
Abstract
Background:
Thyroid hormones (THs) exert a strong influence on mammalian lipid metabolism at the systemic and hepatic levels by virtue of their roles in regulating circulating lipoprotein, triglyceride (TAG), and cholesterol levels, as well as hepatic TAG storage and metabolism. These effects are mediated by intricate sensing and feedback systems that function at the physiological, metabolic, molecular, and transcriptional levels in the liver. Dysfunction in the pathways involved in lipid metabolism disrupts hepatic lipid homeostasis and contributes to the pathogenesis of metabolic diseases, such as nonalcoholic fatty liver disease (NAFLD) and hypercholesterolemia. There has been strong interest in understanding and employing THs, TH metabolites, and TH mimetics as lipid-modifying drugs.
Summary:
THs regulate many processes involved in hepatic TAG and cholesterol metabolism to decrease serum cholesterol and intrahepatic lipid content. TH receptor β analogs designed to have less side effects than the natural hormone are currently being tested in phase II clinical studies for NAFLD and hypercholesterolemia. The TH metabolites, 3,5-diiodo-
Conclusions:
TH-based therapies show clinical promise for the treatment of NAFLD and hypercholesterolemia. Strategies for limiting side effects of TH are being developed and may enable TH metabolites and analogs to have specific effects in the liver for treatments of these conditions. These liver-specific effects and potential suppression of the hypothalamic/pituitary/thyroid axis raise the issue of monitoring liver-specific markers of TH action to assess clinical efficacy and dosing of these compounds.
Introduction
The liver is the hub for regulating serum cholesterol and triglycerides (TAGs) by virtue of its regulation of their biosynthesis and metabolism, packaging and export within lipoproteins, and reuptake via surface receptors for very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL) (1). The intrahepatic lipid content is maintained by a balanced level of lipid anabolic and catabolic processes (1). Thus, derangements in hepatic TAG and cholesterol metabolism can lead to metabolic disorders, such as nonalcoholic fatty liver disease (NAFLD) (2) and hypercholesterolemia (3).
Thyroid hormone (TH) effects on lipid metabolism have been known for over a century (4,5). In the clinical setting, hypothyroidism is associated with weight gain as well as higher serum TAG and LDL cholesterol (LDL-C) levels, whereas hyperthyroidism has opposite effects (6). Recent studies have demonstrated that TH regulation of hepatic autophagy and mitochondrial metabolism are key processes involved in hepatic TAG metabolism (7,8). TH decreases serum cholesterol by its effects on cholesterol synthesis, LDL clearance, and reverse cholesterol transport (RCT). Besides the major biologically active THs, thyroxine (T4) and triiodothyronine (T3), many TH metabolites exist in human serum and tissues at different concentrations. These metabolites are formed by deiodination, decarboxylation, deamination, N-acetylation, sulfation, and glucuronidation and may also have biological activity (9,10) (Table 1). Additionally, TH analogs are being developed that can specifically activate the TH receptor β (THRβ) isoform, the most abundant THR isoform in the liver to minimize TH-associated side effects that occur in the heart and bone that express TH receptor α (THRα) as the predominant isoform (11). Pharmacological strategies are also being developed to preferentially induce TH or TH analog uptake in the liver to minimize any potential off-target effects of THRβ agonist in the central nervous system, hypothalamic/pituitary/thyroid (HPT) axis, and other target tissues (12). In this review, we will provide an overview of the major mechanisms employed by TH to regulate hepatic TAG and cholesterol metabolism, and examine the current status of THs, TH metabolites, and TH analogs as potential therapies for hepatic lipid-associated metabolic diseases.
Thyroid Hormone Metabolites
3,5-T2AM, 3,5-diiodothyronamine; DIT, diiodotyrosine; DITPA, 2,5-diiodothyropropionic acid; T1AM, 3-iodothyronamine; T2, 3,5-diiodo-
TH Regulation of Hepatic TAG and Cholesterol Metabolism
Thyroid hormones
The thyroid gland synthesizes and secretes THs that are essential for the regulation of many metabolic processes throughout the body. The thyroid gland is composed of thyroid follicles, which are the basic units for the concentration of iodide (I−) and the synthesis of the major THs, thyroxine (T4) and T3. In circulation, most T4 and T3 are reversibly bound to serum carrier proteins such as thyroxine-binding globulin (TBG), transthyretin, and albumin (13,14). T4 and T3 then enter cells via plasma membrane transporters that belong to the monocarboxylate transporter 8 (MCT8), organic-anion-transporting polypeptide 1 (OATP1), and the L-type amino acid transporter (LAT) families (15,16). Once inside the cell, THs are activated or inactivated by a family of enzymes known as deiodinases (DIO1, 2, and 3), which control the concentration of T3 within the cell (17). DIO2 deiodinates T4 to generate T3, whereas DIO3 inactivates both T4 and T3 (17). DIO1 converts serum T4 to T3 to act as an intrathyroidal regulator of T3 concentration and the major extrathyroidal source of circulating T3 (17,18). T3 is the major active form of TH and exerts its action by binding to two nuclear hormone receptor isoforms, THRα and THRβ, which act as a ligand-inducible transcription factors that interact with TH response elements (TREs) in the regulatory regions within the promoters as well as enhancers and intronic region of target genes (13,19). THRα is the major isoform in the heart and bone, whereas THRβ is the major isoform in the liver as it may represent >90% of the THRs in the latter tissue.
TH regulation of hepatic TAG synthesis
TH critically regulates various steps involved in hepatic non-esterified fatty acid (NEFA) uptake, de novo lipogenesis, and TAG assembly (Fig. 1). TH positively regulates the expression of fatty acid translocase (FAT/CD36) and fatty acid binding protein (FABP) expression in the liver resulting in increased influx of NEFA into the hepatocytes (20 –22). TH also increases the intracellular pool of NEFA by increasing the transcription and expression of enzymes involved in de novo lipogenesis (Fig. 1). These TH-regulated genes include fatty acid synthase (Fasn) (23), acetyl-CoA carboxy-lase alpha (Acc1; also known as Acaca) (24), malic enzyme (Me) (25), and TH-responsive Spot14 homologue (Thrsp; also known as Spot14) (26). TH can also indirectly control the transcriptional regulation of hepatic lipogenic gene expression by regulating the expression of other transcription factors, such as sterol regulatory element-binding protein 1C (SREBP1C), liver X receptors (LXRs), carbohydrate-responsive element-binding protein (ChREBP), and peroxisome proliferator-activated receptor gamma (PPARγ), which all have crucial roles in hepatic lipogenesis (27). TH stimulation of LXR and ChREBP mediate oxysterol and carbohydrate-driven lipogenesis in the liver (28,29). In contrast, TH negatively regulates the expression of pro-lipogenic transcription factors, such as SREBP1c and PPARγ (30,31). However, despite these effects by TH on genes involved in de novo lipogenesis, chronic TH administration does not cause a net increase in hepatic TAG content in rodents (32). It is noteworthy that TH downregulates stearoyl-CoA desaturase 1 (SCD1) and glycerol-3-phosphate acyltransferase (GPAT) expression and activities to limit the amount of synthesized fatty acids that can be stored as TAG in the liver (33,34).
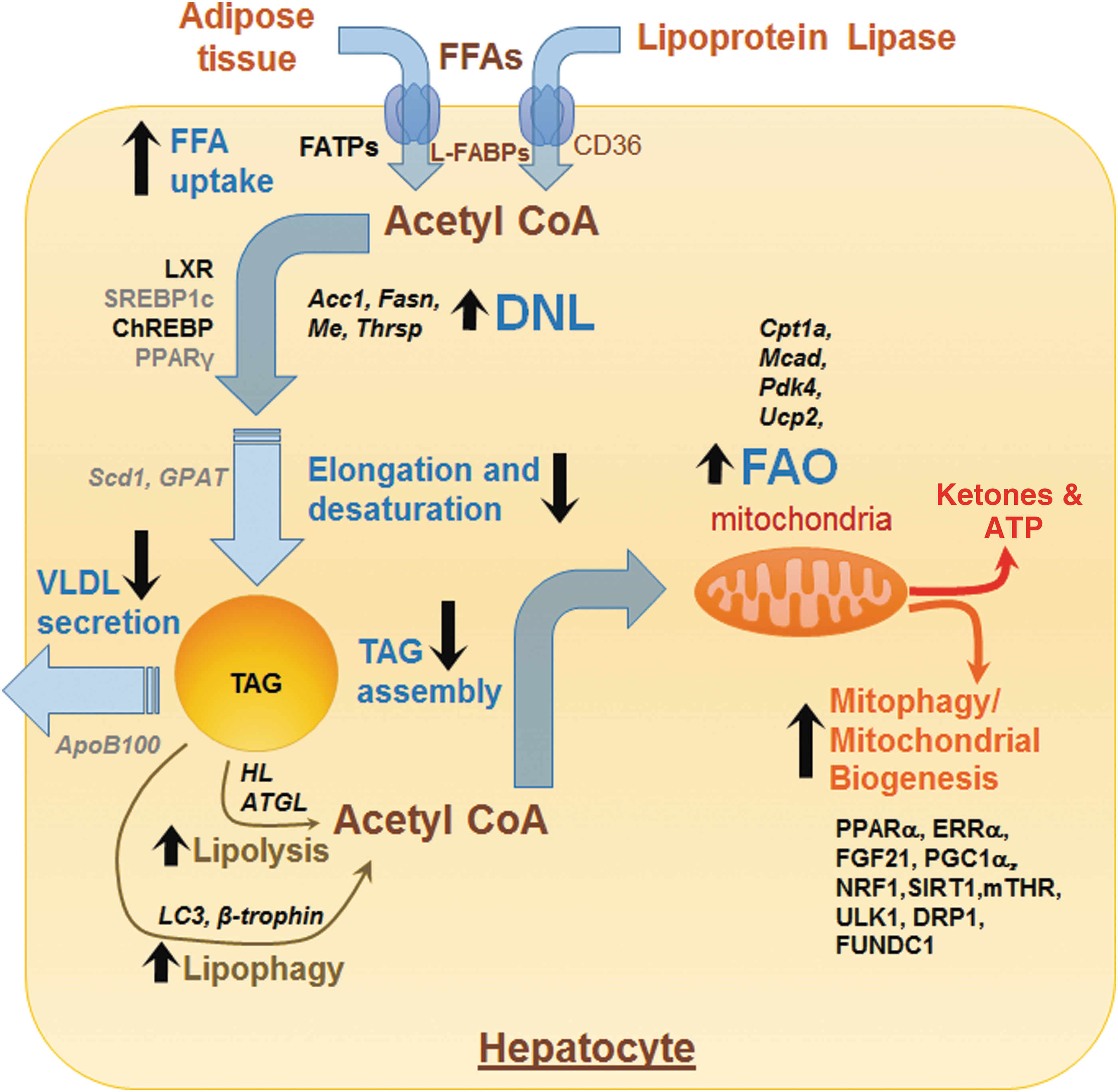
TH and hepatic TAG metabolism. TH regulates several processes involved in TAG anabolism, catabolism, and export. Genes and proteins positively regulated by TH are shown in bold black and those negatively regulated by TH are shown as gray color fonts. Transcription factors and other proteins are shown as running font, whereas genes are italicized. Black arrows represent processes/pathways that are upregulated (upward arrow) or downregulated (downward arrow) by TH. DNL, de novo lipogenesis; FAO, fatty acid oxidation; FFAs, free fatty acids; TAG, triacylglycerol; TH, thyroid hormone; VLDL, very-low-density lipoproteins.
TH regulation of fatty acid β-oxidation
Contrary to the sometimes variable and conflicting reports for TH effects on lipogenesis, TH has a strong effect on increasing hepatic lipid catabolism. The catabolic actions of TH on hepatic lipids are primarily mediated by the mobilization of free fatty acids (FFAs) from stored TAGs and their subsequent β-oxidation (Fig. 1). In this regard, TH increases TAG hydrolysis by stimulating the transcription and activities of both adipose triglyceride lipase (ATGL) and hepatic lipase (35,36). Additionally, TH induces the expression of zinc-α2-glycoprotein in hepatic cells, a protein that may contribute to the lipolytic action of TH (37). Besides its induction of hepatic lipases, TH also stimulates autophagy-lysosomal mediated lipolysis in hepatic cells (8). TH previously was shown to increase the lysosomal activity and lysosomal acid lipases expression (38,39), but the precise mechanism is not well understood. Subsequently, we showed that TH is a potent stimulator of hepatic lipophagy and is required for intracellular TAG hydrolysis and delivery of fatty acids to mitochondria for β-oxidation (8). In support of this notion, we showed that small interfering RNA (siRNA) knockdown of the autophagy gene, ATG5, significantly impairs the induction of hepatic β-oxidation of fatty acids and ketogenesis by TH in mice (Fig. 1) (8). Indeed, it is likely that lipophagy is the predominant mechanism for mobilizing fatty acids from fat droplets acutely, and lipases may contribute only after chronic exposure to TH. In this regard, induction of ATGL and hepatic lipase gene expression increase only after 10 days of TH treatment. Finally, a recent study revealed a critical role for β-trophin (C19orf80; also known as ANGPTL8) in TH-mediated induction of hepatic lipophagy and TAG hydrolysis (40).
Mitochondria are classical targets for TH action in the liver and the major sites for fatty acid catabolism (41). Thus, TH not only regulates the intracellular generation of fatty acids but also their entry into the mitochondria and the expression of mitochondrial enzymes involved in β-oxidation (Fig. 1) (41). Carnitine palmitoyltransferase Iα (CPT1α), a carrier protein that is required for the delivery of long-chain fatty acids (LCFAs) into the mitochondria, is a direct transcriptional target of TH in the liver (42). TH can also regulate the expression of CPT1α indirectly via its stimulation of intrahepatic peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), estrogen-related receptor alpha (ERRα) (43), peroxisome proliferator-activated receptor alpha (PPARα), and fibroblast growth factor 21 (FGF21) levels (44). TH also increases the expression of other mitochondrial enzymes involved in β-oxidation and oxidative phosphorylation, such as medium-chain acyl-CoA dehydrogenase (MCAD) (45), pyruvate dehydrogenase kinase isoform 4 (PDK4) (46), and mitochondrial uncoupling protein 2 (UCP2) (47).
TH also regulates mitochondrial β-oxidation by inducing mitochondria synthesis and maintaining mitochondria quality control (Fig. 1) (48). TH stimulates mitochondria biogenesis by inducing PGC1α gene expression, which in turn stimulates transcription of nuclear respiratory factor 1 (NRF1) and mitochondrial transcription factor A (mtTFA) (48). Furthermore, TH activates SIRT1 to deacetylate PGC1α and increase its ability to bind the regulatory regions of target genes that are involved in mitochondrial synthesis and function (49). In addition to the stimulation of the PGC1α-NRF1-mtTFA axis by TH, we recently showed that TH upregulates a PGC1α-ERRα pathway for mitochondrial biogenesis. ERRα is an orphan nuclear receptor that is induced by PGC1α and stimulates the expression of genes involved in mitochondrial biogenesis and lipid oxidation (43). Indeed, our studies suggest that TH stimulation of PGC1α-ERRα may be the predominant mechanism for stimulating mitochondrial activity since siRNA knockdown of ERRα completely abrogates TH-mediated stimulation of oxidative phosphorylation.
Truncated THRs have been reported within the mitochondria and may regulate transcription from the mitochondrial genome (50). Mitochondrial THRs bind to the D-loop of the mitochondrial DNA and increase its transcription and replication, although the mechanism is not well understood (50). Mitochondrial THRs also interact with the mitochondrial trifunctional protein (MTP), which catalyzes the last three reactions of mitochondrial fatty acid oxidation (hydration, dehydrogenation, and cleavage) on LCFAs (51). Although mitochondrial biogenesis is needed to stimulate hepatic β-oxidation by TH, other mitochondrial homeostatic processes such as mitochondrial fission, fusion, and mitophagy are needed to sustain it (52). In this regard, we earlier showed that TH stimulates protective autophagy of mitochondria (mitophagy) to reduce cellular injury by reactive oxygen species (ROS) (53). We showed that mitophagy itself is induced by excess ROS generation by mitochondria, which then causes intracellular Ca2+ release, activation of calcium/calmodulin-dependent protein kinase kinase 2 (CAMMK2), 5′ AMP-activated protein kinase (AMPK) phosphorylation, and the activation and mitochondrial translocation of Unc-51-like autophagy activating kinase (ULK1) after phosphorylation by AMPK (53). Although excessive ROS production is pathological in severe thyrotoxicosis and can lead to cell death (54), its physiological production may be beneficial in hepatic cellular metabolism via a process known as cellular hormesis. Indeed, we found that there is a dose- and time-dependent increase in ROS production that is linked to TH-induced oxidative phosphorylation in hepatic cells (53). This increase in ROS is important for induction of mitophagy to maintain mitochondrial quality for β-oxidation of fatty acids and oxidative phosphorylation (53). This ROS-induced mitophagy also ensures that damaged mitochondria are swiftly removed from the cell to avoid further oxidative damage and cell death (53).
Besides stimulating β-oxidation of fatty acids, TH also increases lipogenesis, at least acutely. The effects of TH on these two opposing metabolic pathways appear to be paradoxical and are not well understood. It is possible that TH regulation of genes involved in lipogenesis may be temporally regulated (with fatty acid synthesis predominating early and β-oxidation of fatty acids later) and/or dependent on underlying nutrient/energy status/concentration/periportal versus pericanalicular location, and species. However, even with the induction of lipogenesis, it appears that β-oxidation predominates in vivo as β-hydroxybutyrate is detected in serum as early as 3 days after TH administration (8).
TH regulation of hepatic sphingolipid and phospholipid metabolism
One week administration of either T3 or 3,5-diiodo-
TH regulation of cholesterol metabolism
TH regulates hepatic cholesterol metabolism by multiple mechanisms (58,59) (Fig. 2). In rats, TH increases the expression of hydroxymethylglutaryl-CoA reductase (Hmgcr) and farnesyl pyrophosphate synthetase (Fdps) to promote cholesterol biosynthesis in the liver (60). However, TH also negatively regulates hepatic cholesterol secretion by decreasing the expression of both sterol O-acyltransferase 2 (SOAT2) and apolipoprotein B100 (ApoB100), which are required for re-esterification and packaging cholesterol into VLDL and LDL (61 –64). TH decreases SOAT2 by inducing the expression of human miR181d, which decreases the expression of caudal-type homeobox protein 2 (CDX2), a transcription factor that positively regulates SOAT2 gene expression (62).
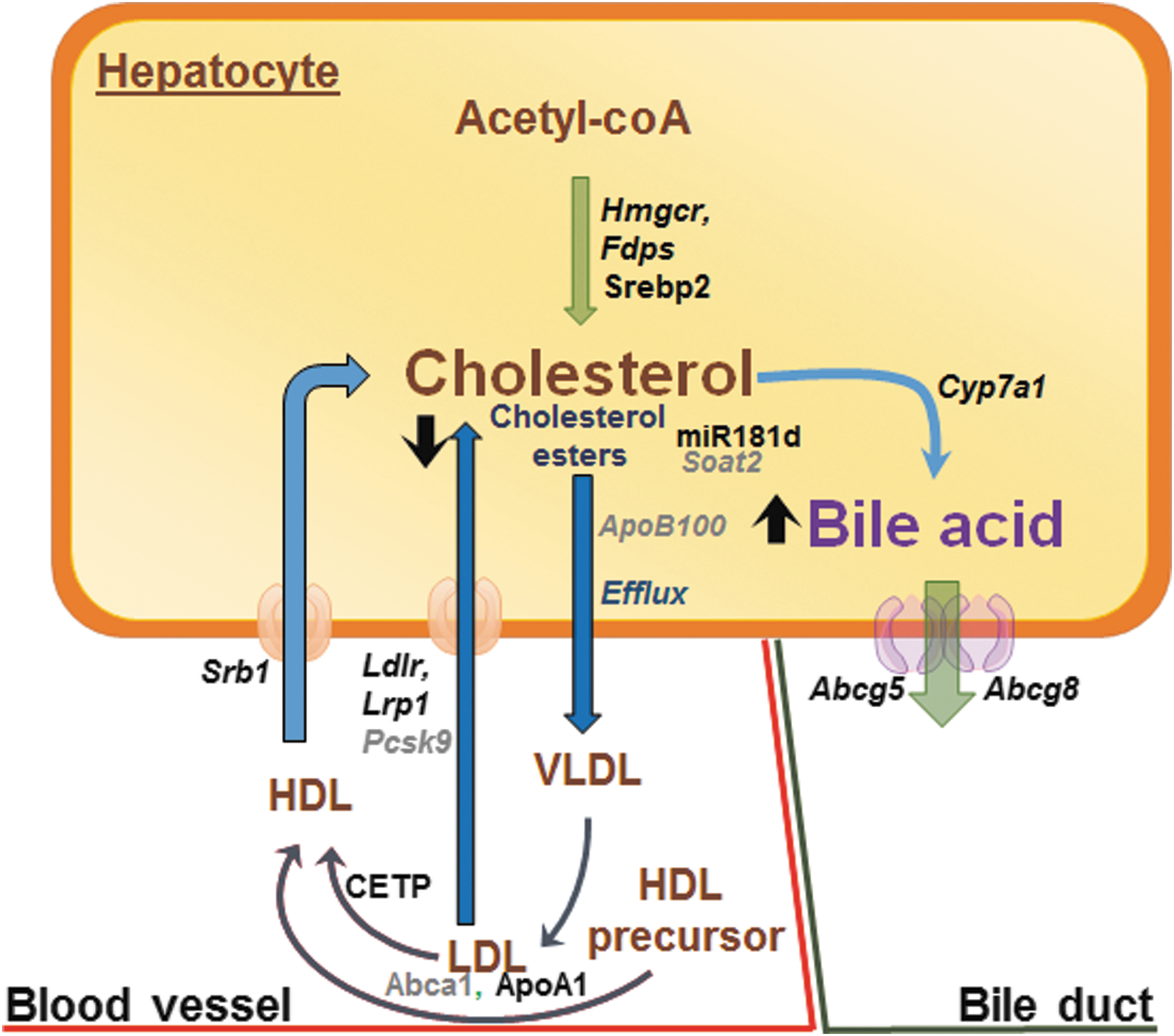
TH and hepatic cholesterol metabolism. TH regulates several processes involved in hepatic cholesterol biosynthesis, export, and degradation. Genes and proteins positively regulated by TH are shown in bold black and those negatively regulated by TH are shown as gray color fonts. Transcription factors and other proteins are shown as running font, whereas genes are italicized. Black arrows represent processes/pathways that are upregulated (upward arrow) or downregulated (downward arrow) by TH. HDL, high-density lipoprotein; LDL, low-density lipoprotein.
TH increases the expression/activity of cholesteryl ester transfer protein (CETP) (65) and expression of hepatic LDL receptor (LDLR) (66) to increase serum cholesterol clearance. LDLR is regulated by SREBP2, which is induced transcriptionally by TH in rodents and humans (67,68). TH also increases the transcription of both mouse and human LDLR-related protein 1 (LRP1), a lipoprotein involved in the removal of TAGs from chylomicron remnants and VLDL. Interestingly, TH can decrease circulating pro-protein convertase subtilisin/kexin type 9 (PCSK9) levels, which may contribute to lower plasma LCL-C levels by enhancing LDLR recycling (69).
TH also has major effects on RCT. TH induces the gene and protein expression of ApoA1 and scavenger receptor class B member 1 (SRB1) to increase cholesterol efflux from peripheral tissues to HDL (70,71). TH also stimulates hepatic lipase to decrease HDL particle size resulting in higher relative levels of lipid-poor ApoA-I in HDL that facilitates transfer of cholesterol from peripheral tissues via the ATP-binding cassette transporter (ABCA1) (60,72). Within the liver, TH increases the expression of rat and human cholesterol 7 alpha-hydroxylase (CYP7A1), the rate-limiting enzyme that converts cholesterol into bile acids in the RCT pathway (60,73). In addition, TH increases the efflux of bile acids in both the liver and intestines, which are the last steps of the RCT pathway, by stimulating ATP-binding cassette subfamily G member (Abcg5/Abcg8) gene transcription (74).
Lessons from THR transgenic models
Knock-in mouse models harboring mutant THRα/β receptors have provided insights on the role of THRs in hepatic lipid metabolism and enlarged our understanding of their systemic and hepatic functions in vivo. A mouse model that expresses a dominant negative mutation in THRβ (THRβPV/PV) mimics the resistance to thyroid hormone (RTH) phenotype in humans and exhibits enlarged livers with hepatosteatosis by 4–5 months of age (30,75). Liver TAGs and expression of several key enzymes involved in lipogenesis (e.g., Scd1, Fasn) also were increased in THRβ−/− mice (76). In contrast, mice expressing a similar dominant negative mutation in the THRα gene locus, THRα1PV/PV, as well as THRα-null mice, showed reduced liver weights and decreased lipid accumulation (30,75,77). It is thought that increased lipogenic enzyme expression and decreased fatty acid β-oxidation activity due to PPARγ expression contributes to the intrahepatic lipid accumulation in THRβPV/PV mice, whereas decreased expression of lipogenic genes reduced liver mass and intrahepatic lipid content in THRα1PV/PV mice (30,75). These observations suggest that the regulation of lipid metabolism in the liver is likely to be THR isoform-dependent; however, it is not known whether these effects are mediated by THR-mediated transcription or by alternative mechanisms that do not necessarily require THR binding to DNA. In this connection, it recently was shown that TH effects on heart rate, body temperature, and lipid metabolism were lost in mice in which classical THR DNA binding ability was lost (76).
TH metabolites in lipid metabolism
The less iodinated version of TH, T2, is present in the picomolar range in human serum and has received attention owing to its bioactive role in animals (78). T2 is thought to modulate cellular function by noncanonical pathways, although THR-mediated signaling has not been totally excluded (79). One of the most profound biological effects of T2 is its regulation of lipid metabolism and insulin sensitivity in hepatic cells (79). T2 directly activated SirT1 to increase the PGC1α activity and increase the expression of CPT1α and genes involved in mitochondrial biogenesis in hepatocytes (80). Proteomic pathway analysis performed in the livers from animals fed a HFD showed that T2 altered pathways involved in fatty acid β-oxidation and amino acid metabolism as well as respiratory chain activity and ROS (81). Besides its induction of mitochondrial pathways, T2 upregulated the levels of ApoB100, the major protein component of VLDL (82), suggesting that T2 stimulated lipoprotein secretion to reduce intrahepatic fat content. Of note, T2 also increased the activity of ATGL in hepatocytes (83). Contrary to T3, T2 suppressed hepatic lipid accumulation by downregulating the expression of lipogenic proteins (84,85).
Thyronamines (TAMs) are endogenous biogenic amines generated from iodinated TH by deiodination and decarboxylation. However, their precise physiological concentrations and functions in the body are still controversial. Scanlan et al. (86) detected endogenous T1AM (3-iodothyronamine) and T0AM at subpicomolar concentrations in rat brain, although it is noteworthy that another group did not identify significant amounts of endogenous TAMs in rat brain and serum, or in human serum and thyroid tissue (87). This issue notwithstanding, TAMs can bind to distinct receptors such as the G protein-coupled receptor TAAR1 at pharmacological doses in the nanomolar range (86). They were found to have rapid and systemic effects such as hypothermia and bradycardia in mice (86) that were opposite to the effects of T3 administration. Therefore, TAMs may be bona fide hormones if produced in sufficient quantities locally; however, the factors that control their synthesis, transport, action, and catabolism are still not known (10). Previous studies indicated that TAMs are able to switch metabolism from carbohydrate to lipid utilization (88,89). T1AM also decreases serum cholesterol and increase serum TAGs in a dose-dependent manner (90). In liver metabolome analysis, T1AM increased fatty acid oxidation and decreased the expression of SIR T4, a negative regulator of fatty acid oxidation, and Ldlrap1, involved in the efficient endocytosis of the LDLR (88,90). Although TAMs previously were shown to exhibit a wide array of effects on thermoregulation, food intake, heart, and tissue metabolism, as well as alter both TH and corticosterone levels, it is not known whether T1AM has a direct in vivo effect on liver lipid metabolism (10). However, in favor of this notion, in vitro and liver perfusion studies have shown uptake of T1AM and stimulation of fatty acid catabolism in hepatocytes (91).
The thyroacetic acid, 3,3′,5,5′-tetraiodothyroacetic acid (Tetrac), circulates in low nanomolar concentrations. The deiodinated form of Tetrac, Triac (3,5,3′,-triiodothyroacetic acid), has a higher affinity for THR than T3 and binds with higher affinity to THRβ than THRα (92). In humans, Triac has a half-life of about 6 hours. Due to its higher affinity for THR and use of different transporters than THs, Triac has been used clinically for patients with RTH syndrome and currently is being tested for patients with MCT8 deficiency (93). When administered to rodents, Triac levels are increased in the liver and induce the expression of T3-regulated target genes (94). To date, there have been no published studies investigating the differences between Triac versus T3 on hepatic lipogenesis, β-oxidation, and VLDL secretion, although it appears to have similar effects as T3 in other tissues (93,94). Therefore, current evidence shows that although Triac and T3 may function differently pharmacokinetically, they share common signaling pathways. Additionally, it should be noted that Triac reduces endogenous TH production by suppressing the HPT axis (reduced thyrotropin [TSH], T4, and T3).
Sulfated and glucuronidated THs have increased water solubility to facilitate their excretion through the bile and gut. However, it is thought that both sulfated and glucuronidated THs can act as a reservoir for total TH within the body and can be converted back to T4 and T3 when necessary (9). Sulfated and glucuronidated TH are thought to be inactive metabolites, and to the best of our knowledge, no bioactive role(s) for the sulfated and glucuronidated TH on lipid metabolism has been described. Similarly, diiodotyrosine (DIT), which is formed by breakage of the thyronine backbone at its ether bond, is involved in disposal of TH but does not have any known direct effects on lipid metabolism (9,95).
TSH and lipid metabolism
TSH is produced by the pituitary gland and stimulates TH production by the thyroid gland. Serum TSH and cholesterol levels have been shown to be positively associated, and TSH may have a direct effect on liver lipid metabolism. Zhang et al. demonstrated the presence of TSH receptor (TSHR) messenger RNA (mRNA) expression in human and rat liver tissue that was identical to that found in thyroid tissue and a thyroid cell line (96). The TSHR was located at the cell membrane of hepatocytes, and bovine TSH and IgG from patients with Graves' disease induced cAMP in hepatic cells in a TSHR-dependent manner (96,97). Recombinant TSH, in the micromolar range, directly upregulated mRNA expression and activity of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), an enzyme involved in cholesterol biosynthesis as well as cholesterol production in human liver cells (93). In thyroidectomized rats, when T4 was supplemented together with increasing concentrations of TSH, there was an increase in HMGCR protein levels and serum cholesterol levels with increasing TSH concentrations (97). Another study from the same group also showed that both thyroidectomized and TSHR−/− mice had repression of bile acid synthesis via SREBP2 induction when supplemented with T4 and bovine TSH. Repression of bile acid synthesis can cause higher intrahepatic and serum cholesterol levels since bile acids are the major means to eliminate excess cholesterol from hepatic cells (97,98).
Recently, a direct relationship between serum TSH and PCSK9 expression and an inverse relationship between serum TSH and LDLR expression were demonstrated (99). PCSK9 inhibitors are effective in lowering serum cholesterol levels via recruitment of LDL-C from circulation by restoring LDLR expression on the cell surface of hepatocytes. Recombinant human TSH alpha/beta heterodimer protein increases PCSK9 mRNA expression and protein levels and impairs LDL-C uptake in human liver cells (99,100). TSHR−/− mice supplemented with T4 also have lower intrahepatic TAG levels both after chow and HFD and lower expression of lipogenic genes than control mice under the same conditions, suggesting that TSH may also regulate intrahepatic lipid metabolism (100). In this context, TSH directly increased lipogenesis by activating SREBP1c via a cAMP/PKA/PPARα signaling pathway associated with AMPK in hepatic cells (101). Recently, it was shown that the administration of recombinant human TSH increases serum apoB, Lp(a), non-HDL cholesterol, and TAGs in patients after thyroidectomy on a stable dose of LT4 (102). After TSH administration, T3 levels were reduced and changes in lipids were inversely associated with the T3 levels, suggesting that TSH may have had an indirect effect on lipid levels (102), perhaps due to effects on the deiodinase activity. Taken together, these foregoing studies suggest that TSH may worsen hypercholesterolemia by increasing cholesterol biosynthesis, reducing cholesterol clearance, lowering LDLR, and worsen NAFLD by increasing lipogenesis, respectively. However, it should be pointed out that studying the direct role of TSH in vivo is challenging in animal models and humans since it will undoubtedly cause alterations in serum T3 and T4 levels (102,103). Even in studies in which thyroidectomized animals or patients are studied, it is likely that restored serum levels of T3/T4 may not fully recapitulate normal serum or tissue levels of TH due to the absorption/pharmacokinetic characteristics of supplemented hormones versus the natural production/removal of endogenous THs.
THs, TH Analogs, and TH Metabolites in NAFLD
Current evidence on TH, TH analogs, and TH metabolites in NAFLD and hypercholesterolemia is summarized for animal models (Table 2) and humans (Table 3).
Effects of Thyroid Hormone, Thyroid Hormone Analogues, and Thyroid Hormone Metabolites in Animal Models of Nonalcoholic Fatty Liver Disease and Hypercholesterolemia on Serum Lipids, Steatosis, Liver Function, Body Weight, Hypothalamic/Pituitary/Thyroid Axis, and Other Side Effects
EDn: dose in nmol/kg/day causing an n % change.
Also significantly changed in 476 nmol/kg/day.
ALT, alanine transaminase; AST, aspartate transaminase; BW, body weight; CD-HFD, choline-deficient high-fat diet; fT3, free triiodothyronine; fT4, free thyroxine; HCD, high-cholesterol diet; HFD, high-fat diet; HFHCD; high-fat high-cholesterol diet; HPT, hypothalamic/pituitary/thyroid; MCD, methionine- and choline-deficient; T4, thyroxine; TC, total cholesterol; TAG, triglycerides; TSH, thyrotropin; WD, western diet.
Effects of Thyroid Hormone, Thyroid Hormone Analogues, and Thyroid Hormone Metabolites in Humans with Nonalcoholic Fatty Liver Disease or Hypercholesterolemia on Serum Lipids, Steatosis, Nonalcoholic Steatohepatitis, Body Weight, Hypothalamic/Pituitary/Thyroid Axis, and Other Side Effects
H-MRS, magnetic resonance spectroscopy; BMI, body mass index; LDL-C, low-density lipoprotein cholesterol; LT4, levothyroxine; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
Nonalcoholic fatty liver disease
NAFLD is a hepatic lipid-related metabolic disorder that is highly associated with diabetes and obesity. The prevalence of NAFLD is rapidly increasing worldwide and currently is thought to afflict 30–40% of all adults in both developed and developing countries (104). NAFLD also increases the risk for developing diabetic complications and cardiovascular diseases later in life (104). NAFLD represents a spectrum of liver derangements starting from benign steatosis that progresses to nonalcoholic steatohepatitis (NASH) characterized by insulin resistance, and liver inflammation and fibrosis (105). Significantly, NASH can progress to cirrhosis and hepatocellular carcinoma in a subset of patients. Although the mechanisms for the pathogenesis and progression of NAFLD are not fully understood, impaired hepatic lipid metabolism appears to have a central role (2).
In early NAFLD, there is an increased uptake of NEFAs inside hepatocytes and de novo lipogenesis followed by intracellular TAG accumulation (106). The major cause for increased NEFA influx in NAFLD is the induction of hormone-sensitive lipase activity in white adipose tissue due to insulin resistance (106). Interestingly, loss of fatty acid transporters such as FATP and CD36 can prevent the development of NAFLD in murine models (107). Besides the increased uptake of NEFAs in early NAFLD, de novo lipogenesis in response to high-carbohydrate feeding leads to the accumulation of TAGs within the liver (108). Furthermore, both TAG lipolysis and mitochondrial β-oxidation of fatty acids are impaired in NAFLD (109,110). The defect in mitochondrial fat oxidation likely leads to increased esterification of FFAs into TAGs as well as synthesis of reactive lipid species such as DAG and ceramides (111) that have been associated with insulin resistance in NAFLD (112). Ceramides are also mediators of mitochondrial damage in hepatocytes and cause lipotoxicity, inflammation, and fibrosis (113). Additionally, TAG export in VLDL is increased during hepatosteatosis but is impaired later in NASH, perhaps due to detrimental changes in phospholipid levels (114).
Cholesterol metabolism is also altered in NAFLD (115). Comparative hepatic and serum lipidomic analysis of normal and NAFLD samples revealed increased hepatic free cholesterol (FC) in NASH (116). The role of dietary cholesterol has been investigated in murine models of NAFLD, and a cholate-deficient and cholesterol-enriched, high-fat atherogenic diet promoted both oxidative stress and experimental NASH (117). Furthermore, increased hepatic FC level was observed in association with the transition from hepatosteatosis to NASH in obese hyperinsulinemic mice (118). Both genetic and nutritional models of hepatosteatosis have implicated mitochondrial FC loading as predisposing animals to tumor necrosis factor- and Fas-induced steatohepatitis and activation of hepatic stellate cells to cause fibrosis (119). So far, there is no approved drug or hormone therapy for NAFLD, and lifestyle management with diet and exercise are the current therapeutic cornerstones for limiting the progression of NAFLD (120).
Effects of TH on NAFLD
Several epidemiological studies conducted across the globe have shown an inverse relationship between hypothyroidism and the incidence of NAFLD (121). Even within the reference range, higher TSH and lower TH levels are associated with a higher prevalence of NAFLD in a dose-dependent manner (122,123). Recent studies in rodents and patients further support this inverse relationship between physiological thyroid status and NAFLD. In mice, mild hypothyroidism increases the prevalence of NAFLD possibly due to both intrahepatic and extrahepatic mechanisms (124). Transcriptome analyses of the liver samples from patients who underwent bariatric surgery for NAFLD showed that several of the major genes that had altered expression are regulated by TH (125). Also, evidence of decreased intrahepatic TH levels in NAFLD was shown in rats and humans (126,127). Other studies also suggested a possible direct role of TSH in stimulating hepatosteatosis since TSHR-knockout mice were protected from steatosis when fed HFD (101).
T3 administration significantly decreases hepatosteatosis and inflammation (128), and it restores mitochondrial function in NASH (7,129). In a recent study, T3 administration in the nanomole per kilogram range to rats fed HFD not only reduced hepatic TAG accumulation but also prevented the generation of toxic lipid species, such as ceramides (55). More recently, a compound formed by chemical hybridization of glucagon and TH showed promising results to directly deliver T3 to the liver (130). This glucagon/T3 conjugate was composed of the amine of T3 covalently linked to the 40-mer glucagon analog through a succinate spacer at the terminal lysine. Glucagon/T3 elicited the transcriptional activity of a TRE reporter in cells expressing the glucagon receptor presumably through hydrolysis of the gGlu spacer to release T3. When the molecule was fluorescently labeled, it was shown to be preferentially accumulated by the liver and to have low uptake in fat, in the heart and pancreas. This uptake correlates with the hepatic expression of glucagon receptor as well as increased intrahepatic T3 levels. Glucagon/T3 treatment improved dyslipidemia, atherosclerosis, body weight, and steatosis in several dietary models of obesity (130). The cardiac side effects of T3 on the heart, in terms of cardiac hypertrophy and reduced ejection fraction, were not observed with treatment with glucagon/T3. Whereas T3 monotherapy at 100 nmol/kg significantly reduces bone volume, glucagon/T3 at equimolar dosage had no such effects. Glucagon/T3 also did not cause any change in serum T3 levels but slightly decreased serum T4. Plasma TSH and hypothalamic thyrotropin-releasing hormone mRNA were normal both with T3 monotherapy and glucagon/T3. Although the side effect profile seemed favorable for this conjugate over T3 monotherapy in mice, it remains to be seen whether this side effect profile and its potential effects on peripheral TH metabolism will enable it to be used clinically in patients. Similarly, chemical conjugation of T3 with antisense oligonucleotide (ASO) targeting nicotinamide N-methyltransferase (NNMT)-ASO or apolipoprotein B (ApoB)-ASO also provides protection from diet-induced obesity and intrahepatic lipogenesis, thereby highlighting hormone/drug conjugation as a novel strategy for countering NAFLD, obesity, and hyperlipidemia (131).
Similar to T3, there have been limited studies on the role of T4 therapy in NAFLD. It has been reported that T4 supplementation reduced the prevalence of NAFLD in patients with subclinical hypothyroidism (132). More recently, we investigated the effects of T4 supplementation in euthyroid male subjects with type 2 diabetes and NAFLD using 1H-magnetic resonance spectroscopy (MRS). Low-dose T4 supplementation (average dose 18.75 μg/day) aimed to result in a low–normal TSH between 0.30 and 1.70 mIU/L for 4 months decreased intrahepatic lipid content as measured by MRS (1H-MRS) (127). This reduction in liver fat content also correlated with an improvement in glycemic control after treatment.
TH analogs as therapies for NAFLD
Several TH analogs/mimetics have been developed to treat hypercholesterolemia, obesity, and/or diabetes. These compounds have THRβ isoform selectivity and/or tissue-specific uptake (e.g., liver) to target their therapeutic effects while mitigating potential side effects. Several studies in rodent NAFLD models have demonstrated consistently the efficacy of TH analogs/mimetics in reducing both hepatosteatosis and liver injury. The THRβ-selective compound GC-1 [sobetirome, 2-(4-((4-Hydroxy-3-(1-methylethyl)phenyl)methyl)-3,5-dimethylphenoxy)acetic acid] was one of the first TH analogs examined as a potential therapy for NAFLD. Perra et al. demonstrated that GC-1 prevented the development of hepatosteatosis and lipo-peroxidation in rodents by feeding a methionine- and choline-deficient diet supplemented with 15 μmol GC-1 per kg diet (128). Similarly, GC-1 also reduced hepatosteatosis in other animal models of obesity such as ob/ob mice and rats fed a HFD (133,134). In addition to GC-1, the THRβ-selective analog KB2115 [eprotirome, 3-((3,5-dibromo-4-(4-hydroxy-3-(1-methylethyl)-phenoxy)-phenyl)-amino)-3-oxopropanoic acid] and the liver-specific analog MB07811 [(2R,4S)-4-(3-chlorophenyl)-2-[(3,5-dimethyl-4-(4-hydroxy-3′-isopropylbenzyl)phenoxy)methyl]-2-oxido-[1,3,2]-dioxaphosphonane] decreased hepatosteatosis in both HFD and ob/ob mice models of NAFLD while also reducing serum TAG levels (133,134). MB07811 is a liver-specific prodrug that undergoes first-pass hepatic extraction and is converted intrahepatically to a negatively charged THR agonist [3,5-dimethyl-4-(4′-hydroxy-3′-isopropylbenzyl(phenoxy)methylphosphonic acid] (MB07344) that has a short biological half-life and, hence, little peripheral side effects. Cable et al. used several experimental models of NAFLD such as Zucker diabetic fatty (ZDF) rats, ob/ob mice, and diet-induced obese mice to demonstrate that MB07811 at micromolar doses markedly reduces hepatosteatosis, as well as plasma FFAs and TAGs (135). This anti-steatotic activity of MB07811 was associated with an increased rate of mitochondrial β-oxidation and stimulation of CPT1α expression (135).
Previously, there were no clinical trials investigating TH analogs for the treatment of NAFLD in humans. Recently, a phase II clinical study for a THRβ-selective analog, MGL-3196, was conducted in patients with biopsy-proven NASH. These patients had a statistically significant reduction in hepatosteatosis after 12 and 36 weeks of treatment as measured by 1H-MRS (136). Also, the first results of a phase II study on the effects of Viking 2809 (MB07811) on NAFLD and hypercholesterolemia showed decreased hepatic lipid content and serum LDL-C (137). Definitive results from these studies are awaited to fully assess the effects of these compounds on the HPT axis and other potential side effects in humans.
In general, it appears that TH analogs can have suppressive effects on the axis, although the effects may depend on the specific compound and the dose used. In terms of axis suppression, GC-1 and KB2115 suppress serum TSH and T4 levels (133,138). MB07811 and another THRβ-selective thyromimetic KB141 also suppress serum T4, although MB07811 did not have any significant effects on serum TSH at a lower concentration (3 mg/kg) due to its conversion to a short-lived active metabolite in the liver (139). In contrast to T3, GC-1 (64 days, 15 μg/kg) did not reduce bone mineral density (140) and had little effect on heart rate in monkeys (141). Since serum TSH, T3, and T4 levels are not reliable markers for thyrotoxicity in patients who take such compounds, clinical assessment of patients before and during therapy is imperative. Patients with cardiac disease or osteoporosis will likely need to be disqualified beforehand. Patients who are started on such drugs will also need to be monitored for tachycardia and atrial arrhythmias, bone loss (e.g., bone densitometry, markers for bone resorption such as urine hydroxyproline and deoxypyridinoline, and serum osteopontin and bone sialoprotein), and hepatic damage (liver enzymes such as alanine transaminase [ALT] and aspartate transaminase [AST]). Efficacy of treatment can be measured by changes in fat content (measured by MRS, magnetic resonance imaging-proton density fat fraction [MRI-PDFF], or liver biopsy), liver function tests (ALT/AST), serum fibrosis markers (Pro-C3), and histological changes of inflammation and fibrosis in liver biopsy. It may also be necessary to titrate and optimize the dose to serum markers known to be regulated by TH such as sex hormone binding globulin (SHBG), ferritin and cholesterol levels, with SHBG likely having the highest specificity (142). It is also possible that serum metabolomic markers such as serum acylcarnitines or branched chain amino acids may prove to be useful markers for TH action (143).
TH metabolites as therapy for NAFLD
Recently, the TH metabolite, T2, received attention as a potential therapy for NAFLD owing to its bioactive role in animals (78). To mimic hepatosteatosis in vitro, primary cultures of rat hepatocytes were exposed to an oleate/palmitate mixture in lipid-load hepatocytes. After lipid loading hepatocytes in vitro, the addition of T2 caused a significant reduction in hepatocyte lipid content and lipid droplet diameter, as well as reduced activity of acyl-CoA oxidase (ACOX), a rate-limiting enzyme of peroxisomal β-oxidation (144). Furthermore, T2 reversed hepatic fat accumulation in a rodent model of NAFLD and it prevented hepatosteatosis at a dose of 476 nmol/kg (80,145). Recently, using a targeted metabolomics approach, we compared the effects of T2 and T3 on the early metabolic adaptation in the livers of rats fed HFD (55). We found that both T2 and T3 strongly induce autophagy and intrahepatic acylcarnitine flux, while preventing the generation of sphingolipid/ceramides in rats fed HFD (55). Interestingly, although both T2 and T3 decrease hepatic fat content, only T2 rescues the impairment in Akt and mitogen-activated protein kinase (MAPK/ERK) signaling pathways caused by HFD (55). Interestingly, T2 suppresses hepatic lipid accumulation by downregulating the expression of lipogenic proteins (85). T2 also seems to have a favorable safety profile compared with T3, although it suppresses the axis by decreasing serum TSH and T3/T4 in rodents in a dose-dependent manner (64,146,147). However, no change in the HPT axis was observed in two patients given T2 for 3 weeks, thus, it is not known whether this suppression also occurs in humans (148). Finally, a synthetic T2 mimetic, TRC150094, reduced hepatic steatosis and increased mitochondrial respiration in obese Zucker rats and rats fed HFD and did not cause any changes in the axis (149,150). However, a phase II clinical study with TRC150094 in obese men with mild steatosis failed to show a beneficial effect on intrahepatic lipid content measured by 1H-MRS after 4 weeks of treatment (151). In this study, there was a small but significant increase in serum free thyroxine (fT4), but no changes in serum TSH or free triiodothyronine (fT3) were observed. Taken together, the foregoing studies suggest that T2 improves NAFLD in animal models; however, it remains to be seen whether these beneficial effects are able to be translated to humans and whether there may be some additional side effects that need to be further considered.
Finally, there has been only limited study of the TH metabolite Triac in NAFLD. Cable et al. showed that the administration of Triac was equally effective as T3 as well as the THRβ receptor agonists GC-1, and MB03744 for lowering intrahepatic TAGs in rats (135). No effects on the HPT axis were discussed in this study.
TH, TH Analogs, and TH Metabolites in Hypercholesterolemia
Hypercholesterolemia
Hypercholesterolemia is a key clinical manifestation of metabolic syndrome that occurs with high prevalence in developed countries (World Health Organization 2008 global prevalence of raised total cholesterol). Clinical studies have shown that inappropriately increased cholesterol biosynthesis and/or clearance are major contributing factors for hypercholesterolemia. Notably, hypercholesterolemia increases the risk for atherosclerosis and ischemic heart disease (152).
Atherosclerosis is due to the deposition of oxidized LDL-C that damages the endothelial cell wall to induce inflammation, recruitment of macrophages (to remove oxidized cholesterol), and smooth muscle cell proliferation (152). Over time, this process leads to atherosclerotic plaque formation and causes narrowing of the affected blood vessel lumen (152). Statins currently are the major class of drugs used to treat hypercholesterolemia and reduce serum cholesterol levels by inhibiting the activity of HMGCR, the key rate-limiting enzyme in cholesterol biosynthesis. This leads to a decrease in intrahepatic cholesterol concentration and stimulation of LDLR expression by the SREBP2/INSIG pathway, leading to lower serum cholesterol levels (153). Despite their efficacy and safety, some patients still experience side effects or resistance to statins (154); thus, there is strong clinical and research interest in developing novel drugs that can decrease cholesterol biosynthesis and/or increase clearance in the liver.
Effects of TH on hypercholesterolemia
Clinical studies have shown that hyperthyroid patients have decreased serum cholesterol levels, and hypothyroid patients have elevated levels (155). With the discovery of the inverse relationship between TH status and serum LDL-C in the early 1950s, early intervention studies using L-T4 and D-T4 significantly lowered LDL-C in humans (156,157). However, it soon became apparent that clinical use of these TH compounds to treat hypercholesterolemia was not feasible due to their serious adverse effects on the heart, bone, and muscle (158). Recently, a TH and glucagon conjugate induced a greater decrease in serum cholesterol when compared with equimolar doses of GC-1 and KB-2115 (130). As mentioned previously, side effects of this compound appeared to be less than T3 monotherapy due to relative selectivity for the liver.
TH analogs for hypercholesterolemia
Despite these considerations, the promising results of TH led to the development of other liver-selective compounds (e.g., L-94901, CGH-509A, CGS-23425, and T-0681) that showed efficacy in lowering serum levels of LDL-C in animal studies (138,159). GC-1 (sobetirome) is the first-generation THRβ agonist that lowers serum cholesterol and TAG levels in animal models of obesity, and it reduces aortic artery plaque formation in apolipoprotein E (APOE)-deficient mice and acts in an LDLR-independent manner (70,160,161). Other THRβ-selective thyromimetics, KB-141 (162) and KB2115 (163) (eprotirome), decrease plasma levels of cholesterol in both rodents and primates, primarily through its stimulation of RCT. The liver-selective prodrug, MB07811, is effective in reducing serum levels of LDL-C and total cholesterol in rabbits, dogs, and monkeys (139,164).
In humans, a phase I clinical study with GC-1 (sobetirome) reduced serum levels of LDL-C by more than 40% in healthy participants (165). Also, KB2115 (eprotirome) further decreased serum levels of LDL-C, TAGs, and lipoprotein(s) in patients with familial hypercholesterolemia, but its clinical development was discontinued due to induction of liver enzymes and cartilage/bone side effects in dogs (163,166). In both studies, there were decreases in serum fT4, but there were no changes in TSH levels. Currently, both MB07811 and MGL-3196 are undergoing phase II trials for the treatment of hypercholesterolemia (167,168).
TH metabolites as therapy for hypercholesterolemia
The TH metabolite, T2, can reduce serum cholesterol in rodents (80), possibly via non-LDLR-mediated pathways (64). There has been only one small pilot study of T2 in two human subjects, and it showed that T2 reduced serum cholesterol (148). Although cholesterol-lowering effects of Triac have not been observed in animals, human studies have shown reduction in serum cholesterol levels in euthyroid and hypothyroid patients and reduction in serum TAG levels in hypothyroid subjects (11,169). Even though the cholesterol-lowering effects of Triac may be more potent than T3, the use of Triac in hypercholesterolemia has been limited by its more pronounced side effects on bone and a short half-life requiring dosing multiple times per day.
The TH metabolite, 2,5-diiodothyropropionic acid (DITPA), was originally developed as a potential therapy for congestive heart failure (CHF) due to its inotropic effects. In human studies, decreased serum levels of total cholesterol, LDL-C, and TAGs in patients with CHF treated with DITPA were observed (170). However, this drug has not been further pursued clinically due to intolerable side effects (especially bone), suppression of TSH, and questionable clinical benefit (171). T1AM significantly decreases body weight and serum cholesterol but increases serum TAG in spontaneously overweight female (CD-1) mice that were treated for 7 days (90). In a model of polycystic ovary syndrome liver, TAG and cholesterol were decreased by T1AM, an effect that should be further investigated in an established NAFLD model (172).
In conclusion, TH regulates hepatic and systemic cholesterol and lipid homeostasis through its effects on their metabolism, circulating lipoprotein levels, and intrahepatic TAG and cholesterol concentration. TH also regulates the expression and activities of key enzymes involved in hepatic lipid anabolic and catabolic pathways. Interestingly, defects in intrahepatic deiodinase expression and lower intrahepatic TH concentration may be hallmarks of NASH and suggest that treatment with TH, TH metabolites, or TH mimetics as lipid-modifying drugs may reduce hepatosteatosis, inflammation, and fibrosis in NAFLD. TH also lowers serum cholesterol by virtue of its effects on cholesterol synthesis, LDLR expression, and RCT. From a clinical point of view, it can be argued that patients with hepatosteatosis or hypercholesterolemia in the context of subclinical hypothyroidism may benefit from levothyroxine supplementation to ameliorate these abnormalities. Careful clinical studies will need to be performed to determine whether these interventions will indeed improve the clinical outcomes of such patients. Additionally, there is a need for liver-specific markers of TH status to assess intrahepatic TH action, which may be a better determinant of proper dosing for NAFLD than serum TH and TSH levels. It is also possible that the expression of these markers may change during the course of the disease. Identifying the appropriate liver serum markers for TH action as well as inflammation and fibrosis could be useful in determining the type of patients and stage of NAFLD for which TH or TH agonist treatment would be most appropriate.
Concerning strategies to treat NAFLD and cholesterol with TH, TH metabolites, or TH analogs, the biggest challenge is limiting their side effects while achieving maximal beneficial effect on the liver. Strategies designed to achieve this aim include tissue- or cell-specific delivery of such compounds to the liver, cell-specific activation of prodrugs, preferential uptake by the liver, or isoform-specific activation of THRβ expressed in the liver. TH metabolites or mimetics that have one or more of these properties will likely limit the extrahepatic side effects of the drug, particularly in tissues such as the bone and heart that express primarily THRα and are prone to the side effects of hyperthyroidism. However, it should be noted that specific compounds might specifically address noncanonical pathways mediated by THRs. It is also important to point out that TH analogs and metabolites may have variable effects in suppressing the axis and that careful consideration of clinical parameters, for example, tachycardia, weight loss, heat intolerance, and tremor, as well as serum markers of hepatic TH action such as SHBG should be considered in clinical studies and may be useful in assessing the thyroid status once patients are taking such drugs. Another important clinical consideration is when to treat patients, particularly given the wide range of effects of TH on lipid metabolism, autophagy, mitochondrial turnover, ceramide metabolism, and even fibrosis (156). Clinical studies for NAFLD are needed to examine the ability of TH metabolites or mimetics to prevent the progression of NASH as well as their abilities to reverse established NASH. Likewise, it remains to be seen whether TH-like compounds can be used as monotherapy for hypercholesterolemia or should be given in combination with statins. In summary, given the important actions of TH in maintaining metabolic homeostasis within the liver and systemically, selective hepatic targeting with TH metabolites and analogs may represent a novel and exciting therapy for the treatment of liver-associated metabolic diseases, such as NAFLD and hypercholesterolemia.
Footnotes
Author Disclosure Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding Information
The authors are thankful to the funding agencies (Ministry of Health, Ministry of Education, and Ministry of Trade, Singapore, and Agency for Science, Technology and Research [A*StaR]) for the grants CIRG/1340/2012, National Medical Research Council (NMRC)/Clinician Scientist Award (CSA) Grant/MH95:03/1–8 (awarded to P.M.Y.), Khoo Postdoctoral Fellowship Award (awarded to E.B.), NMRC/OFYIRG/0002/2016 (awarded to B.K.S.), and Wellcome Trust/DBT India Alliance-Intermediate Fellowship IA/I/16/2/502691 (awarded to R.A.S.).