variant is considered to be probably benign. The M918T pathogenic variant is qualified as the most aggressive variant in the ATA classification. In this study, another type of substitution was found at codon 918: M918V. Although this variant was also predicted to be deleterious by the three bioinformatics algorithms used, functional testing showed that it has a low oncogenic potential (26). As reported in a recent study showing a moderate risk of MTC associated with the M918V variant, this patient was diagnosed with MTC at just 56 years of age (28). The M918T pathogenic substitution has been shown to modify the three-dimensional structure of the protein, creating new hydrogen bonds and stabilizing its close conformation (29,30). Unpublished data show that valine, a small amino acid that does not create new hydrogen bonds, does not alter the conformation of the protein. Based on these arguments, this variant is believed to be pathogenic. In contrast to M918T, the M918V variant is associated with a moderate risk of aggressive MTC in this patient.
Concerning the combined mutations, each variant was classified as moderate risk by the ATA. For the concomitant presence of V804M and R844L, or K603Q and L790F, there is no clinical evidence that suggests a need for upgrading the risk from “moderate” to “high.” For the patient carrying D631Y and Y791F, the diagnosis of PHEO was made earlier than in his father who carried D631Y alone. However, in the absence of information about his mother, and because of the controversy about the pathogenicity of Y791F, there are insufficient data to support a change in risk category.
Overall, nine variants have been classified as probably pathogenic, and six as probably benign. It is known that MTC occurs at a variable age and aggressivity, depending on the specific pathogenic variant. The clinical follow-up of these patients and further phenotype-genotype information will be required to specify the MTC risk profile. As recommended for the patients carrying pathogenic RET variants, serum calcitonin assays and ultrasound examinations can serve as an indicator for MTC in patients carrying variants not yet classified by the ATA (1,31,32).
The present results show that the distribution of mutations was significantly different between patients with or without PHEO. Patients with PHEO had predominantly mutations in exons 10 or 11, and these concerned cysteine residues. These tumors are known to produce catecholamines, and about one-third of the tumors are bilateral at initial diagnosis. The more dramatic clinical behavior explains why the mean age of diagnosis is lower when PHEO is the first manifestation of MEN 2A. Thus, the ATA recommended that screening for PHEO should begin by 11 years of age for children carrying a 634 pathogenic variant and by 16 years of age in children with other pathogenic variants.
The polymorphic RET alleles G691S (rs1799939), L769L (rs1800861), S836S (rs1800862), and S904S (rs1800863) have sometimes been considered as disease modifiers. The role that the RETG691S polymorphism plays in the susceptibility to MTC has been investigated, and a meta-analysis based on 11 studies, combining 968 MTC cases and 2115 controls, confirms the involvement of this RET variant in MTC (15), but no involvement in risk of PHEO has been reported. In silico analysis has shown that the G691S SNP changes the phosphorylation pattern that may enhance signal transduction. Moreover, two functional studies (33,34) have demonstrated that two pathogenic variants (S891A and K666Q), qualified as “moderate” risks according to the ATA classification, display a higher oncogenic potential when they are associated with the G691S substitution. This study investigated whether G691S modifies the age at diagnosis for patients carrying pathogenic variants with low oncogenic potential, but no modifier effect was found. However, it was found that the G691S variant increases the risk of PHEO independently of the 634 RET pathogenic variant. According to some studies, G691S should be described as a risk modifier in MEN2. The additive effect of RET polymorphisms on sporadic MTC and PHEO has been investigated, including multiple RET variants, that is, G691S, L769L, S836S, and S904S (8,11). The results showed that three or more polymorphic alleles were not associated with age at diagnosis or tumor size, but increased the risk of lymph node and distant metastases at diagnosis of sporadic MTC. In the case of PHEO, the presence of two RET variants was associated with increased susceptibility to PHEO and age-related penetrance. No link was found between the number of SNPs and the age at diagnosis of MTC, but the susceptibility for PHEO increased. This study is retrospective and evaluated the potential role of the four RET SNPs found in the eight analyzed exons. The results suggest that RET G691S or a combination of SNPs may affect the development of PHEO in MEN2. These results need to be confirmed by family screening to identify an intra-familial co-segregation of these SNPs with PHEO.
The main limitations of this work may be related to the retrospective nature of the collection of data from multiple centers and the heterogeneity of the observation due to the varying method of patient management over the 10-year period of the study. Moreover, it is likely that subjects carrying mutations with a low transforming activity may not have been diagnosed if they had no family histories and had not yet developed MEN2.
Finally, the results offer a more comprehensive vision of the distribution of RET variants in France, and, to the authors' knowledge, this is the first exhaustive study of RET screening over a 10-year span in France. The clinical description is reported of 36 patients carrying non-ATA-classified RET variants, and they are classified according to the recommendations of the ACMG. The results suggest that RET SNPs such as G691S, or a combination of SNPs, may be associated with the development of PHEO. This finding may have significant implications in the management of patients, particularly for the diagnosis of PHEO in patients carrying the G691S SNP. With the advent of next-generation sequencing, comprehensive analysis of the RET exome or the entire locus may be extended to all patients with MEN2 so that phenotype–genotype correlations for rare or unknown variants could be usefully implemented for the management of patients.
Footnotes
Acknowledgments
We are grateful to Mr Kanaya Malkani and Ms Caroline Jacques for their critical reading of the manuscript. This work was supported by the University Hospital of Angers, INCa, Groupes d'étude des tumeurs endocrines (GTE), OncoGENetic on NeuroEndocrine Tumors (TENGEN) Société Francaise d'endocrinologie (SFE), INSERM, CNRS.
Author Disclosure Statement
All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.