
Abstract
Background:
Tick-borne bacteria and protozoa can cause a variety of human and animal diseases in China. It is of great importance to monitor the prevalence and dynamic variation of these pathogens in ticks in ever-changing natural and social environment.
Materials and Methods:
Ticks were collected from Heilongjiang and Jilin provinces of northeastern China during 2018–2019 followed by morphological identification. The presence of Rickettsia spp., Anaplasma spp., Ehrlichia spp., Borrelia spp., Babesia spp., and Theileria spp. was examined by PCR and Sanger sequencing. The obtained sequences were subjected to phylogenetic analysis through Mega 7.0. Statistical analysis was performed using SPSS 24.0.
Results:
A total of 250 ticks from 5 species of 3 genera were collected. Ixodes and Haemaphysalis ticks carried more species of pathogens than Dermacentor, and the pathogens detected in Haemaphysalis japonica varied significantly among different sampling sites. The infection rates of Rickettsia spp., Anaplasma spp., Ehrlichia spp., Borrelia spp., Babesia spp., and Theileria spp. were 41.2%, 0, 2.0%, 7.2%, 1.2%, and 7.2%, respectively. Twelve pathogens were identified, among which Rickettsia raoultii (29.6%), Candidatus Rickettsia tarasevichiae (9.2%), and Theileria equi (4.4%) were the three most common ones. Rickettsia had its dominant vector, that is, R. raoultii had high infection rates in Dermacentor nuttalli and Dermacentor silvarum, Ca. R. tarasevichiae in Ixodes persulcatus, and Rickettsia heilongjiangensis in H. japonica. Interestingly, unclassified species were observed, including a Rickettsia sp., an Ehrlichia sp., a Borrelia sp., and a Babesia sp. Coinfections with different pathogens were identified in 9.2% of all tested ticks, with I. persulcatus most likely to be coinfected (23.8%) and Rickettsia spp. and Borrelia spp. as the most common combination (16.7%).
Conclusions:
The results of this study reflect high diversity and complexity of pathogens in ticks, which are useful for designing more targeted and effective control measures for tick-borne diseases in China.
Introduction
Ticks belong to the order Parasitiformes and are divided into three families, Ixodidae, Argasidae, and Nuttalliellidea, with over 900 recorded species (Barker and Murrell 2004, Guglielmone et al. 2010). As strictly hematophagous ectoparasites, ticks can transmit pathogens, such as bacteria, protozoa, and viruses, to humans and animals during feeding, causing substantial public health and veterinary problems (de la Fuente et al. 2008, Dantas-Torres et al. 2012, Fang et al. 2015). Different tick species have been shown to harbor diverse pathogenic microorganisms (Yu et al. 2015).
Tick-borne diseases are a growing threat to human health in China. Since 1982, 33 emerging tick-borne pathogens, transmitted by nearly 30 different tick species, have been reported (Fang et al. 2015). The majority of them were bacteria and protozoa, for example, Rickettsia spp., Anaplasma spp., Ehrlichia spp., Borrelia spp., Babesia spp., and Theileria spp., which can cause a variety of human and animal diseases, including rickettsiosis, anaplasmosis, ehrlichiosis, Lyme disease, tick-borne relapsing fever, babesiosis, and theileriosis (Song et al. 2018, Hou et al. 2019, Wang et al. 2019, Li et al. 2020). Rickettsia spp. were the most common pathogens among them.
Rickettsia spp. are obligate intracellular Gram-negative bacteria that can cause human rickettsioses, usually characterized by fever, headache, asthenia, anorexia, nausea, rash, and eschar at the tick-biting sites (Santibáñez et al. 2013, Fang et al. 2017). At least 10 species of tick-borne Rickettsia have been identified over the past 30 years in China, including Rickettsia raoultii, Rickettsia heilongjiangensis, and Candidatus Rickettsia tarasevichiae, the causative agents of human rickettsioses, which were widely present in the associated ticks and detected in specimens from tick-bitten patients in northeastern China (Fournier et al. 2003, Cao et al. 2008, Jia et al. 2013a, 2014, Wu et al. 2013, Liu et al. 2016).
Northeastern China has large areas of forest and various forest types, providing suitable habitats for the survival and reproduction of ticks. Dermacentor silvarum, Dermacentor nuttalli, Haemaphysalis concinna, Haemaphysalis japonica, and Ixodes persulcatus ticks, the major species known to transmit tick-borne diseases, are highly prevalent in this region (Chen et al. 2010, Yu et al. 2015, Zhang et al. 2019). The pathogens carried by ticks will change with the natural environment and socioeconomic development, which needs to be monitored continuously. Moreover, one tick species can harbor diverse pathogens, and several tick-borne diseases often coexist in the same natural foci (Labuda and Nuttall 2004). Humans and animals bitten by coinfected ticks would have more complicated conditions. However, there are currently limited studies on the cocirculation of multiple pathogens in ticks in northeastern China. In this study, we investigated Rickettsia spp., Anaplasma spp., Ehrlichia spp., Borrelia spp., Babesia spp., and Theileria spp. and their coinfections in ticks collected in northeastern China from 2018 to 2019, aiming to achieve a more comprehensive understanding of the prevalence and dynamic variation of tick-borne pathogens.
Materials and Methods
Tick collection
From April to June in 2018–2019, ticks were collected in seven counties of northeastern China, including Huanan, Saertu, Ranghulu, Acheng, Shangzhi, and Wuchang located in Heilongjiang Province, and Wangqing in Jilin Province (Fig. 1). These collection sites were selected based on their diverse landscape features ranging from the forested highlands of Changbai Mountains (Huanan and Wangqing) to low foothill areas (Acheng, Shangzhi, and Wuchang) and plains (Saertu and Ranghulu). Questing ticks were collected by flagging vegetation of woodland or grassland, and infesting ticks were collected from body surfaces of goats and cattle. All ticks were identified morphologically to species level and different developmental stages by an entomologist. They were stored at −80°C until nucleic acid extraction.
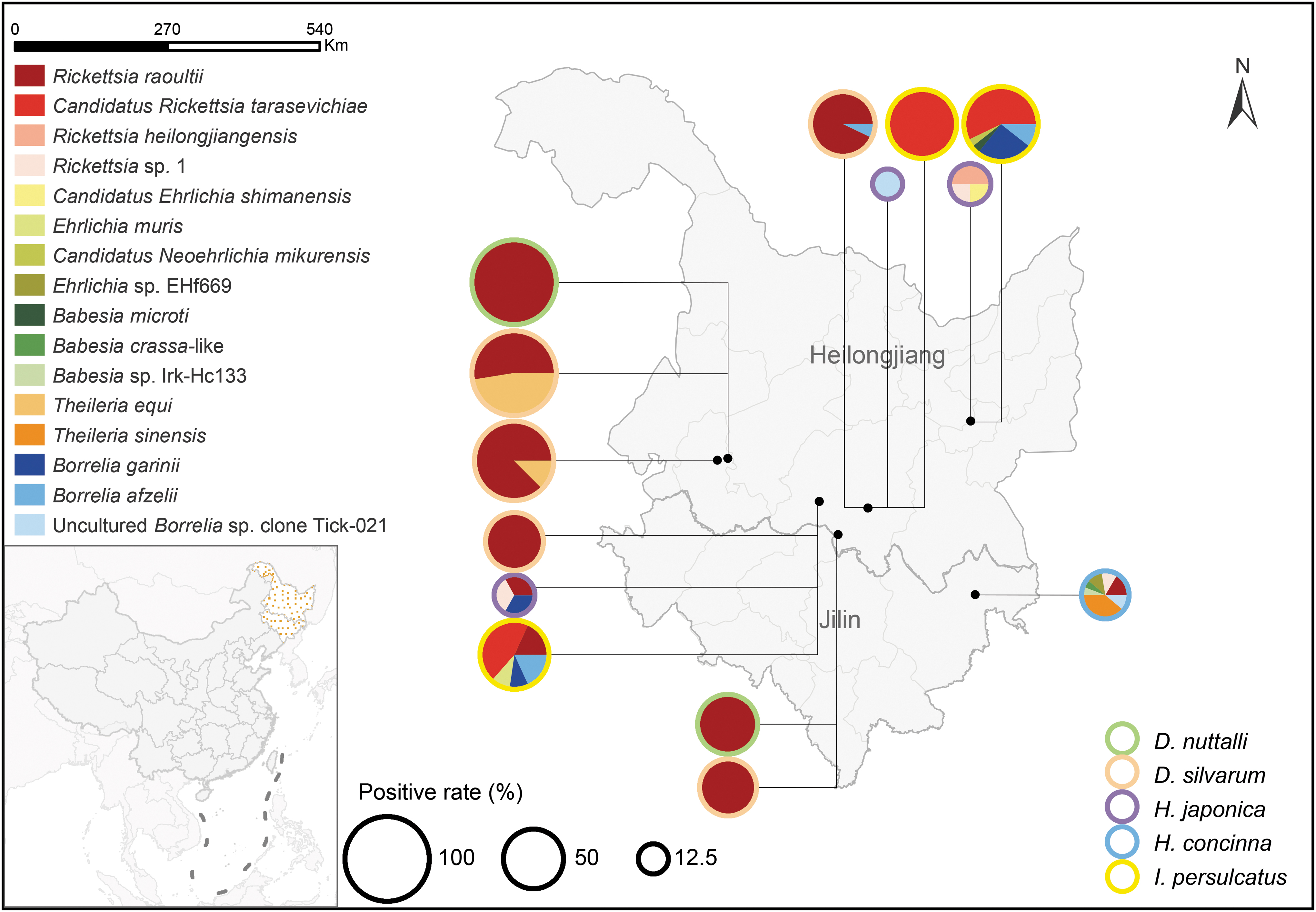
Distribution and composition of detected pathogens in five tick species from seven sampling sites in northeastern China. The color and size of the circle represent the tick species and the overall positive rate of pathogens in each tick species at each sampling site, respectively. The composition of pathogens is indicated by pie charts with different colors. Color images are available online.
Molecular examination of tick-borne pathogens
Total nucleic acid was extracted from each tick individually using a Viral RNA/DNA Extraction Kit (TaKaRa, Dalian, China) according to the manufacturer's instructions, and then used as a template in PCR assays. The primers are listed in Supplementary Table S1. Four sets of primers for amplifying the 17-kDa antigen gene (17-kDa), citrate synthase gene (gltA), 16S rRNA gene, and outer membrane protein A gene (ompA) were used to identify the species of Rickettsia (Anstead and Chilton 2013, Jia et al. 2013a). Babesia/Theileria spp. and Borrelia miyamotoi were examined by conventional PCR targeting the 18S rRNA and 16S rRNA genes, respectively (Jiang et al. 2015, 2018). Anaplasma spp. and Ehrlichia spp. were examined using seminested primers, Eh-out1/3-17 and Eh-out1/Eh-out2, targeting the 16S rRNA gene (Wen et al. 2002). Anaplasma capra and Borrelia burgdorferi sensu lato (s.l.) were examined by nested PCR targeting the gltA and 5S-23S rRNA genes, respectively (Chu et al. 2008, Li et al. 2015). PCR conditions were as described (Wen et al. 2002, Chu et al. 2008, Anstead and Chilton 2013, Jia et al. 2013a, Jiang et al. 2015, 2018, and Li et al. 2015); a blank and a negative control were included in each amplification.
Sequencing and phylogenetic analyses
All PCR-positive products were purified and then directly sequenced with a 3730 DNA Sequencer (Applied Biosystems, Foster City, CA, USA). The obtained sequences were compared with those available in GenBank using BLAST (
Statistical analysis
Differences in the prevalence of pathogens in different tick species were evaluated with SPSS software (version 24.0, Chicago, IL, USA) by Chi-square test or Fisher's exact test with Yates' correction. A p value of <0.05 was considered statistically significant.
Nucleotide sequence accession numbers
The sequences obtained in this study were deposited into GenBank with the accession numbers MT830896, MT830897, and MT843243–MT843272.
Results
Tick samples
A total of 250 ticks (180 females, 67 males, and 3 nymphs) belonging to 5 species of 3 genera were collected from Heilongjiang and Jilin provinces of northeastern China (Supplementary Table S2). The majority (233, 93.2%) of them were free living. Only a few (17) fed ticks were sampled. The species of ticks were morphologically determined as D. silvarum (102, 40.8%), H. concinna (47, 18.8%), I. persulcatus (42, 16.8%), H. japonica (38, 15.2%), and D. nuttalli (21, 8.4%), with D. silvarum as the dominant species.
Overall distribution of detected pathogens
In general, 12 pathogens and 4 sequence variants were detected from 124 ticks (49.6%) in 7 counties. The overall prevalence in I. persulcatus, D. silvarum, D. nuttalli, H. concinna, and H. japonica was 73.8%, 57.8%, 52.4%, 31.9%, and 21.1%, respectively. D. nuttalli and D. silvarum carried one and three pathogens, respectively, predominated with R. raoultii, while I. persulcatus, H. japonica, and H. concinna harbored the most varied species of pathogens (Fig. 1). The pathogens carried by H. japonica were different in Huanan (R. heilongjiangensis, Rickettsia sp. 1, and Candidatus Ehrlichia shimanensis), Shangzhi (uncultured Borrelia sp. clone Tick-021) and Acheng (R. raoultii, Rickettsia sp. 1, and Borrelia garinii) counties (Fig. 1). The specific pathogens detected in ticks were described as follows.
Characterization of the known pathogens
Rickettsia was the most frequently detected pathogen with an overall prevalence of 41.2%, and was the only pathogen identified in all five surveyed tick species (Table 1). The infection rates of Rickettsia spp. in Ixodes (59.5%, 25/42) and Dermacentor ticks (55.3%, 68/123) were almost comparable (χ 2 = 0.229, df = 1; p = 0.632), which were significantly higher than that in Haemaphysalis ticks (11.8%, 10/85) (χ 2 = 46.294, df = 2; p < 0.001). DNA of R. raoultii, Ca. R. tarasevichiae, and R. heilongjiangensis were identified. R. raoultii (29.6%, 74/250) was the most prevalent Rickettsia species and detected in all tick species (Fig. 1). The infection rates of R. raoultii in both D. silvarum (55.9%, 57/102) and D. nuttalli (52.4%, 11/21) were strikingly higher than those in other tick species. DNA of Ca. R. tarasevichiae (9.2%, 23/250) and R. heilongjiangensis (0.8%, 2/250) were only present in I. persulcatus and H. japonica, respectively (Fig. 1). The representative sequences obtained in this study were phylogenetically analyzed based on 360-nt gltA and 430-nt ompA sequences, respectively (Fig. 2a, b). R. raoultii sequences in this study were identical to the isolate N42 from a D. silvarum tick (MN550897) and a human isolate MDJ4 (JX885458). The deduced gltA gene sequences of Ca. R. tarasevichiae completely matched the sequence from I. persulcatus available in GenBank (JX996054). The gltA and ompA genes of R. heilongjiangensis were 100% identical to a human isolate MDJ5 (JX945522) and the strain HLJ-054 from ticks (AF179362), respectively.
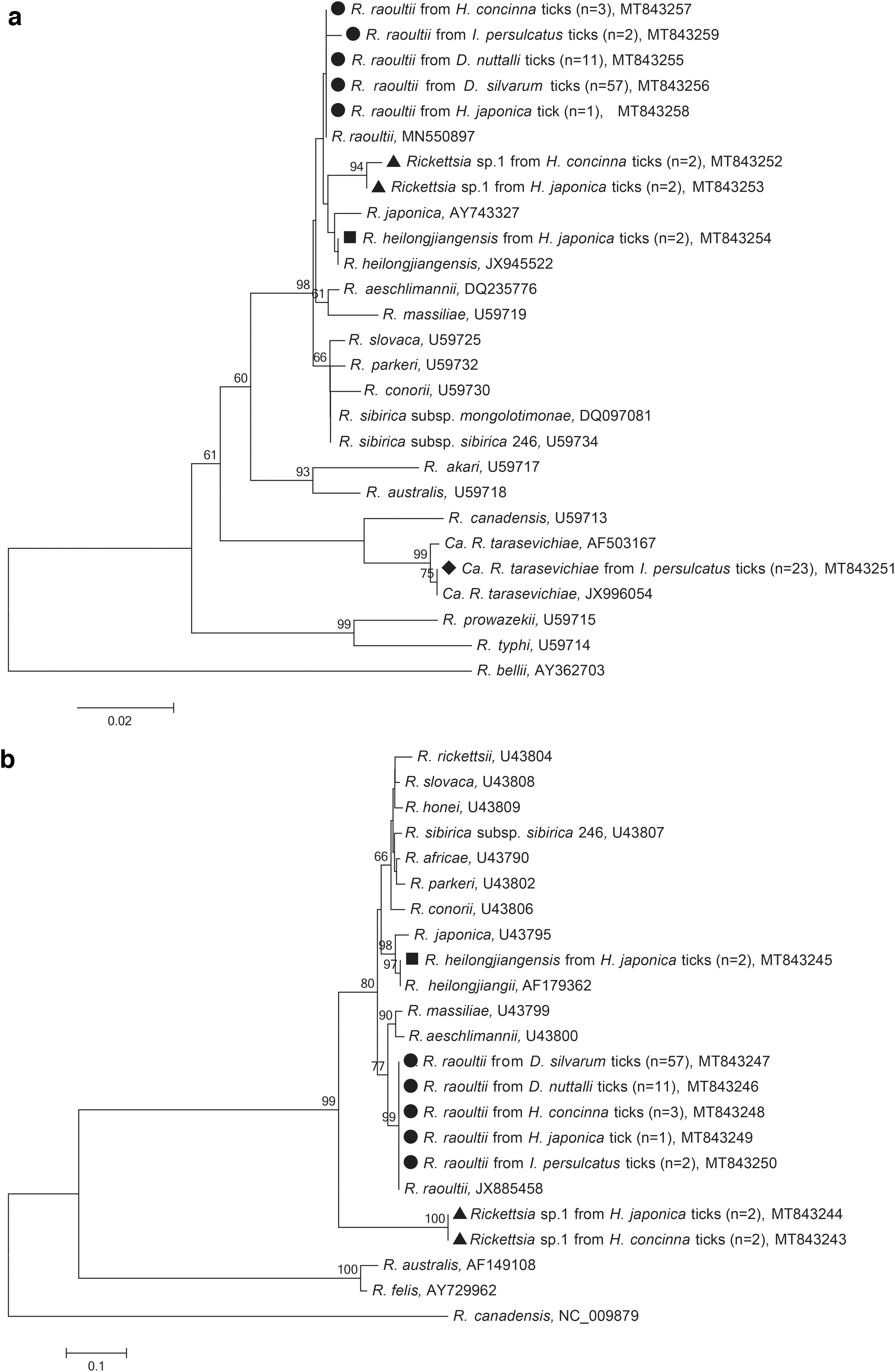
Phylogenetic analyses of Rickettsia spp. based on partial (360 nt) citrate synthase gene
Prevalence and Coinfections of Detected Pathogens in Different Tick Species
R, Rickettsia spp.; A, Anaplasma spp.; E, Ehrlichia spp.; Ba, Babesia spp.; T, Theileria spp.; B, Borrelia spp.
Significant differences in the prevalence of pathogens in five tick species (p < 0.05).
Borrelia was detected with an overall prevalence of 7.2%. Except for D. nuttalli, ticks from 4 species, including 13 (31.0%) I. persulcatus, 2 (5.3%) H. japonica, 2 (4.3%) H. concinna, and 1 (1.0%) D. silvarum were positive (Table 1). The difference in infection rates among tick species was statistically significant (χ 2 = 43.646, df = 4; p < 0.001), and the frequency of positive I. persulcatus was strikingly higher compared with the other tick species. DNA of B. garinii and Borrelia afzelii were identified. Four representative sequences obtained in this study were included in phylogenetic analysis based on 260-nt 5S-23S rRNA sequences (Fig. 3a). The sequences of B. garinii were 98.4% identical to each other and clustered with the B. garinii strains VH4 (DQ150544) and Ip-5831 (AM748065) (Fig. 3a). The detected B. afzelii sequences were 100% identical to each other and to those found in Suneus murinus (MK333413) and Mus caroli (MK333414) of China (Fig. 3a).
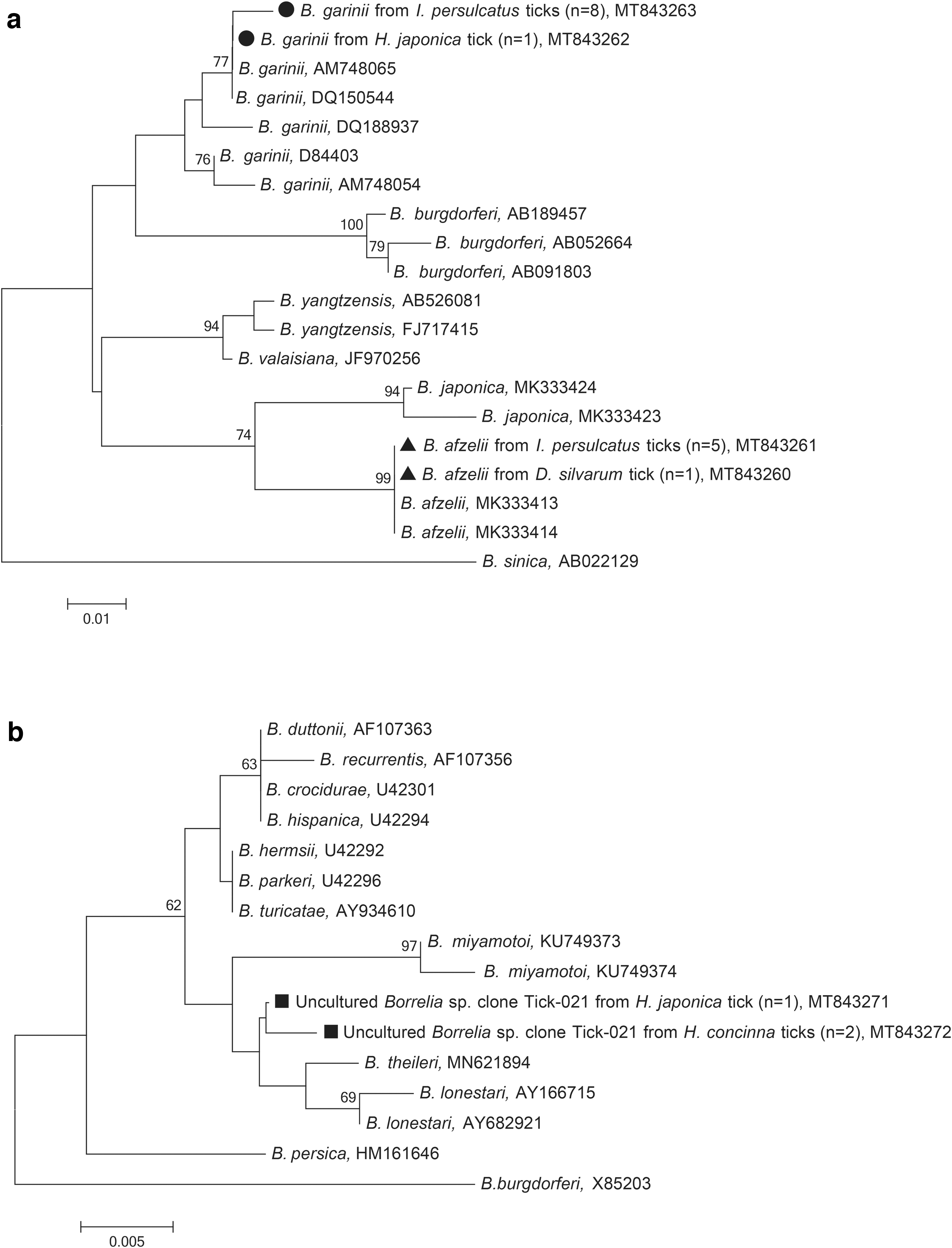
Phylogenetic analyses of Borrelia burgdorferi s.l. and Borrelia miyamotoi based on partial (260 nt) 5S-23S rRNA
Ehrlichia was detected with an overall prevalence of 2.0%. Except for Dermacentor spp., ticks from other three species, including two (4.8%) I. persulcatus, two (4.3%) H. concinna and one (2.6%) H. japonica, were positive (Table 1). The difference in infection rates among tick species was not statistically significant (χ 2 = 5.420, df = 4; p = 0.247). DNA of Ca. E. shimanensis, Ehrlichia muris, and Candidatus Neoehrlichia mikurensis were identified. The representative sequences obtained in this study were included in phylogenetic analysis based on 545-nt 16S rRNA sequences (Fig. 4a). The detected Ca. E. shimanensis sequence exhibited a close relationship with a previous strain TS37 (AB074459). The sequence of E. muris was 100% identical to those of E. muris strains detected in mouse from Japan (NR_121714) and Eothenomys kageus from the United States (NR_025962). The obtained Ca. N. mikurensis sequence was 99.8% identical to the sequences identified in Ixodes ricinus ticks from Estonia (KU535862), and in humans from China (JQ359045), which were phylogenetically distinctive from the isolates of Ca. N. mikurensis from Apodemus draco (JQ359051) and Ixodes ovatus (AB074460) (Fig. 4a).
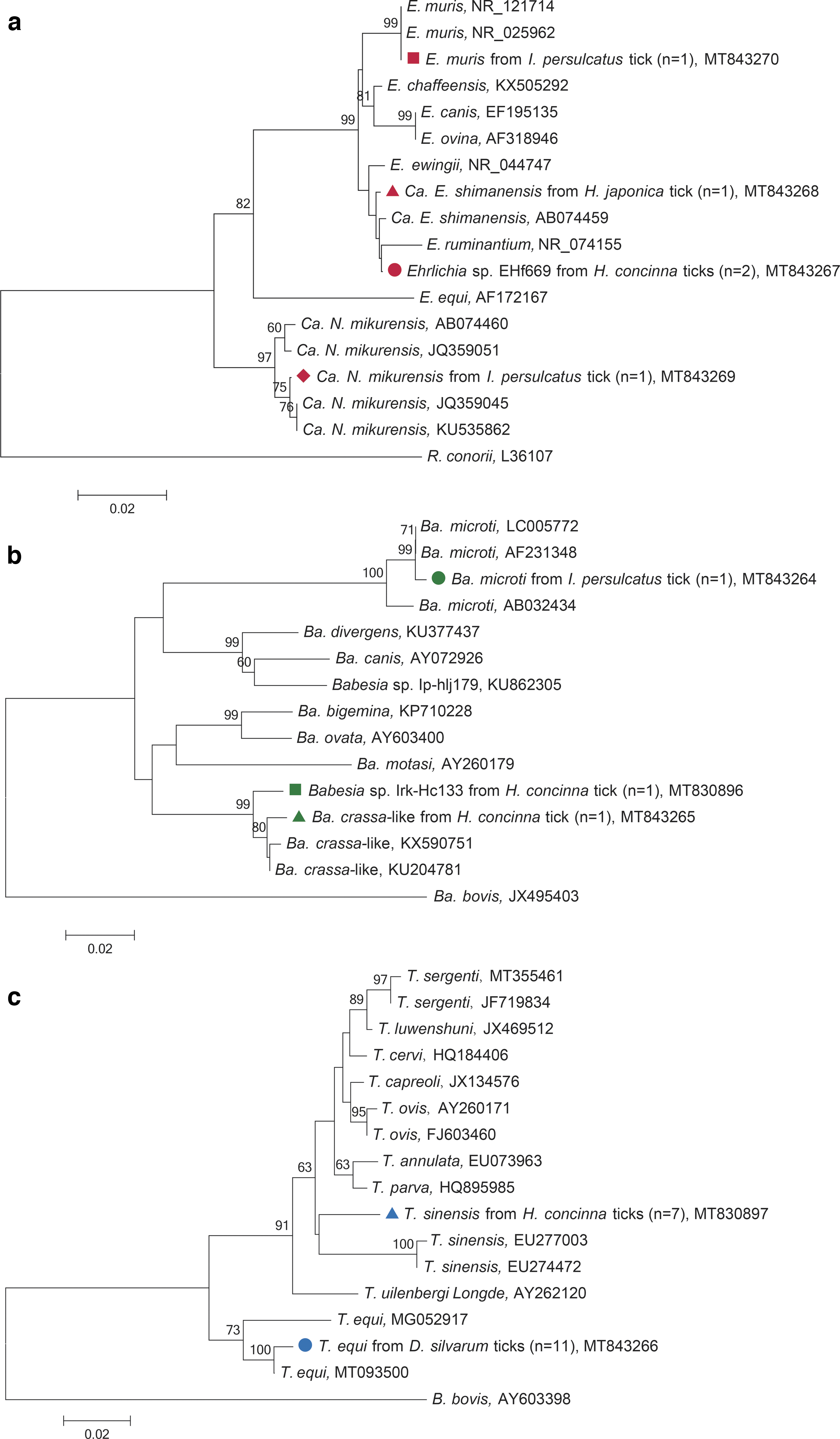
Phylogenetic analyses of Ehrlichia spp., Babesia spp., and Theileria spp. based on 545-nt 16S rRNA
Babesia was detected with an overall prevalence of 1.2%. Only two tick species were positive, including two (4.3%) H. concinna and one (2.4%) I. persulcatus (Table 1). The difference in infection rates among tick species was not statistically significant (χ 2 = 6.126, df = 4; p = 0.190). DNA of Babesia microti and Babesia crassa-like were identified. The obtained Ba. microti 18S rRNA sequence were 99.7% identical to those detected in I. persulcatus from Mongolia (LC005772) and human from the United States (AF231348). Phylogenetic analysis based on 380-nt 18S rRNA sequences showed that Ba. crassa-like identified in this study was clustered in the same clade with those previously detected in H. concinna (KU204781) and patients (KX590751) from northeastern China (Fig. 4b).
Theileria was detected with an overall prevalence of 7.2%. Eleven (10.8%) D. silvarum and 7 (14.9%) H. concinna were positive (Table 1). The difference in infection rates among tick species was statistically significant (χ 2 = 13.905, df = 4; p = 0.008), and the frequency of positive D. silvarum was strikingly higher compared with the other tick species. DNA of Theileria equi and Theileria sinensis were identified. Two representative sequences obtained in this study were included in phylogenetic analysis based on 415-nt 18S rRNA sequences (Fig. 4c). The obtained sequences of T. equi were 98.1% identical to the isolate JL08 from horses in China (MT093500) and clustered with the isolate BR_S5 (MG052917), which formed a haplotype distinct from other Theileria spp. strains (Fig. 4c). The sequences of T. sinensis were clustered with those of the known strains in the tree (Fig. 4c).
The presence of Anaplasma spp. in the 250 ticks was also investigated and the results were all negative.
Uncharacterized species of pathogens
An unclassified Rickettsia species (provisionally designated it as “Rickettsia sp. 1”), was found in H. japonica and H. concinna. The gltA genes of “Rickettsia sp. 1” had 99.4% identity with each other and 99.2% identity to an unclassified Rickettsia species detected from Haemaphysalis flava in Japan (JQ697957) (Fig. 2a). The ompA genes of “Rickettsia sp. 1” were 100% identical with each other, which formed a separate clade different from the other validated Rickettsia species (Fig. 2b).
The Ehrlichia sequence variant Ehrlichia sp. EHf669 and the Babesia sequence variant Babesia sp. Irk-Hc133 were both found in H. concinna ticks, which had over 99% similarity with the unclassified sequences from Haemaphysalis ticks in Japan (AY309969), and H. concinna in Russia (KJ486563), respectively.
The B. miyamotoi sequence variants uncultured Borrelia sp. clone Tick-021 were 99.4% identical to each other and matched the one detected in H. longicornis (MG798792), forming a separate branch on the phylogenetic tree (Fig. 3b).
Coinfections
Of the total 250 ticks, 23 (9.2%) showed the presence of coinfections. No simultaneous infection with three or more pathogens was found.
Frequent coinfections were observed in I. persulcatus, D. silvarum, and H. concinna. Among these species, the highest prevalence (16.7%) was found for Rickettsia spp. + Borrelia spp. infections in I. persulcatus. In addition, coinfections of Rickettsia spp. + Theileria spp. in D. silvarum, Rickettsia spp. + Babesia spp. in H. concinna, Borrelia spp. + Ehrlichia spp., Borrelia spp. + Babesia spp. in I. persulcatus, and Borrelia spp. + Theileria spp. in H. concinna were also identified (Table 1).
Discussion
It has been reported that tick-borne pathogens may change with the local natural environment and socioeconomic conditions, such as rapid urbanization, land use and climate changes, and so forth (Kilpatrick and Randolph 2012, Fang et al. 2015). Thus, active surveillance on ticks from the natural foci is very necessary for the public health authority. In this study, Rickettsia was the most frequently detected pathogen (prevalence, 41.2%), more common than that previously reported in northeastern China (1.53–32.25%) (Cao et al. 2008, Sun et al. 2014, Liu et al. 2016), indicating that Rickettsia represents an increasing public health problem in this area.
The composition of Rickettsia spp. was different from earlier studies. Previously, R. heilongjiangensis was prevalent in D. silvarum and Haemaphysalis ticks in northeastern China during 1985–2005 (Wu et al. 2005). However, in this study, R. heilongjiangensis was only detected in H. japonica with a low infection rate. Rickettsia sibirica strain BJ-90, the pathogen of Siberian tick typhus, was previously reported in tick-bitten patients of northeastern China (Jia et al. 2013b), but was not detected in this study. R. raoultii and Ca. R. tarasevichiae, mainly transmitted by Dermacentor and I. persulcatus, respectively, have become the prevalent Rickettsia species in this area. Similarly, although several species of Anaplasma, including Anaplasma phagocytophilum, A. capra, and Anaplasma bovis, have been reported in Ixodes, Dermacentor, and Haemaphysalis ticks in northeastern China (Cao et al. 2003, Jiang et al. 2011, Li et al. 2015, Wei et al. 2016), no Anaplasma spp. was detected in this study. This may reflect the geographical and seasonal variations of infected ticks.
There are nearly 37 million hectares of forest land in northeastern China, accounting for 27% of the national forest area. To date, ∼21 tick species of seven genera have been recorded in northeastern China, including Ixodes, Dermacentor, Haemaphysalis, Hyalomma, Rhipicephalus, and Argas (Chen et al. 2010). A variety of pathogens carried by ticks were identified in this region (Fang et al. 2015). People's life and work in northeastern China are closely related to forest areas, and the local tourism has rapidly developed in recent years, which increased the chances of people being bitten by ticks. Many tick-bitten patients have been reported in this region (Jia et al. 2013a, 2013b, 2014, Jiang et al. 2015, 2018, Li et al. 2015). In this study, we collected five species of hard tick, including D. silvarum, H. concinna, I. persulcatus, H. japonica, and D. nuttalli. All of them can act as vectors to transmit pathogens to humans and animals. The infection rates of pathogens were much higher in I. persulcatus, D. silvarum, and D. nuttalli, suggesting that we should pay more attention to local Ixodes and Dermacentor ticks. Notably, the pathogens carried by H. japonica varied dramatically in different sampling sites. Thus, it is necessary to collect H. japonica from different sites for pathogen surveillance and take more targeted control measures for specific tick-borne diseases in each site.
Eleven bacteria and five protozoa, as well as multiple coinfections were identified in this study, reflecting high diversity and complexity of pathogens in northeastern China. The most frequent coinfections were observed in I. persulcatus; Rickettsia spp., and Borrelia spp. might be more likely to coexist with other pathogens. Previously, several tick-bitten patients coinfected with two tick-borne pathogens, including Rickettsia spp. with Borrelia spp. have been reported in northeastern China (Liu et al. 2019). The results from this study demonstrate the existence of the biological basis, and indicate that humans and animals in this area were at a higher risk of coinfections, leading to more complicated clinical manifestations and misdiagnosis. Considering the diversity of tick species and tick-borne pathogens identified in this study, the clinicians should be aware of the clinical symptoms of multiple tick-borne diseases as to differential diagnosis.
The agents transmitted by ticks can cause many diseases, with various clinical manifestations and even death. In this study, a variety of human and animal pathogens were detected. R. raoultii can cause human tick-borne lymphadenopathy (TIBOLA) with necrotic erythema, eschar, and cervical adenopathy (Parola et al. 2009), and several TIBOLA cases have been reported in northeastern China (Jia et al. 2014). Ca. R. tarasevichiae and R. heilongjiangensis, the pathogen of human rickettsiosis and Far-eastern spotted fever, respectively, have also been detected in tick-bitten patients in northeastern China (Wu et al. 2005, Jia et al. 2013a). E. muris, Ca. N. mikurensis, B. garinii, B. afzelii, Ba. microti, and Ba. crassa-like, the etiological agents of human ehrlichioses, Lyme borrelioses, and babesioses were identified in this study as well as previous reports (Dumler and Bakken 1995, Ni et al. 2014, Zhou et al. 2014, Silaghi et al. 2016, Wei et al. 2016, Jia et al. 2018). The expense of diagnosis and treatment of these tick-borne diseases, added with decreased life quality, represent a significant economic burden for individual patients, their families, and the Chinese authority. Additionally, two pathogenic Theileria spp., T. equi, and T. sinensis, which were rarely reported in northeastern China, were detected in this study. Infections of these two pathogens in livestock usually result in weight loss, abortion, and even death (Hao et al. 2020), increasing the economic burden on the breeding industry and farmers. However, some other tick-associated bacterial pathogens, such as Coxiella burnetii, Francisella tularensis, and Bartonella spp., as well as tick-borne viruses cannot be detected by PCR with primers and conditions used in this study, which needs to be examined in further studies.
In recent years, more and more emerging tick-borne pathogens have been reported and raised the public concerns. In this study, several sequence variants have been found. For example, a potential new Rickettsia species “Rickettsia sp. 1,” an Ehrlichia sp. variant related to Ehrlichia sp. EHf669, a Borrelia sp. variant related to uncultured Borrelia sp. clone Tick-021, and a Babesia sp. variant related to Babesia sp. Irk-Hc133 were identified. These findings suggest that emerging pathogens still pose a significant health risk to humans. More high-throughput technology should be applied to analyze the tick-borne microbiome and to understand the distribution and composition of new agents.
Conclusions
We conducted a molecular survey on Rickettsia spp., Anaplasma spp., Ehrlichia spp., Borrelia spp., Babesia spp., and Theileria spp. in five tick species in northeastern China from 2018 to 2019. Ixodes and Haemaphysalis ticks harbored the most species of pathogens, and the pathogens carried by H. japonica varied differently in different sampling sites. Twelve pathogens and four potential new variants, as well as multiple coinfections were identified, reflecting high diversity and complexity of pathogens in ticks. These results are useful for designing more targeted and effective control measures for tick-borne diseases in China.
Footnotes
Acknowledgment
The authors thank Dr. Yi Sun from Beijing Institute of Microbiology and Epidemiology for his contribution in tick identification.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (grant nos. 81621005 and 81773492) and State Key R&D Program of China (grant no. 2019YFC1200501).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.