
Abstract
The thyroglobulin (TG) gene encodes a protein required for thyroid hormone synthesis and iodine storage. Deleterious TG mutations produce congenital hypothyroidism (CH) often presenting with undetectable serum TG. Alu elements, common throughout the human genome, have a poly(dA) region in the 3′ end of the strand. Herein two of four siblings of a consanguineous Sudanese family with CH, goiter, high initial serum thyrotropin, and undetectable TG were found to have a novel frameshift insertion of an Alu element within an exon of the TG gene: c.7909ins p.Y3637Ffs. This report demonstrates a novel Alu element insertion within TG causing CH.
Introduction
The thyroglobulin (TG) gene is crucial for thyroid hormone (TH) synthesis. Consisting of 48 exons, a properly formed and folded TG protein is necessary to form THs, thyroxine (T4) and triiodothyronine (T3), as well as for storage of iodine (1,2). Deleterious mutations reported in TG are associated with congenital hypothyroidism (CH), usually presenting with a goiter and occur along three regions of TG as well as the ChEL domain that is important for proper protein structure and function (1,2). Such mutations previously reported include missense, splicing, single nucleotide insertions or deletions, multiple and large nucleotide deletions, and an imperfect DNA inversion (1,2).
Alu elements are the most common type of transposable elements in humans and have been well documented with a distinctive poly(dA) region at the 3′ end of the strand with varying length (3). Most documented Alu elements are ∼300 bp in length, with a wide variety of disease associations including hemophilia, breast cancer, and cystic fibrosis (4). We present a family with CH due to the insertion of an Alu element within an exon of the TG.
Methods
Written informed consent was obtained from all patients and from their parents for those under the age of 18 years. Genomic DNA from whole blood was extracted using Qiagen QIAamp® DNA Blood Mini Kit (Hilden, Germany) for each patient and blood was also drawn for thyroid function tests (TFTs). TFTs were performed on the Immulite 1000® (Siemens, Munich, Germany), and whole-exome sequencing (WES) (Agilent SureSelect Human All Exon V6 Kit; Novogene) was used to analyze and identify potential causative mutations. The mutation was confirmed using Sanger sequencing (Abi 3730xl DNA Analyzer; Genewiz) to determine the genotype of each family member. Forward primer sequence of GAAGGGAAAGTCTTGGTTTTGG and reverse primer sequence of CAGTGGCTGGTGATTATAGTGTC were used to confirm the mutation by Sanger sequencing.
Case Report
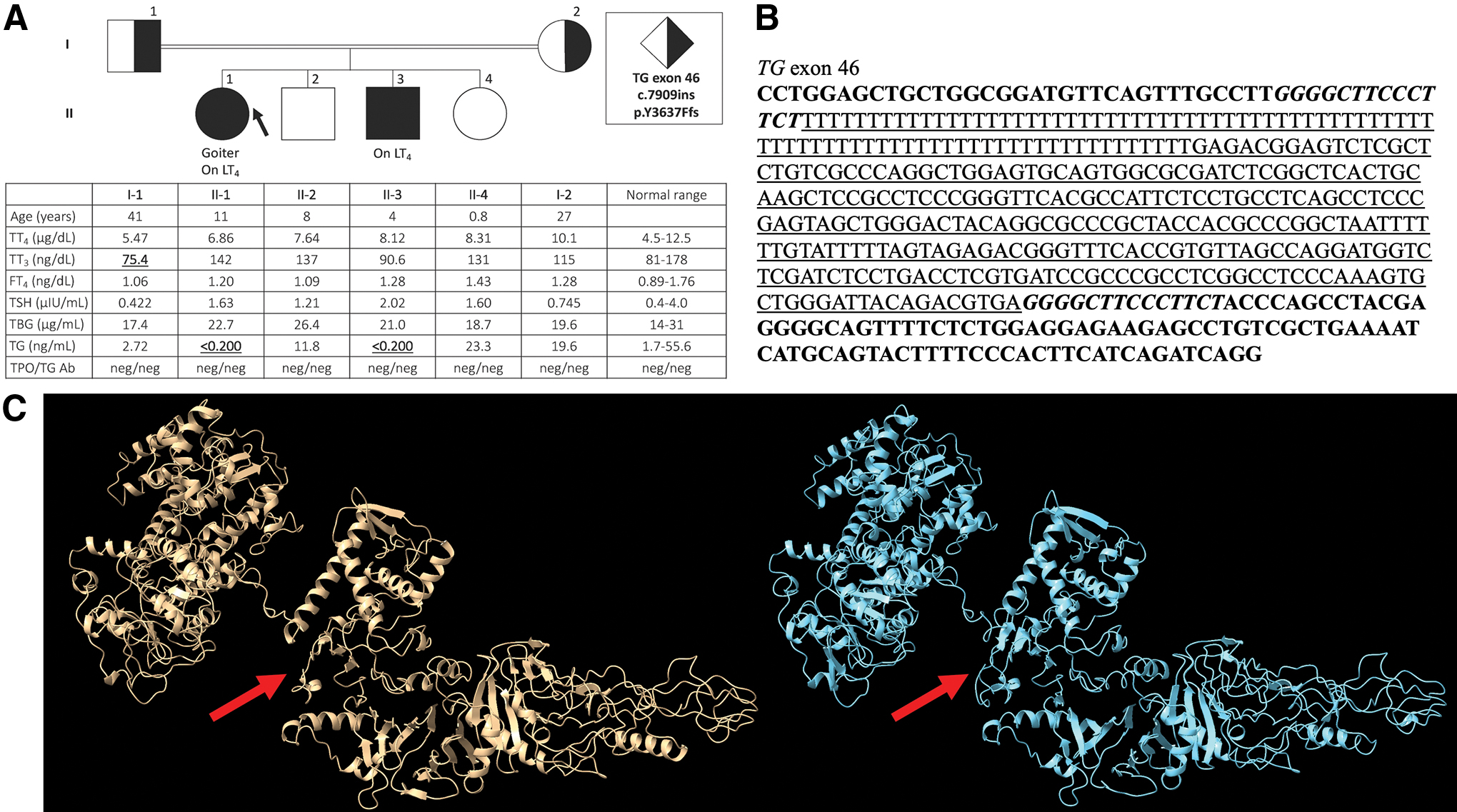
The proband was a Sudanese female born to consanguineous parents. The diagnosis of CH was delayed until age 3 years when she presented with goiter and high thyrotropin (TSH) levels. After starting levothyroxine (LT4) treatment, her TSH normalized. At the age of 11 years, both T4 and T3 normalized with treatment (Fig. 1), but the goiter persisted, and TG levels were undetectable. Her younger brother was diagnosed at 40 days of age because of the high clinical suspicion of CH and confirmed to have an elevated serum TSH. Similar to his sister, TSH normalized on LT4 treatment, but TG levels were undetectable. Both parents and two other siblings were unaffected and reported no thyroid issues (Fig. 1A). WES demonstrated a novel frameshift insertion of 345 bp along with a duplication of 14 bp, c.7909ins p.Y3637Ffs, in exon 46 of the TG gene (Fig. 1B). Search of the database Dfam revealed close homology of 83.2% for the insertion to the AluYk2 family. The proband and her affected brother were confirmed to be homozygous for this novel mutation, whereas both parents were found to be heterozygous and the two unaffected siblings were wild type (Supplementary Fig. S1).
(
Discussion
TG has previously been linked to many cases of CH (1,2) and there is one instance of an Alu element being reported in the TG gene (5). However, no previous cases involving an Alu element linked to CH have been reported as the previous Alu element reported within TG was found within an intron (5). The Alu element present in this case occurs within an exon and is marked by a poly(dT) tract from the 5′ end to the 3′ end that is ∼81 bp in length. Following the poly(dT) tract, 264 bp are also inserted and a duplication of 14 bp is also present before the wild-type sequence ensues [Fig. 1B, C (6 –8)]. This insertion within an exon of TG would be expected to significantly alter the protein shape and folding due to its location within the important ChEL domain, the large number of amino acids inserted, and premature termination (Fig. 1C). This alteration of TG is illustrated on the TFTs (Fig. 1A), wherein both children homozygous for the mutation have undetectable levels of TG. This finding is similar to previous reports of deleterious TG mutations causing the protein to undergo a different confirmation in the endoplasmic reticulum, ultimately trapping it within the endoplasmic reticulum and rendering it inefficient due to inability to synthesize TH and store iodine (9). This report documents the first case of CH caused by insertion of an Alu element within an exon of TG crucial for proper TH synthesis.
Footnotes
Acknowledgments
The authors thank Prof. Gilbert Vassart, Université libre de Bruxelles, for arranging access to the samples.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by funds from the Esformes Thyroid Research Fund, Evelyn Rosenfield, and National Institutes of Health grant MD010722 to R.W., grant DK15070 to S.R., and grant DK110322 to A.D.
Supplementary Material
Supplementary Figure S1