
Abstract
Background:
Inherited medullary thyroid carcinoma (MTC) is primarily caused by RET mutations that are commonly localized in exons 5, 8, 10, 11, and 13–16. In this study, we report pedigrees for individuals with MTC that harbor a germline S409Y variant within exon 6 of the RET proto-oncogene.
Methods:
Targeted sequencing was used to diagnose four apparently sporadic MTC index cases carrying the germline RET S409Y (c.1226 C>A) variant. Subsequently, 27 relatives of these individuals underwent clinical and genetic assessments and/or thyroid surgery. Furthermore, in silico analyses and in vitro assays were performed to predict or verify the potential oncogenic activity of the S409Y variant.
Results:
Overall, 15 of 31 participants were found to carry the RET S409Y variant. Of these, 6 presented with isolated MTC (mean age 50.2 years; range 41–75 years), of which 3 presented with neck lymph node metastases and 2 presented with distant liver or lung metastases. Among the remaining 9 carriers, 3 (mean age 56 years; range 41–76 years) had elevated serum calcium-stimulated calcitonin (sCtn) or concurrent marginally elevated serum calcitonin (Ctn) levels, whereas the other 6 (mean age 37.5 years; range 14–52 years) exhibited typical Ctn/sCtn levels (p < 0.05). None of the 15 carriers in these 4 families presented clinical evidence of pheochromocytoma, hyperparathyroidism, or Hirschsprung's disease. In silico analyses revealed that S409Y was a “possibly damaging” mutation that could affect the RET protein inter-domain interface. An in vitro assay revealed that the phosphorylation level of RET tyrosine 905 was relatively higher in the RET S409Y mutant than in wild-type (WT) RET. Moreover, transfection of HEK 293 cells with S409Y enhanced the phosphorylation activity of AKT, ERK pathways, and it increased cell proliferation compared with WT RET, but to a lesser degree than that for the RET C618Y and C634Y mutations.
Conclusions:
This study demonstrates that the novel germline RET S409Y variant is likely pathogenic and is associated with lower penetrance of MTC than that for the C618Y and C634Y mutations. Individuals with S409Y should be managed using a personalized approach, and additionally, “at-risk” family members should be evaluated. Additional studies are needed to elucidate the correlation between the S409Y mutation and multiple endocrine neoplasia type 2-specific tumors.
Introduction
Multiple endocrine neoplasia type 2 (MEN2) is a rare autosomal-dominant inherited cancer syndrome comprising two distinct subtypes: MEN2A (OMIM 171400) and MEN2B (OMIM 162300) (1). These neoplasias are primarily caused by germline mutations of the RET proto-oncogene (OMIM 164761) (1 –3). Sequencing of RET over the past 25 years identified 195 RET variants, of which ∼50% are p athogenic mutants (1,4). Identification of RET mutations as a cause of MEN2 represents a paradigm for precision medicine, which instructively prevents and predicts a disease course, guides the successful personalized management of MEN2-specific tumors, and facilitates recognition of apparently “sporadic” medullary thyroid carcinoma (MTC) harboring an RET germline mutation (1 –3,5 –7).
However, the benign or pathogenic transforming activity of most new RET germline mutations detected as having a potential association with sporadic MTC is unclear. Thus, appropriate functional and careful follow-up family studies require validation to avoid misinterpretation and irreversible clinical outcomes (8 –11).
Germline mutations in RET exons 5, 8, 10, 11, and 13–16 are most likely to be involved in MEN2, although several RET variants in exons 2, 7, 9, 12, 18, and 19 have also recently been detected. Notably, no RET variants in exon 6 have been found (1 –4). In this study, we report a rare and novel serine-to-tyrosine substitution at codon 409 within exon 6 of RET (S409Y; c.1226 C>A; NM_020975.4) identified by gene targeted and next-generation sequencing (targeted sequencing) in four pedigrees having isolated MTC clinical phenotypes. We also used in silico analyses to make functional predictions and conduct in vitro analyses of the potential oncogenic activity of the extracellular RET S409Y mutation.
Materials and Methods
Participants
In the past decade, 287 cases of MTC (216 “sporadic” and 71 MEN2) diagnosed histopathologically were subjected to genetic screening at the 903rd PLA Hospital and Zhejiang Cancer Hospital (Hangzhou, China). Clinically, 4 patients with MTC (FAII-4, FBII-1, FCII-1, and FDII-1) from four unrelated Chinese pedigrees were detected as having the RET S409Y variant. We further investigated 27 relatives of these 4 families (I-1, I-2, II-1–3, II-5–6, III-1–10, and IV-1, IV-2 in FA; I-1, I-2, II-2, II-3, and III-1, III-2 in FB, III-1 and IV-1 in FD; Fig. 1 and Table 1). All relatives examined lived in Zhejiang Province, China.
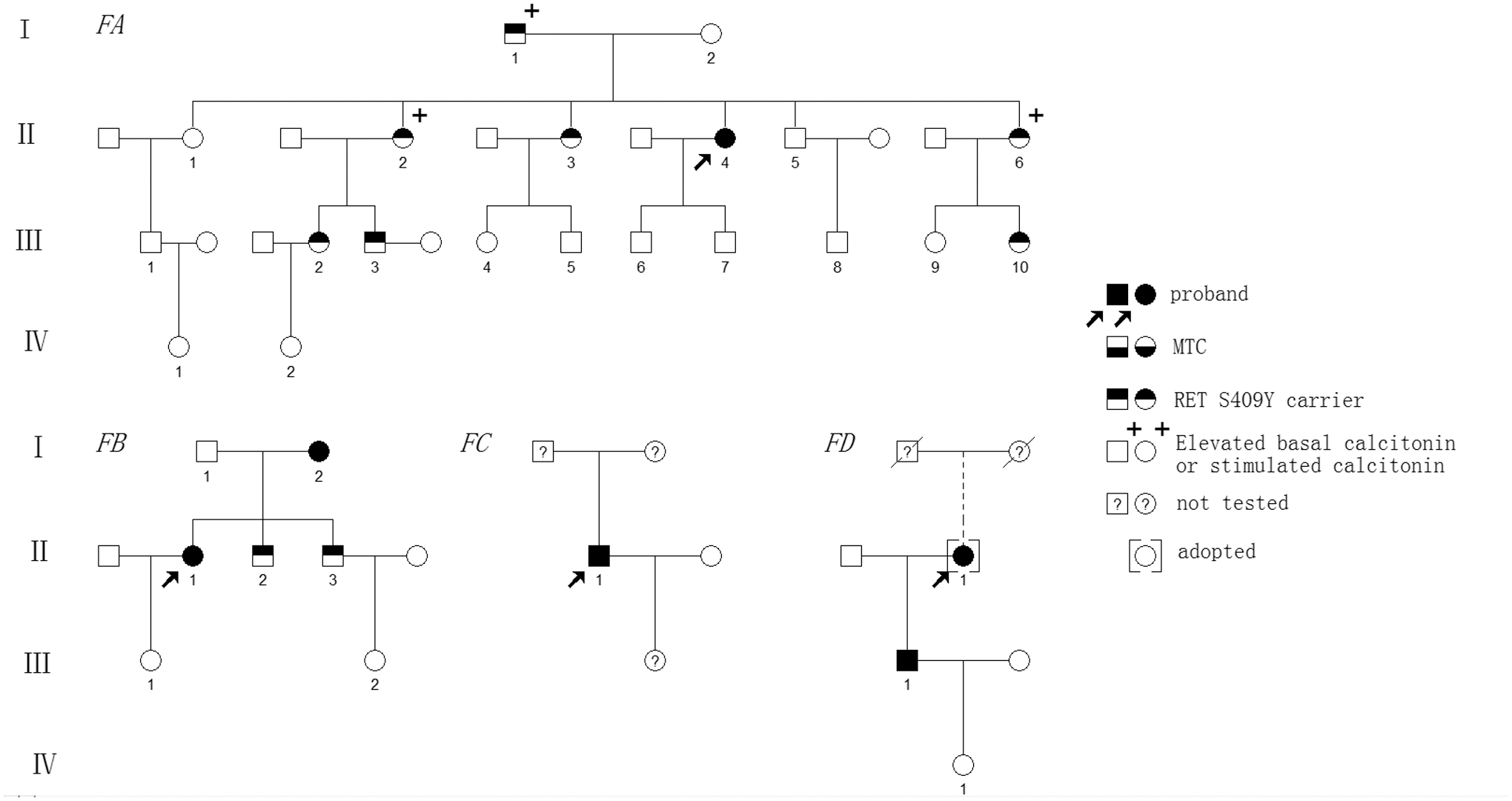
Genealogies of four families with the RET S409Y mutation and associated MTC. Numbers indicate family members. Circles and squares indicate females (F) and males (M), respectively. MTC, medullary thyroid carcinoma.
Clinical Characteristics of RET S409Y Mutation Carriers in the Four Chinese Pedigrees with Associated Medullary Thyroid Carcinoma
Clinical suspected MTC or C cell hyperplasia.
Clinical suspected benign thyroid nodule.
−, negative; ADM, age at diagnosis of MTC; BiLND(VI), bilateral level VI lymph node dissection; FA, family A; FB, family B; FC, family C; FD, family D; L/R, left or right thyroid nodule; M/F, male/female; MBiND, modified bilateral neck dissection; MTC, medullary thyroid carcinoma; NA, not available; Pre-Ctn, pre-surgery basal serum calcitonin (normal for males and females is <8.4 and <5.0 pg/mL, respectively); sCtn, calcium-stimulated calcitonin (normal <100 pg/mL); TNM, tumor-node-metastasis; TT, total thyroidectomy; US, ultrasound; WW, watchful waiting.
In addition, we matched 200 unrelated healthy individuals and a total of 218 MEN2 patients from 71 families that carried pathogenic RET mutations (213 MEN2A with C609R, C611Y, C618G/R/S/Y, C620R/S, C634F/G/R/S/W/Y, L790F, V804M, S891A, M918V, S891A/R525W, V292M/R67H/R982C, 634F/V292M/R67H/R982C, and 634Y/V292M/R67H/R982C, and 5 MEN2B with M918T) as controls. The study protocol was approved by the Ethics Committee of the 903rd PLA Hospital, and written informed consent was obtained from all study subjects or their legal guardians.
Clinical approach
All 31 individuals (including the 4 index cases after screening positive in RET screening for the presence of S409Y) underwent clinical and biochemical/imaging examinations, as described previously (1,2,10). The biochemical examinations included determination of serum basal calcitonin (Ctn; normal <8.4 pg/mL [males] and <5.0 pg/mL [females]), carcinoembryonic antigen (CEA; normal <5.0 ng/mL), parathyroid hormone, serum calcium, and metanephrine/normetanephrine levels. Imaging examinations involved Doppler ultrasound (US), computed tomography (CT), magnetic resonance imaging (MRI), and emission CT, if indicated. Among 15 carriers of the RET S409Y variant, 13 (FCII-1 and FDII-1 were excluded) participated in additional serum calcium-stimulated calcitonin (sCtn; normal <100 pg/mL) testing, as reported previously (10).
After confirming RET mutations and elevated Ctn levels, prophylactic or surgical thyroidectomy was performed for diagnosis of MTC. Furthermore, tumor staging was performed using the American Joint Committee on Cancer version 7 TNM (tumor–node–metastasis) classification system (12). The subjects were followed-up until June 2018.
RET screening using targeted sequencing
Peripheral blood genomic DNA samples obtained from 31 individuals in 4 families were prepared for targeted sequencing using an Illumina HiSeq 2000 Analyzer. Briefly, a custom capture array (NimbleGen; Roche) was designed to capture all exons and splice sites, as well as immediately adjacent intron sequences for 10 genes (RET, VHL, SDHA, SDHB, SDHC, SDHD, SDHAF2, NF1, MAX, and TMEM127) associated with hereditary pheochromocytoma (PHEO) or paraganglioma according to GeneReviews (NCBI). The methods used for DNA target capture, enrichment, elution, and targeted sequencing were previously described (6,13,14). The targeted sequencing results were further validated by Sanger sequencing using an ABI 3700 Genetic Analyzer (Perkin-Elmer, Fremont, CA).
Pathogenic mutation confirmation
In silico analyses
In this study, in silico analyses were performed using public databases and web-based software to assess the functional significance of the RET S409Y variant. The pathogenic potential of the RET S409Y variant was estimated computationally using an algorithm to assess interspecies sequence variations with Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v-2 (PolyPhen-2), Mendelian Clinically Applicable Pathogenicity (M-CAP), and VAAST Variant Prioritizer (VVP) to estimate the impact of the RET S409Y variant on protein function. We also matched the abovementioned 19 different RET pathogenic point mutations within exons 10, 11, 13–16, and a recurrent single nucleotide polymorphism (SNP; G691S within exon 11) identified among the MEN2 cohort controls at our hospital (1,9,13,15 –18).
In vitro assays
Cell lines and cell culture
Human embryonic kidney (HEK 293) and HEK 293T cells were purchased from the cell library of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco). All cells were incubated at 37°C with 5% CO2 (Thermo Fisher Scientific, Inc., Waltham, MA).
RNA extraction, RET gene cloning, and point mutations
Total RNA was extracted from HEK 293T cells using TRIzol reagent (Invitrogen, Grand Island, NY) according to the manufacturer's instructions. Reverse transcription was conducted using an M-MLV Kit (Promega, Madison, WI), and the full-length open reading frame of the RET9 gene was amplified and subcloned into the pLenti-enhanced green fluorescent protein lentivirus vector and the pFLAG-CMV2 plasmid. The RET9 point mutations S409Y, C618Y, and C634Y were generated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions.
Packaging of RET gene lentivirus and infection of cell lines
HEK 293T cells were plated on 10-cm plates precoated with poly-
Western blotting assay
Total cellular protein was extracted using RIPA lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS [sodium dodecyl sulfate]) containing complete ethylenediaminetetraacetic acid-free Protease Inhibitor Cocktail Tablets (Roche). The lysate protein concentration was determined by the bicinchoninic acid method (Beyotime Biotechnology, Shanghai, China). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were transferred to a nitrocellulose membrane (Millipore, Temecula, CA). All membranes were blocked in 5% bovine serum albumin for 1 hour at room temperature and then incubated with the following primary antibodies: anti-ERK1/2 (no. 4695), anti-phospho-ERK1/2 T202/Y204 (no. 4370), anti-AKT (no. 2920), anti-phospho-AKT S473 (no. 4060), anti-β-actin (no. 3700) (Cell Signaling Technology, Danvers, MA), anti-RET (no. ab134100), and anti-phospho-RET Y905 (no. AF8055) (Affinity, Cincinnati, OH). Horseradish peroxidase-conjugated goat anti-rabbit/mouse IgG antibodies (Jackson ImmunoResearch, West Grove, PA) were used as secondary antibodies, and anti-actin mouse monoclonal antibody was used as an internal control. All samples were detected using the electrochemiluminescence method (Millipore).
Colony formation assay
To assess the effect of RET9 mutations on cell proliferation, HEK 293 cells expressing WT and mutated RET genes were seeded into 6-well plates and incubated for 14 days. The resulting colonies were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet (Sigma) at 37°C for 30 minutes and counted.
Statistical analysis
Each experiment was repeated independently in triplicate wells three times. All data were analyzed using either one-factor analysis of variance (ANOVA) for more than three groups or Student's t-test for comparison of two independent treatments. For all tests, differences were considered significant at p < 0.05 (*p < 0.05, ***p < 0.001; no markings indicate the absence of significant differences).
Results
Clinical features and MTC phenotypic data
Family A
The index case was a 47-year-old female (FAII-4; Fig. 1) who was diagnosed with MTC when she was 41 years old and was inappropriately treated by right total thyroidectomy at another hospital due to the presence of a palpable solid mass in the neck and cystic masses at other sites. Her initial pathology report was available for confirmation of right-sided MTC (1.0 cm). One month after the first surgery, preoperative findings collected at our hospital showed elevated Ctn (8.45 pg/mL) and CEA in the normal range (2.86 ng/mL). Both US and CT revealed a hypoechoic nodule in the left thyroid lobe (0.5 cm × 0.4 cm × 0.4 cm). Subsequently, the patient underwent a left total thyroidectomy with bilateral level VI lymph node dissection. Left-sided MTC was confirmed histopathologically (LN+/resected, 0/2; T1aN0M0). Her Ctn then dropped to undetectable levels (<2 pg/mL). Germline RET testing of this patient revealed the presence of the S409Y variant.
In 2015, previously hesitant family members agreed to participate in biochemical testing, imaging studies, and RET screening. Of these, 12 of 19 family members exhibited normal Ct levels and US images, but 7 (FAI-1, FAII-2, FAII-3, FAII-6, FAIII-2, FAIII-3, and FAIII-10) were found to carry the RET S409Y variant. Of these 7 S409Y carriers, 1 female (FAII-2) had a slightly elevated Ctn level (5.36 pg/mL), 2 (FAI-1 and FAIII-2) exhibited a small thyroid nodule on US but had undetectable or normal Ctn levels, and the remaining 4 (FAII-3, FAII-6, FAIII-3, and FAIII-10) had normal Ctn measurements and no thyroid nodules. Further sCtn testing revealed that 3 (FAI-1, FAII-2, and FAII-6) of the 8 S409Y carriers (index case and family members) showed elevated sCtn levels of 253, 106, and 152 pg/mL, respectively, which suggested that these 3 carriers could present with MTC or C cell hyperplasia (CCH), whereas another carrier (FAIII-2) was suspected of having a benign thyroid nodule (Table 1). However, these individuals declined further treatment, including fine-needle aspiration (FNA) examination, and selected a watchful waiting approach.
Family B
The index case was a 55-year-old female (II-1; Fig. 1) diagnosed with bilateral MTC at the age of 50 years when she detected palpable neck masses. A biochemical examination revealed elevated Ct (>2000 pg/mL) and CEA (316.66 ng/mL) levels. Both US and CT revealed multicentric hypoechoic nodules with calcifications in the neck and multiple quasi-circular masses (maximum size 3.1 cm) in the liver but normal alpha-fetoprotein levels (1.91 ng/mL; normal <5.0 ng/mL). She underwent a total thyroidectomy with bilateral level VI lymph node dissection and modified lateral neck dissection at another hospital. Histopathological examination revealed bilateral MTC (left 2.5 cm; right 1.5 cm) with lymph node metastases (LN+/resected, 14/21; T2N1bMx).
At 1 month after the surgery, Ctn and CEA levels determined at our hospital had declined to 1215 pg/mL and 192.02 ng/mL, respectively. Further emission CT revealed metabolic activity in the neck and multiple bones, including the seventh, eighth, and ninth thoracic vertebrae, ninth rib, second and third lumbar vertebrae, sacroiliac joint segments, and the liver. Tumor metastases of the bone and liver were assessed by MRI (T2N1bM1). This patient declined further treatment and selected a watchful waiting approach. At 65 months after her initial surgery, the patient had progressive disease, with Ctn and CEA of 6980 pg/mL and 798 ng/mL, respectively, and enlarged multiple nodules in the liver (maximum size 4.5 cm) and bilateral neck lesions (maximum size 1.8 cm). She was found to be positive for S409Y on mutational analysis of the RET gene.
Meanwhile, the index patient's 75-year-old mother (FBI-2) was positive for S409Y and had Ctn of 33.9 pg/mL and two nodules (0.6 cm) in both thyroid lobes; her two brothers (FBII-2 and FBII-3) were also S409Y carriers yet had undetectable Ctn levels (<2 pg/mL). sCtn testing detected peak values of 2160 pg/mL for the patient's mother and normal levels (range 28.1.7–82.3 ng/L) for the mother's brothers. Subsequently, the patient's mother (FBI-2) underwent a total thyroidectomy with bilateral level VI lymph node dissection. A histopathological examination revealed bilateral MTC (LN+/resected, 0/4; T1aN0M0; Table 1). The mother's two brothers (FBII-2 and FBII-3) are currently under observation.
Family C
The index case was a 55-year-old male (FCII-1; Fig. 1) who was diagnosed with MTC at 49 years old after bilateral thyroid lesions were detected during a routine physical examination. His Ctn and CEA levels were elevated at 357.47 pg/mL and 21.6 ng/mL, respectively. Both US and CT revealed two hypoechoic nodules in both thyroid lobes. FNA was performed on both thyroid nodules, and a cytological examination revealed features suggestive of MTC. He agreed to undergo a total thyroidectomy with bilateral level VI lymph node dissection. A histopathological examination revealed bilateral MTC (left 0.7 cm; right 0.8 cm), with lymph node metastases (LN+/resected, 2/5). After 1 month, his postoperative Ctn and CEA levels declined to 50.5 pg/mL and 7.48 ng/mL, respectively (T1aN1M0; Table 1). RET testing revealed that this index patient carried a germline S409Y variant; however, his parents and 27-year-old daughter have not been tested.
Family D
The index case was a 63-year-old female (FDII-1; Fig. 1) diagnosed with bilateral MTC at 44 years old, with a 2-year history of a palpable neck mass in the right thyroid. US revealed two hypoechoic nodules in both thyroid lobes. Accordingly, a right total and left subtotal thyroidectomy with regional neck lymph node dissection was performed. Her initial pathology report from a local hospital confirmed bilateral MTC (left 0.5 cm; right 1.8 cm) with cervical lymph node metastases (LN+/resected, 7/15; T1bN1bMx). In 2010, 10 years after the initial surgery, she agreed to undergo a second “bilateral lateral neck dissection” due to multiple enlarged lymph nodes (1.5–3.0 cm) in the bilateral supraclavicular region and elevated CEA levels (454.5 ng/mL). In addition, bilateral cervical lymph node MTC metastases (LN+/resected, 8/16) were suggested by histopathological examination. In 2015, 15 years after the initial surgery, this patient underwent a second surgery to treat a relapse of enlarged neck masses (3.0–4.0 cm) revealed by US and CT examinations and elevated CEA levels (46.7 ng/mL). A histopathological examination revealed neck lymph node MTC metastases (LN+/resected, 4/6) and left thyroid remnant lobe hyperplasia, with acidophilic degeneration elsewhere.
In 2018, she visited our hospital and presented with neck masses present for the previous 6 months. On presentation, her Ctn and CEA levels were 8700 pg/mL and 424.07 ng/mL, respectively. Both US and CT scanning revealed multi-hypoechoic nodule in the right neck
This patient was an orphan who was adopted. Thus, the endocrinopathy history of her biological parents was unclear. RET testing revealed that this patient carried a germline S409Y variant, as did her only son (FDIII-1, 41 years old). Her son had a slightly elevated Ctn at 26.9 pg/mL and his CEA was within the reference range. Both US and CT revealed that he had bilateral thyroid nodules and he underwent total thyroidectomy with bilateral level VI lymph node dissection. Histopathological examination revealed bilateral MTC (LN−/resected, 0/4; T1aN0M0). His postoperative Ctn/sCtn levels decreased to undetectable levels (Table 1), and his daughter tested negative in the screening for S409Y and MTC.
Other MEN2-associated diseases and surgical outcomes
All 15 RET S409Y carriers in this study exhibited no evidence of PHEO, hyperparathyroidism (HPT), Hirschsprung's disease (HD), cutaneous lichen amyloidosis (CLA), corneal nerve thickening, or other endocrine tumors during follow-up (Fig. 1 and Table 1).
Of these 15 S409Y carriers, 6 (mean age 56.2 years; range 41–76 years) underwent thyroid surgery; 3 (FAII-4, FBI-2, and FDII-1) had undetectable Ctn, 2 (FBII-1 and FDII-1) had higher Ctn levels (>3000 pg/mL), and 1 (FCII-1) declined further assessment after initial thyroidectomy. Among the 9 remaining S409Y carriers, 6 exhibited no abnormal Ctn or sCtn values (mean age 37.5 years; range 14–52 years) and 1 (FAII-2) exhibited slightly elevated Ctn and sCtn levels (5.8 and 106 pg/mL, respectively). Meanwhile, 3 affected individuals (FAI-1, FAII-6, and FAII-2) reported elevated sCtn levels (>100 pg/mL; mean age 57 years; range 42–77 years). A comparison of the mean age of MTC patients with and without elevated Ctn/sCtn levels revealed ages of 56.4 and 37.5 years, respectively (p < 0.05; Table 1).
Furthermore, none of the 16 individuals who tested negative for the S409Y variant had clinical manifestations of MEN2, MTC, PHEO, or HPT (Fig. 1).
Identification of RET variants
A heterozygous missense variant within exon 6 of RET, c.1226 C > A (S409Y), was verified in the 4 probands and 11 other family members (Fig. 1 and Table 1). Meanwhile, 6 other recurrent exonic SNPs were detected: c.135G>A (A45A), c.1296A>C (A432A), c.2037C>T (P679P) and c.2071G>A (G691S), c.2307T>G (L769L), and c.2712C>G (S904S) that were within exons 2, 7, 11, 13, and 15, respectively. None presented evidence of other RET mutations. All available information identified by targeted sequencing was validated by Sanger sequencing, but there was no evidence to associate these 6 SNPs with the MTC phenotype (1 patient [FCII-1] carried RET G691S alone) and none of the 418 controls were positive for S409Y.
Functional studies of the RET mutation
In silico analyses
The American Thyroid Association (ATA) risk categories (2015) include 19 known mutants. For the ATA-MOD (moderate risk), C609R, C611Y, C618G/R/S/Y, C620R/S, L790F, V804M, S891A, M918V are present; for ATA-H (high risk), C634F/G/R/S/W/Y; and for ATA-HST (highest risk), M918T. In this study, the new missense variant S409Y and the recurrent SNP G691S of RET were observed (Table 2). In silico analyses by SIFT, M-CAP, VVP, and PolyPhen-2 indicated that 17 of the 19 mutants (except for V804M and S891A, by VVP) were definitive pathogenic mutations compared with S409Y, which was considered a “possibly damaging” or “damaging” mutation by SIFT, M-CAP, and VVP, but by PolyPhen-2 was categorized as a “benign” variant. The G691S variant was consistent with a specific polymorphism as indicated by SIFT, PolyPhen-2, and VVP (Table 2).
In Silico Analysis of the Functional Significance of RET Variants
Suggested as an ATA-MOD.
Definitive pathogenic 2 mutants, V804M and S891A, but benign by VVP.
Not available.
ATA, American Thyroid Association; B, benign; D, damaging; H, high risk; HST, highest risk; M-CAP, Mendelian Clinically Applicable Pathogenicity; MOD, moderate risk; Pred, prediction; SIFT, Sorting Intolerant From Tolerant; SNP, single nucleotide polymorphism; T, tolerated; VVP, VAAST Variant Prioritizer.
In vitro assays
We selected two mutants (ATA-MOD, C618Y and ATA-H, C634Y) from our hospital's MEN2 cohort as positive controls for in vitro studies. Figure 2 summarizes the in vitro functional characteristics of HEK 293 cells transfected with RET9 WT, S409Y, C618Y, or C634Y.
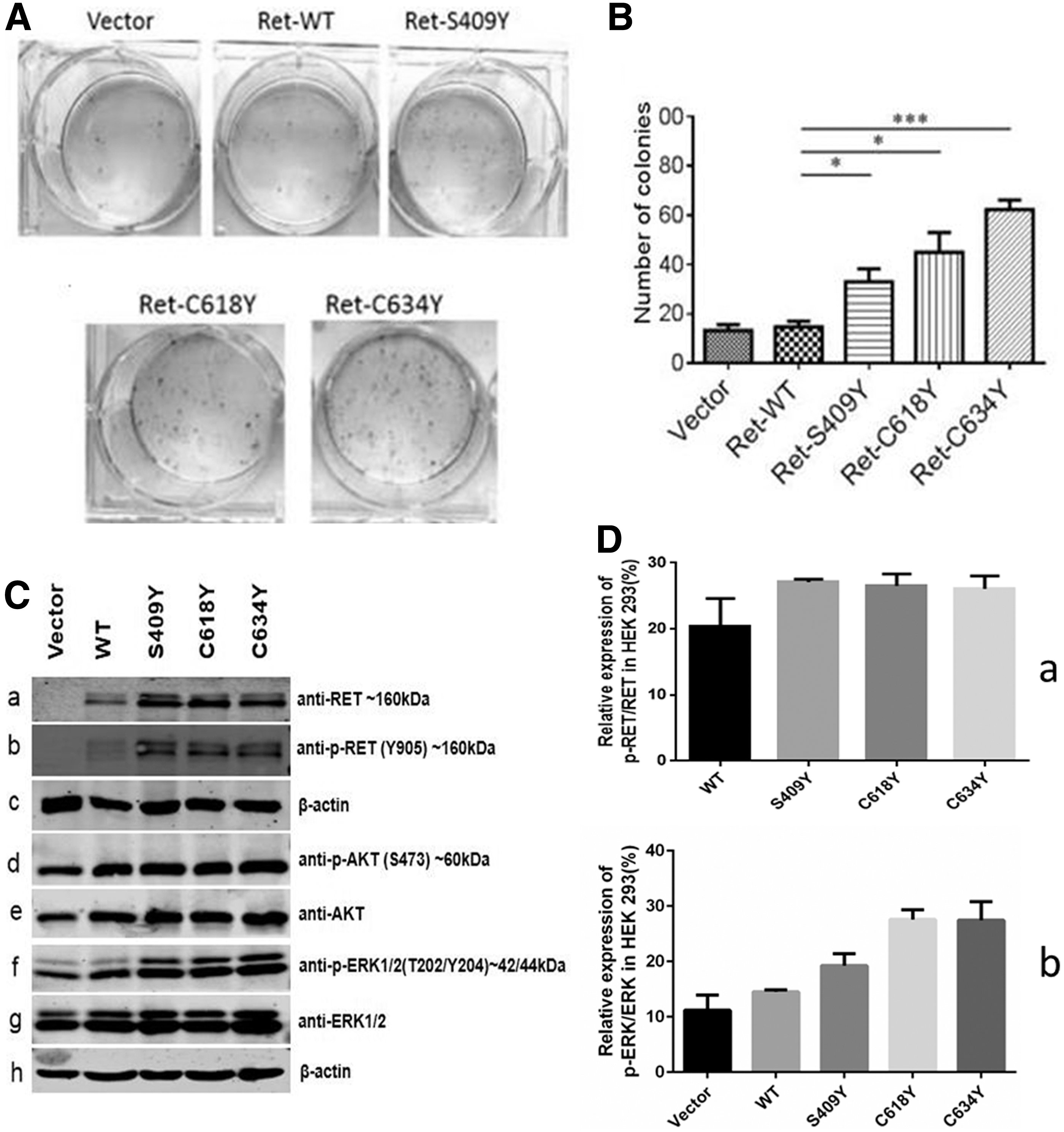
In vitro assays performed to characterize the RET9 S409Y mutation. (
In colony formation assays, cells transfected with RET9 S409Y, C618Y, or C634Y formed significantly more colonies than cells expressing WT RET9, although the increase associated with S409Y was lower than that observed for the C618Y and C634Y mutants (Fig. 2A, B).
Western blotting indicated that HEK 293 cells do not express endogenous RET proteins (Fig. 2C-a). After transfection with plasmids carrying RET9 WT, S409Y, C618Y, or C634Y, Western blotting showed the presence of double bands representing partially glycosylated and glycosylated RET proteins, with the former predominating in WT RET9 cells (Fig. 2C-a). Phosphorylation of RET at tyrosine 905 was observed in cells expressing RET9 S409Y, C618Y, and C634Y and was present at higher levels than those observed for RET9 WT (Fig. 2C-b, D-a). Moreover, all three mutants had increased levels of AKT (S407; Fig. 2C-d) and ERK1/2 (T202/Y204; Fig. 2C-f, D-b) phosphorylation compared with RET9 WT. Collectively, these data suggest that the S409Y mutant activates the AKT and ERK pathways, but to a lesser degree than that observed for the C618Y and C634Y mutants (Fig. 2C, D). Nonreducing SDS-PAGE confirmed that RET9 C618Y and C634Y were present as dimers, but the S409 mutant was not (data not shown).
Discussion
This study describes the first analysis of an extracellular RET mutation S409Y (c.1226 C>A) that cosegregates with MTC alone in four Chinese families (Fig. 1 and Table 1) and could represent a novel mutation with a defined genotype–phenotype relationship in familial MTC (FMTC; OMIM 155240), which contains four variants: classical MEN2A, MEN2A with CLA, MEN2A with HD, and FMTC (1).
Germline RET mutation analysis currently plays an integral role in the clinical management of MTC (1 –3). Thus, in this study, targeted sequencing based on the RET screening results served as a rapid and cost-effective approach for the genetic diagnosis of this monogenic disorder (14,19). Unexpectedly, among 216 “sporadic” MTCs, we found a rare non-hot spot variant in exon 6 of RET, S409Y, which was carried by the 4 index cases. None of the 200 healthy individuals and 218 patients having RET-defined MEN2 who served as controls carried S409Y. Moreover, examination of S409Y variant frequency listed in the Catalog of Somatic Mutations in Cancer, the Single Nucleotide Polymorphism Database, the Exome Aggregation Consortium, the 1000 Genomes Project database, and the University of Utah MEN2 database (4) showed that the S409Y variant had not been previously described and thus could be considered as a novel variant. In our series, 6 of the 15 affected individuals presented clinically with apparently isolated MTCs, yet of the 9 other carriers, 3 exhibited slightly elevated sCtn and/or Ctn levels, which might support suspicion of affected micro-MTC or CCH (Table 1) (20). These results suggest that the rare variant S409Y is not a polymorphism but is instead a disease-causing mutation, a notion which is also supported by in silico analysis using the SIFT method suggesting that it is a “possibly damaging” mutation and the M-CAP method which scores it as a “damaging” mutation. Moreover, analysis using the VVP method indicated that the S409Y mutation is associated with Mendelian diseases with high sensitivity and pathogenic variants with high specificity (Table 2) (15 –17).
Meanwhile, two other patients were reported to have other amino acid changes at RET residue 409. A 50-year-old male German patient with post-resistant metastatic melanoma after vemurafenib monotherapy was found to carry the somatic mutation RET S409F (c.1226C>T), a mutation that was not present before therapy (21), whereas a non-Finnish European was found to have a germline RET S409C (c.1226C>G) mutation. Additionally, the absence of somatic mutations of KRAS, NRAS, or HRAS in formalin-fixed and paraffin-embedded tissues of MTC from our 4 index cases was identified by additional exome sequencing (data not shown). The existence of these other potentially pathogenic mutations, S409C/F, at codon 409 within exon 6, and lack of RAS driver oncogenes, also favor the hypothesis of the pathogenetic nature of the RET S409Y variant (22,23).
Residue 409 of RET lies at the C-terminal end of the four extracellular cadherin-like domains (CLD-4) in the receptor and outside the cysteine-rich domain (CRD) (3). This location differs from that for MEN2 patients in which RET mutations were detected in the extracellular CRD or the intracellular tyrosine kinase domain (TKD) (1 –4). Accumulating evidence revealed that the previously rarely reported three germline pathogenic mutations, L56M in exon 2 (CLD-1), V292M in exon 5 (CLD-3), and 505_506del in exon 7 (CLD-4) lying upstream of the CRD (exons 1–7), are all related to the classical MEN2A phenotype and are associated with a milder form of MTC and/or PHEO, but the absence of HPT. Interestingly, L56M is also associated with HD (4,24 –26). In contrast, in this study, 9 affected subjects (mean age 52.1 years) having elevated levels of Ctn/sCtn presented or were suspected of having MTC/CCH alone, had no evidence of either PHEO or HPT, and HD. Meanwhile, the remaining 6 affected subjects (mean age 37.5 years) had Ctn/sCtn in the reference range.
The serine-to-tyrosine substitution at position 409 could affect the structural organization of RET CLD-4 and CRD and in turn impact inter-domain interfaces that are essential for proper folding of the CLD4 and CRD domains. In addition, the S409Y mutation could affect autophosphorylation of the intracellular TKD that could promote continuous activation of downstream signal transduction pathways resulting in uncontrolled cell proliferation and malignant transformation (24,25). In this study, transfection of HEK 293 cells with RET S409Y enhanced the phosphorylation activity of the AKT and ERK pathways as well as cell proliferation relative to cells expressing RET9 WT in vitro, but this enhancement was relatively weaker than that observed for cells expressing the more frequently occurring C618Y and C634Y mutations (Fig. 2A–D). These results support our clinical data indicating that the RET S409Y mutation is associated with low and late, but nonetheless a marked risk of MTC, which is similar to previously reported patterns for L56M, V292M, and 505_506del RET mutations and suggests that the S409Y mutation should also be treated as an ATA-MOD mutation (1,24,25). Nevertheless, especially in the FB family, the proband (FBII-1, 50 years; T2N1bM1) exhibited MTC at an earlier age with a more aggressive presentation than that observed for her mother (FBI-2, 75 years old; T1aN0M0; Table 1). This finding could reflect a diverse manifestation of MTC and a unique clinical pathological process, although it remains unclear whether S409Y behaves similarly to the RET SNP G691S that segregates with a more aggressive MTC phenotype or whether it is associated with the development of PHEOs (27,28). Despite the diverse clinical manifestations of MTC phenotypes, individual genetic backgrounds and environmental mutagens could also modify disease susceptibility and clinical phenotype (9,24,25,27 –32). Furthermore, our in vitro results suggest that S409Y is a gain-of-function mutation. However, the constitutive RET S409Y activation occurs independent of dimerization, in contrast to that for extracellular CRD mutations, such as C618Y and C634Y, but instead through aberrant homodimerization (33,34).
Typically, RET screening is recommended for any patient with MTC, as well as for at-risk family members. This screening has remarkably improved management of MEN2 patients and serves as a paradigm for the individualized management of other genetic diseases. For asymptomatic carriers of RET mutations, the use of biomarkers such as Ctn/sCtn is critical to identify optimal surgical windows of opportunity. A cautious approach is likely warranted when managing individuals with the RET S409Y mutation (ATA-MOD), as was the case for the 6 individuals in this study (FAII-3, FAIII-2, FAIII-3, FAIII-10, FBII-2, and FBII-3; Fig. 1 and Table 1) who had no evidence of MTC or had Ctn/sCtn levels that were within normal limits. In this scenario, watchful waiting or strict follow-up monitoring and psychological support should be considered, unless there is evidence of MEN2-specific tumors (1,2,7,35 –40). For the other 3 affected individuals (FAI-1, FAII-2, and FAII-6) who had slightly elevated Ctn/sCtn levels, US-guided FNA should be conducted. If cytological examination show features that are associated with MTC/CCH, prophylactic thyroidectomy with avoidance of radical procedure(s) is appropriate (1,2,7,35 –38). Notably, all 6 patients with apparent MTC underwent thyroid surgery; 1 was lost during follow-up (FCII-1) and 3 (FAII-4, FBI-2, and FDIII-1; T1aN0M0) underwent total thyroidectomy with bilateral level VI lymph node dissection. Subsequently, their Ctn levels were in the “reference range” and were either undetectable or only slightly detectable after initial surgery, suggesting that these 3 patients have an excellent prognosis (1 –3,41 –43). Meanwhile, the other 2 (FDII-1 and FBII-1) who had MTC regional or distant metastasis (T1bN1bM1 and T2N1bM1) exhibited consistently higher postoperative Ctn levels (>3000 pg/mL). Although recurrent aggressive surgery for MTC metastasis was performed for these patients, their prognosis remained poor (1 –3,41 –43), which highlights the importance of establishing consensus guidelines for the early diagnosis and treatment to improve the management of MTC patients also in China.
In conclusion, this study demonstrates that the novel RET S409Y mutation has potentially pathogenic effects and is associated with a relatively later age of onset and a lower penetrance of MTC, in addition to the absence of PHEO and HPT. Nonetheless, individuals carrying the S409Y mutation should be managed using an instructive personalized approach, and additionally, “at-risk” family members should be identified to further elucidate the correlation between the S409Y mutation and MEN2-specific tumors.
Footnotes
Acknowledgment
The authors thank all the patients and family members who agreed to participate in this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (81472861), the Key Project of Zhejiang Province Science and Technology Plan, China (2014C03048-1), and the Medical Science and Technology Project of Zhejiang Province, China (2014KYB219; 2017196976).