
Abstract
Lyme disease is considered as one of important tick-transmitted zoonosis in northeastern China, where the causative agents, the Borrelia burgdorferi sensu lato (s.l.) complex, remain poorly characterized. The purpose of the present study was to determine the prevalence and genospecies of B. burgdorferi s.l. in ticks in northeastern China. In May, 2015, a total of 2785 unfed adult ticks were collected in the Jilin and Heilongjiang provinces of northeastern China, with the predominant tick species of Ixodes persulcatus (59.9%), followed by Haemaphysalis concinna (14.8%), Haemaphysalis longicornis (8.9%), Dermacentor nuttalli (9.4%), and Dermacentor silvarum (7.0%). Only I. persulcatus was tested positive for Borrelia spirochetes DNA by PCR, targeting the 5S-23S rRNA intergenic spacer and 16S rRNA genes, with a prevalence of 1.9%. Phylogenetic analysis based on the partial 5S-23S rRNA intergenic spacer and 16S rRNA genes showed that these positive samples were grouped into four pathogenic genospecies for humans, including Borrelia garinii (2.8%), Borrelia afzelii (0.2%), Borrelia bavariensis (0.1%), and Borrelia bissettii (0.1%). These results showed that B. garinii is the predominant genospecies and I. persulcatus is the main tick host and carrier in northeastern China. To our knowledge, B. bissettii were detected for the first time in China.
Introduction
Lyme borreliosis (LB), caused by different genospecies of the Borrelia burgdorferi sensu lato (s.l.) complex, is an important emerging tick-borne zoonosis in Asia, Europe, and North America, which can lead to various clinical symptoms, ranging from relatively benign skin lesions to severe cardiac, arthritic, and neurologic manifestations (Rudenko et al. 2011). There are a number of B. burgdorferi s.l. genospecies, many of them related to human LB (Strnad et al. 2017). The disease is caused predominantly by B. burgdorferi sensu stricto (s.s.), Borrelia afzelii, Borrelia garinii, and Borrelia bavariensis (Margos et al. 2009). Additionally, other genospecies Borrelia bissettii (Rudenko et al. 2009), Borrelia lusitaniae (Collares-Pereira et al. 2004), Borrelia spielmanii (Fingerle et al. 2008), and Borrelia valaisiana (Diza et al. 2004), are occasionally detected in humans; however, their pathogenicity is still unclear (Stanek et al. 2011). Of these, three genospecies are primarily responsible for human diseases in different geographical regions, that is, B. burgdorferi s.s. is the primary pathogen of LB in the United States (Dunn et al. 2014), whereas B. garinii and B. afzelii cause LB in Asia and Europe (van Duijvendijk et al. 2016). Infections with different genospecies may lead to different clinical manifestations: B. burgdorferi s.s. especially results in arthritogenic infections, B. garinii is primarily neurotropic, and B. afzelii particularly causes skin lesion (Hasle et al. 2011).
In China, since the first LB case was described in the Heilongjiang province in northeastern China in 1986 (Takada et al. 1998), six genospecies, including B. garinii, B. afzelii, B. burgdorferi s.s., B. valaisiana, Borrelia sinica, and B. valaisiana-related genospecies, have been identified, of which B. garinii, B. afzelii, and B. valaisiana-related genospecies are associated with human diseases (Fang et al. 2015, Zhai et al. 2017). The major endemic areas in China include the northeastern forest regions, where over 3 million people are estimated to be at risk of tick bite annually, and ∼30,000 people suffer from LB (Wu et al.
Materials and Methods
Ticks collection
In May, 2015, ticks were collected by flagging vegetation in forestry regions of northeastern China, where the forest coverage rate reaches 40% and a forestry area accounts for 15% of China, thus, there are abundant wild animal resources and a wide range of natural focal diseases. The sampled ticks were identified by the morphological classification (Chen et al. 2010), and confirmed by PCR, targeting the 16S ribosomal RNA as described previously (Liu et al. 2016).
Detection of Borrelia spirochetes DNA by PCR
Ticks were pooled, ∼12 ticks per pool, based on the tick species and sampling sites. The genomic DNA of each pool was extracted using the TIANamp Genomic DNA Kit (Tiangen, Co., Ltd, Beijing,
Phylogenetic analysis
The PCR products were sequenced in both directions with the specific primers, and the obtained sequences were analyzed by BlastN and aligned with ClustalW (Thompson et al. 1994). The aligned sequences (GenBank® accession nos: MG557639–MG557646, MK121659–MK121673) were further analyzed by comparing with the sequences of different genospecies (Supplementary Table S1). We included 20 different genospecies of Borrelia from different hosts or geographic origin in phylogenetic analyses using the software MEGA version 5.1. The neighbor-joining method was used to generate a phylogenetic tree. The reliability of branches in the tree was evaluated by bootstrapping analysis with 1000 replicates, and the bootstrap value >60% was considered significant. The relapsing fever Borrelia is the closed common ancestor for B. burgdorferi s.l., thus, Borrelia hermsii, the pathogen of relapsing fever, was used as outgroup for the rooted tree in the study (Fukunasa et al. 1996, Pritt et al. 2016).
Statistical analyses
The prevalence of B. burgdorferi s.l. infection in ticks was calculated by using the program PooledInfRate Excel Add-In version 4.0 (Biggerstaff 2005). Infection rates for pooled ticks were analyzed using maximum likelihood estimation (MLE) method with 95% confidence intervals (CIs) with one-sample analysis for unequal pool sizes and expressed as the MLE of infection rate per 100 ticks. MLE calculations are based on the number of pools, pool sizes (number of individuals per pool), and the number of positive pools (Hepworth and Murtagh 2005). Statistical analyses were performed using SPSS version 17.0 (SPSS, Inc., Chicago, IL). p value <0.05 was considered statistically significant.
Results
Totally, 2785 ticks were collected in the Jilin and Heilongjiang provinces, including 215 Dermacentor nuttalli, 166 Dermacentor silvarum, 101 Haemaphysalis longicornis, and 393 Ixodes persulcatus in Jilin, as well as 47 D. nuttalli, 29 D. silvarum, 146 H. longicornis, 1276 I. persulcatus, and 412 Haemaphysalis concinna from Heilongjiang, respectively. Except for the absence of H. concinna in Jilin, all the tick species were found in both provinces (Table 1). The predominant tick species in northeastern China was I. persulcatus (59.9%), followed by H. concinna (14.8%), H. longicornis (8.9%), D. nuttalli (9.4%), and D. silvarum (7.0%).
Distribution of Tick Species in Jilin and Heilongjiang Provinces, Northeastern China
CI, confidence interval; nd, no data of this tick species.
A total of 45 tick pools out of 1669 I. persulcatus (1.9%, 95% CI: 1.4–2.4%) were tested Borrelia positive, whereas other tick species were negative. To identify the Borrelia species in these positive samples, partial 5S-23S rRNA ITS (330 bp) and 16S rRNA (514 bp) gene sequences were further obtained and sequenced (GenBank accession numbers: MG557639–MG557646, MK121659–MK121673). After BLAST on the NCBI website, sequences in the GenBank database most similar to the query sequences were retrieved and used for phylogenetic analysis, revealing that these sequences could be clustered into four clades, belonging to B. garinii, B. afzelii, B. bissettii, and B. bavariensis, respectively (Figs. 1 and 2).
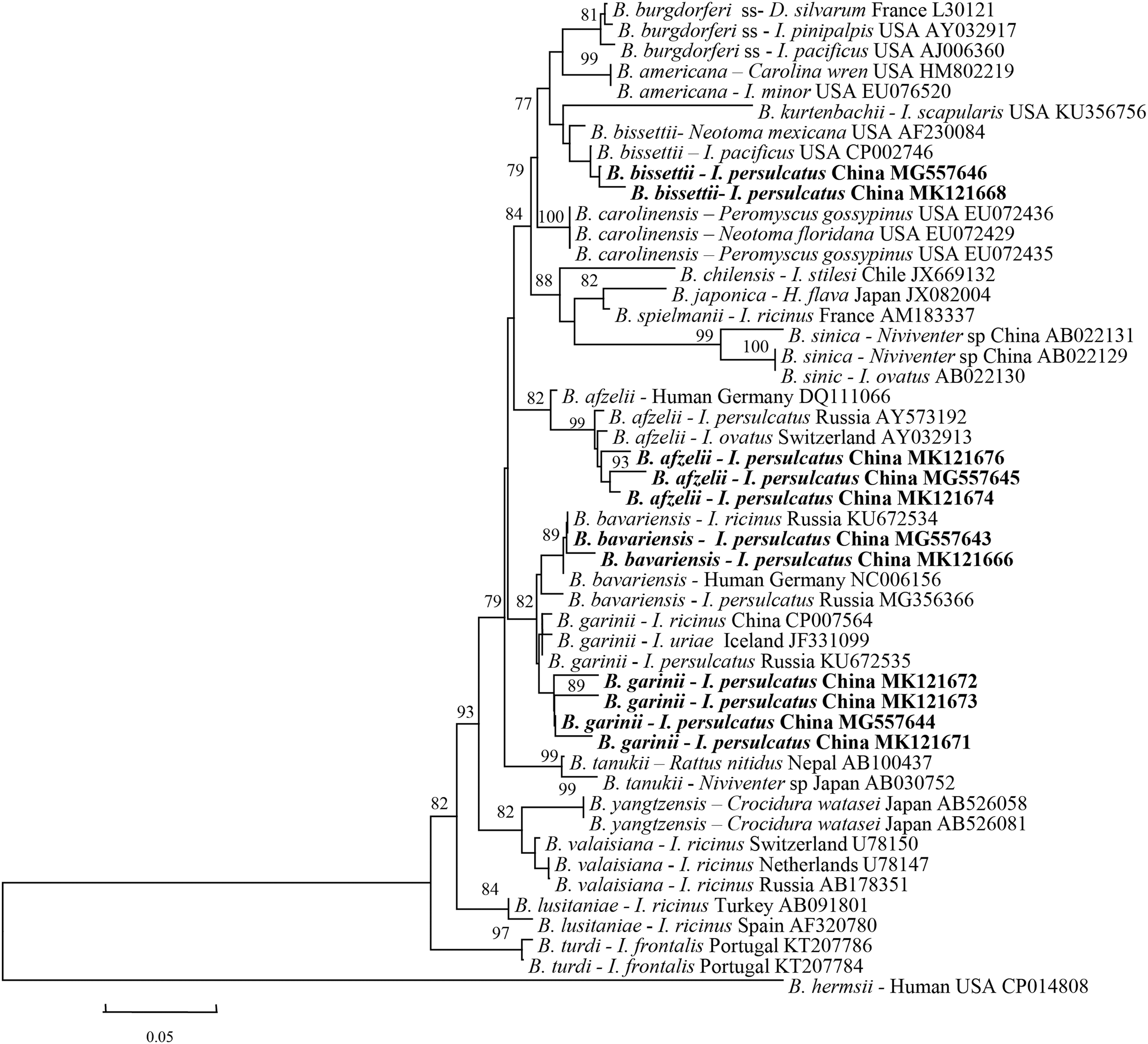
Phylogenetic analysis of the partial 5S-23S rRNA ITS (330 bp) from Borrelia in Ixodes persulcatus of northeastern China. The phylogenetic trees were constructed by the Neighbor-Joining method using the Kimura's 2-parameter model. A total of 300 positions were included in the final analysis. Sequences are identified by their strain name and the origin, followed by the GenBank accession number. The partial 5S-23S rRNA gene ITS of Borrelia hermsii was used as outgroup. The detected Borrelia sequences in this study are marked in bold. The scale bars indicate 0.05 substitutions per site. ITS, internal transcribed spacer.

Phylogentic analysis of the partial 16S rRNA gene (514 bp) from Borrelia in I. persulcatus from northeastern China. The phylogenetic trees were constructed by the Neighbor-Joining method using the Kimura's 2-parameter model. A total of 500 positions were included in the final analysis. The partial 16S rRNA gene ITS of B. hermsii was used as outgroup. Sequences are identified by their strain name and the origin, followed by the GenBank accession number. The detected Borrelia sequences in this study are marked in bold. The scale bars indicate 0.05 substitutions per site.
The genospecies distribution of B. burgdorferi s.l. complex showed the obvious regional differences. For example, B. bissettii was detected in Yichun of Heilongjiang, B. bavariensis was found in Dunhua of Jilin and Suifenhe of Heilongjiang, whereas B. afzelii was detected in Dunhua of Jilin, and Jiamusi and Suifenhe of Heilongjiang (Table 2).
Detection of Borrelia DNA in Ixodes persulcatus by Nested PCR from Jilin and Heilongjiang Provinces, Northeastern China
The prevalence of infection in ticks was calculated by MLE using the program PooledInfRate.
The prevalence in Suifenhe was expressed as the minimum infection rate, as the MLE is not applicable for calculation of the infection rate when all pools are positive.
MLE, maximum likelihood estimation.
The infection rate of B. burgdorferi s.l. in I. persulcatus by MLE in Jilin and Heilongjiang provinces was estimated to be 2.7% (95% CI: 1.3–4.9%) and 3.4% (95% CI: 2.4–4.1%), respectively. B. garinii was shown to be the predominant spirochete species in both Jilin and Heilongjiang provinces, with a prevalence of 2.0% (95% CI: 0.9–3.9%) and 2.8% (95% CI: 2.0–3.8%), respectively. These results demonstrated that I. persulcatus may be the main host and carrier for the Lyme disease, and four genospecies, including B. garinii (2.8%, 95% CI: 2.0–3.8%), B. afzelii (0.2%, 95% CI: 0–0.5%), B. bavariensis (0.1%, 95% CI: 0–0.4%), and B. bissettii (0.1%, 95% CI: 0–0.4%), were found in northeastern China. No coinfection of genospecies was detected in the pooled samples, which may be associated with low infection rates in this study.
Discussion
LB spirochetes is transmitted by infected ticks of the genus Ixodes (Rudenko et al. 2011). In China, I. persulcatus is the principal vector in the north, whereas I. granulatus and I. sinensis are the main vectors in the south (Wu et al. 2013). The infection rates are variable from many geographic locations in China, ranging from 19.44% in I. granulatus in Zhejiang (Hou et al. 2015), 25.6% in I. granulatus in Guizhou (Geng et al. 2010), to 33.8% in I. persulcatus in Heilongjiang (Cao et al. 2003). The principal vector of Europe is Ixodes ricinus, whereas the main vector in North America is Ixodes scapularis (Steere et al. 2016). Different infection rates are also observed in different countries, including 36% in I. scapularis in Canada (Scott et al. 2017), 33% I. scapularis in Southwestern Virginia (Herrin et al. 2014), 34.5% of I. persulcatus and 19.5% in Ixodes ovatus in Japan (Murase et al. 2013), 58% in I. ricinus in Poland (Hubalek and Halouzka 1998), 19.5% in I. ricinus in Germany (Schwarz et al. 2012), and 7.9% in I. ricinus in Italy (Aureli et al. 2015). Furthermore, the pathogen has also been reported in other unexpected vector, H. longicornis in China and Japan, with a low prevalence of 5.2–7.7% (Murase et al. 2013, Hou et al. 2015).
A total of six genospecies, including B. garinii, B. afzelii, B. burgdorferi s.s., B. valaisiana, B. sinica, and B. valaisiana-related genospecies, have been identified in China (Chu et al. 2008). Of these, B. garinii, B. afzelii, and B. valaisiana-related genospecies have been found in humans in Heilongjiang province (Wu et al. 2013; Liu et al. 2012), and B. garinii and B. burgdorferi s.s. have been detected in sika deer (Cervus nippon) from Jilin Province (Zhai et al. 2017). B. garinii and B. afelii have been detected in I. persulcatus in Liaoning and Inner Mongolia, which resemble those of Borrelia isolates from Japan and Far Eastern Russia (Li et al. 1998, Takada et al.
Previous reports showed that B. burgdorferi s. l. infection rates in I. persulcatus ranged from 2.1% to 33.8% in Heilongjiang during 2003–2011 (Cao et al. 2003, 2011). The present study showed a 10-fold difference compared with that of I. persulcatus (33.8%) in Heilongjiang (Cao et al.
In previous studies, the strain of B. bavariensis was termed B. garinii strain PBi (Baranton et al. 1992). The obtained B. bavariensis16S rRNA and 5S-23S rRNA ITS gene sequences were 99–100% identical to the PBi strain (GenBank accession numbersNR074854 and CP000013). B. bavariensis is widely distributed in Europe and Asia (Margos et al. 2013). European B. bavariensis strains are frequently associated with neuroborreliosis (Tveitnes and Oymar 2015). Furthermore, some related evidence in the majority of Japanese patients genetically grouped into B. bavariensis, indicated that B. bavariensis could cause LB in humans (Gatzmann et al. 2015). The Chinese strain NMJW1 from Inner Mongolia, currently assigned to B. garinii in GenBank, is genetically grouped into B. bavariensis (Margos et al. 2017).
Both common B. garinii and B. afzelii genospecies leading to LB in China has been identified in many tick species, such as I. persulcatus and H. longicornis (Zhang et al. 2014, Hou et al. 2015). However, only I. persulcatus was tested positive in this study, showing a pivotal role of the tick in transmission of LB spirochetes in northeastern China.
There were some limitations to this study. First, the low numbers of some tick species in each region may reduce the probabilities of getting positive results. For example, B. valaisiana-related genospecies has been detected in humans in Heilongjiang (Ni et al. 2014), but we did not find the pathogen in the present study. Second, the collected ticks were adult rather than larva and nymph, which brings into question whether B. burgdorferi s.l. spirochetes can be transstadially transmitted in I. persulcatus. Third, the pooled samples were unfit for detecting coinfection of different genospecies.
In summary, five tick species, including I. persulcatus, H. concinna, H. longicornis, D. nuttalli, and D. silvarum, were in the forestry regions of northeastern China, with the dominant tick species being I. persulcatus. We detected four genospecies of B. burgdorferi s.l. complex in I. persulcatus, including B. garinii, B. afzelii, B. bavariensis, and B. bissettii, in which B. bissettii was detected for the first time in China. Further studies are needed to determine the public health risk of these novel genospecies in the studied areas.
Footnotes
Acknowledgments
The authors thank Prof. Feng Wei and Prof. Mingxin Song for help in collecting tick samples. This study was supported financially by the National Key R&D Program of China (2016YFC1201602, 2017YFD0500104, and 2017YFD0501702) and the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2014-LVRI-03).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.