
Abstract
The emergence and spread of infectious diseases, particularly zoonotic diseases originating from wildlife, pose significant threats to global health and economy. This review examines the pivotal role of ticks as vectors in transmitting pathogens to humans, livestock, and wildlife and the use of molecular techniques in their identification. Tick infestations result in economic losses through reduced animal productivity, anemia, and quality deterioration of hides. Furthermore, ticks serve as reservoirs for a wide range of pathogens including viruses, bacteria, fungi, protozoa, and nematodes, contributing to the transmission of diseases such as Crimean–Congo hemorrhagic fever, tick-borne encephalitis, and African swine fever among others. The interface between wildlife, livestock, and humans facilitates the transmission of zoonotic pathogens, exacerbated by nomadic and pastoralist lifestyles that promote interactions between wildlife and domestic animals. This movement of animals across landscapes enhances the dispersion of tick vectors, increasing the risk of pathogen exposure for diverse populations. Historically, tick identification in sub-Saharan Africa has relied on morphological characteristics despite limitations such as species overlap and variability. Molecular techniques offer a more precise means of species identification, providing critical data for effective tick and pathogen management strategies. Integrating molecular approaches into tick research enhances our understanding of tick diversity, distribution patterns, and pathogen dynamics. This knowledge is essential for developing targeted interventions to mitigate the impact of tick-borne diseases on public and veterinary health worldwide.
Introduction
The emergence of infectious diseases is increasing worldwide, leading to significant global economic losses (Blancou 2005). Many of these are zoonotic diseases originating from wildlife. In recent years, numerous pathogen crossovers from wildlife have occurred, resulting in emerging and reemerging diseases. Examples include Hantavirus Pulmonary Syndrome, Nipah virus disease, Hendra virus disease, and East Coast fever caused by Theileria parva in cattle (Dewart 2000).
Ticks, among other vectors, contribute to substantial economic losses due to their adverse impacts on human, livestock, and wild animal health (Estrada-Peña and de la Fuente, 2014). Tick infestations can lead to a reduction in animal body weight, limit animal production, and induce anemia, particularly in heavily infested animals (Walker et al. 2003). Moreover, tick bites may cause irritation, resulting in reduced hide quality (Walker et al. 2003). Certain tick species, such as Rhipicephalus evertsi, Ixodes rubicundus, and Hyalomma truncatum, inject toxins into animals, causing paralysis (Estrada-Peña and de la Fuente 2014). Ticks also serve as reservoirs and vectors of various pathogens, including viruses, bacteria, fungi, protozoa, and nematodes, which can cause diseases in humans, livestock, and wild animals (Estrada-Peña et al. 2013).
Several tick-transmitted pathogens, including viruses, pose threats to human and animal health (Estrada-Peña and de la Fuente 2014). These viral diseases include Crimean–Congo hemorrhagic fever virus (CCHFV) disease, Kyasanur forest disease virus, severe fever with thrombocytopenia syndrome virus disease, tick-borne encephalitis virus disease, Heartland virus disease, African swine fever virus disease, Nairobi sheep virus diseases, and Louping ill virus disease (Estrada-Peña and de la Fuente 2014). The interaction between humans, livestock, and wildlife promotes the spread of zoonotic pathogens. Nomadic and pastoralist lifestyles, particularly those at the wildlife–livestock interface, accelerate the exposure and sharing of previously isolated pathogens (Labuda and Nuttall 2004).
Areas at the wildlife, livestock, and human interface are characterized by poor animal husbandry and grazing practices, exerting great pressure on land resources. This pressure often necessitates the continuous movement of large numbers of animals, especially cattle and goats, in search of pasture. As a result, livestock frequently share pastures with wild animals, leading to significant human–livestock–wild animal contacts. This migration pattern facilitates the movement of potential ticks across great distances, increasing the risk of exposure to tick-borne pathogens for domestic animals, wild animals, and human populations (Labuda and Nuttall 2004).
Studies aiming to quantify and identify tick species in sub-Saharan Africa are limited, with information primarily restricted to morphological features (Kwak et al. 2014). However, morphological characterization of ticks based on phenotypic criteria has limitations (Ganajli et al. 2011). Morphological identification is challenging, especially for larvae and nymphs of closely related species, and may result in misidentification due to overlapping distinguishing characteristics and high intraspecific variability (Kerario et al. 2017). Additionally, morphological identification is not well applicable to damaged ticks (Rumer et al. 2011); traits may vary with size, age, and population polymorphism (Anderson et al. 2004).
This review provides an overview of ticks and their identification using molecular techniques. Molecular data on ticks offer reliable information to entomologists, farmers, and other stakeholders for the rational control of ticks and tick-borne pathogens.
Key Characteristics of Ticks
Ticks, ectoparasites of mammals, birds, and reptiles, primarily feed on blood. With around 800 species worldwide (Estrada-Peña et al. 2013), ticks are obligate ectoparasites falling under the subphylum Chelicerata, class Arachnida, subclass Acari, and suborder Ixodida. This order comprises three recognized families: Ixodidae (hard ticks), Argasidae (soft ticks), and Nuttalliellidae (Walker et al. 2003).
Ixodidae, the largest group with ∼600 species, feature a hard cuticle, while Argasidae, comprising about 150 species, possess a smooth cuticle. Nuttalliellidae stand out with only one known species (Walker et al. 2003). Hard ticks generally range from 3 to 18 mm in size and include notable genera such as Hyalomma, Rhipicephalus, Amblyomma, Haemaphysalis, and Dermacentor. Common genera of Argasidae encompass Antricola, Ornithodorus, Argas, and Otobius (Walker et al. 2003).
Tick Species and Associated Tick-Borne Diseases in Sub-Saharan Africa
Gene Markers in Tick Molecular Studies: Cellular Locations and Resolution Levels
COI, cytochrome oxidase subunit 1; ITS, internal transcribed spacers; rRNA, ribosomal RNA.
A tick’s body is divided into two main parts: the anterior capitulum and the posterior idiosoma. The anterior capitulum bears the palp, a segmented sensory organ, and the chelicera, highly sclerotized appendages. Among different genera, the shape of the basis capitula, fused coxae of the palp, varies (Walker et al. 2003). The hypostome, an extension found ventrally and anteriorly of the basis capitula, is present. The legs, situated in a podosoma connected to the main body at the coxa, consist of the trochanter, femur, genu, tibia, and tarsus. Haller’s organs, chemoreceptors used to locate hosts, are found on the tarsi (Wall and Shearer 2008).
Figure 1 illustrates the anatomical features of a tick, from the anterior chelicera to the posterior anal plate: chelicerae, paired mouthparts used for gripping the host’s skin during feeding; palps, structures aiding in environmental sensing and potentially assisting in feeding; hypostome, anchoring the tick to the host’s skin during feeding, often with backward-facing barbs; capitulum, containing the mouthparts and visible above the host’s skin during feeding; scutum, present in some species, serving as a shield covering part of the body, usually absent or reduced in females; idiosoma, constituting the main body, including the abdomen and fused cephalothorax; legs, eight legs with segments and claws for locomotion and attachment; anal plate, located in the posterior region, involved in reproduction and waste elimination.
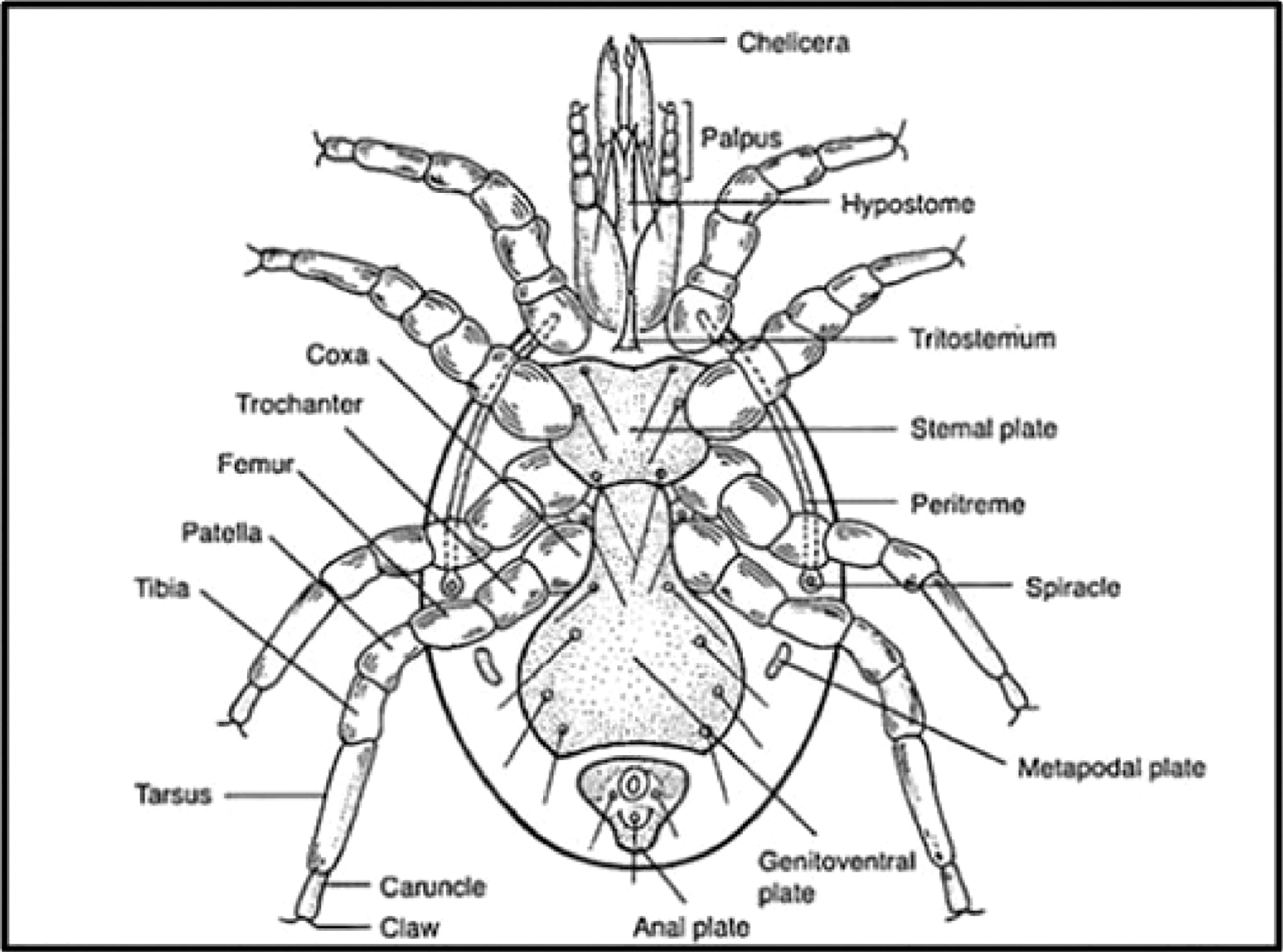
Morphological structure of a tick.
Developmental Stages of Ticks
Ticks undergo a fascinating journey through four developmental stages: eggs, larvae, nymphs, and adults. The life cycle begins when a female tick lays eggs, typically in the environment where the tick’s host frequents. These eggs give rise to larvae, which are tiny, six-legged creatures. In most cases, larvae and nymphs feed on smaller and medium-sized hosts to grow and molt into the next stage. Nymphs, larger than larvae and with eight legs, require a host to further develop into adults. Upon feeding, nymphs molt into adults, equipped with fully developed reproductive organs and ready to mate. The stages can vary depending on the species, with various life cycles observed.
These cycles include the one-host cycle, where the tick remains on the same host for all stages, feeding and molting until the female lays eggs and dies. Examples of such species include some Rhipicephalus spp. In the two-host cycle, the larva feeds on a host, molts into a nymph, feeds again on the same or another host, molts into an adult, and seeks a new host for mating. This cycle characterizes species such as some Hyalomma spp. In the three-host cycle, each developmental stage feeds on a different host. After each blood meal, the tick drops off, molts in the ground, and seeks a new host (Jongejan and Uilenberg 2004). Species such as Amblyomma spp. exemplify this cycle, with a survival period of ∼3 years (Walker et al. 2003). Understanding these stages and cycles is vital for managing tick populations, as ticks are vectors for diseases affecting humans and animals (Soneshine 1991).
Figure 2 illustrates the sequential developmental stages of ticks, with dashed lines and arrows indicating the progression. Each stage represents a pivotal phase in the tick’s life cycle. Egg Stage: The life cycle initiates with the deposition of eggs by adult female ticks. These eggs are typically laid in sheltered environments such as leaf litter or soil. Larva Stage: Following an incubation period, the eggs hatch into larvae. Larvae, characterized by six legs, primarily feed on the blood of a host, usually small mammals or birds. Nymph Stage: Upon completing their blood meal, larvae molt into nymphs. Nymphs, possessing eight legs, seek larger hosts for another round of blood feeding. This stage is critical for tick growth and development. Adult Stage: After maturing as nymphs, ticks undergo another molt, emerging as adults. Adult ticks exhibit sexual dimorphism, with females typically larger than males (Walker et al, 2003). They seek out larger hosts, including mammals such as deer or domestic animals, for mating and another blood meal to support egg production (Jongejan and Uilenberg 2004). The dotted lines with arrows represent the continuous progression of tick development, emphasizing the cyclical nature of their life cycle. Understanding these stages is crucial for effective tick population management and the prevention of tick-borne diseases (Soneshine 1991).
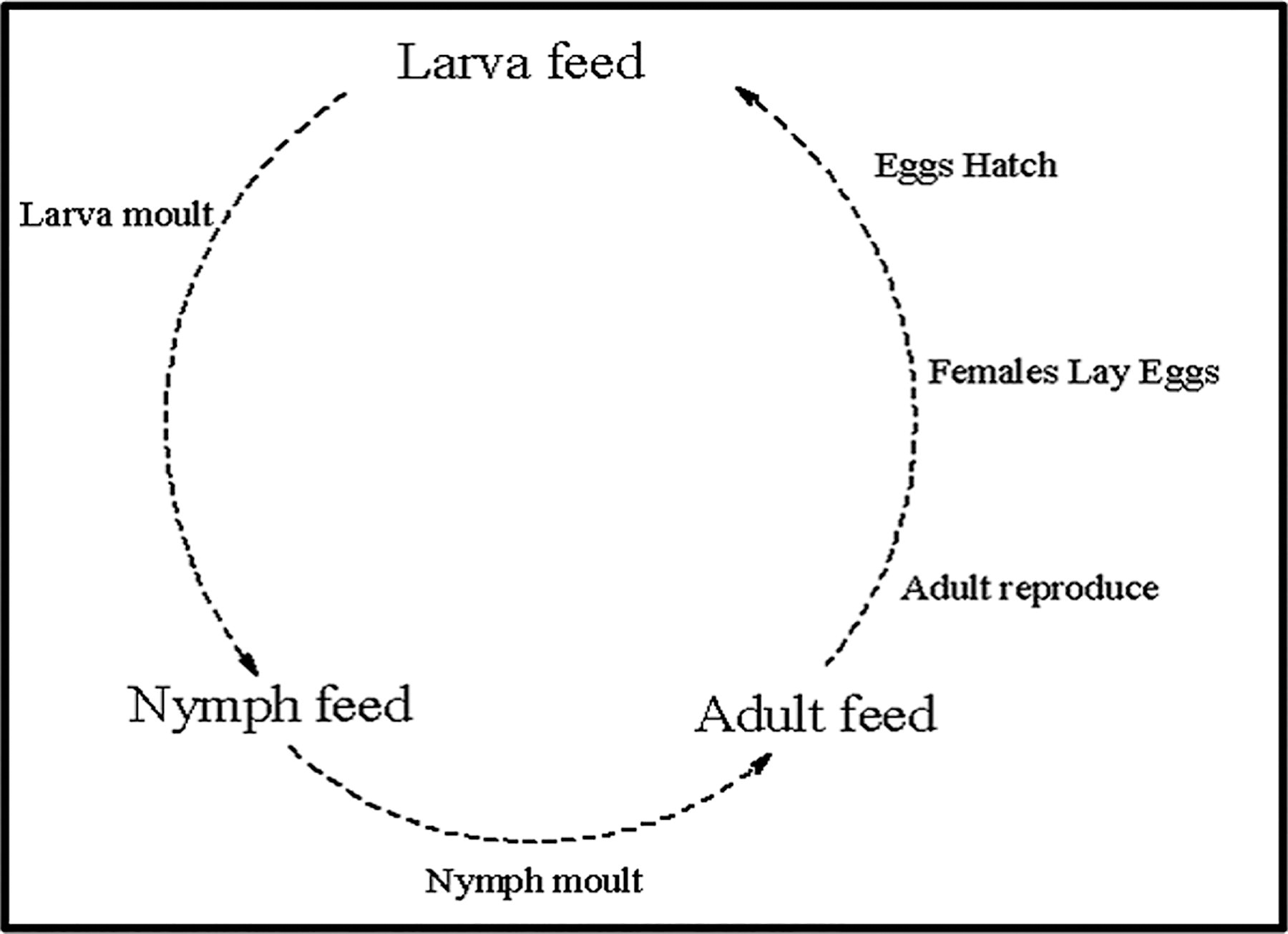
Tick developmental stages.
Factors Influencing the Survival and Abundance of Ticks
Survival and abundance of ticks are influenced by a myriad of biological and environmental factors beyond their reliance on hosts for nutrition (Keesing et al. 2006). Ticks have evolved sophisticated mechanisms to adapt to the immune systems of their hosts, including the production of immunomodulatory molecules that suppress host defenses and facilitate successful feeding (Wikel 2018). Climate also plays a critical role in shaping tick populations. Temperature, humidity, and precipitation levels directly impact tick survival, development, and questing behavior (Estrada-Peña et al. 2012). For instance, increased temperatures can accelerate tick development and activity, while prolonged periods of high humidity are essential for preventing desiccation, a major threat to tick survival (Sonenshine 1991). Furthermore, weather patterns influence vegetation growth, which, in turn, affects host availability and habitat suitability for ticks (Ostfeld and Brunner 2015).
Tick control measures, such as the application of acaricides, are essential for managing tick populations and reducing the risk of tick-borne diseases. However, the efficacy of these control methods can be compromised by the development of resistance in tick populations (Stone et al. 2018). Additionally, the indiscriminate use of acaricides may have adverse environmental consequences, highlighting the importance of integrated pest management strategies that incorporate alternative control measures (Eisen and Dolan 2016).
Predators also play a significant role in regulating tick populations. Various vertebrate and invertebrate species, including birds, mammals, and arthropods, feed on ticks at different life stages (Allan et al. 2010). For example, certain bird species are known to consume large numbers of ticks, thereby reducing tick abundance in their habitats (Dunn et al. 2017). Likewise, predatory arthropods such as ants and mites may prey on tick eggs and larvae, exerting additional pressure on tick populations (Birkhofer et al. 2017).
Nonparasitic life stages of ticks, such as eggs and engorged females, also face challenges for survival. Weather conditions, particularly temperature and humidity levels, can influence the viability of tick eggs and the survival of engorged females during off-host periods. Moreover, the availability of suitable oviposition sites and environmental substrates for egg development is crucial for the successful reproduction of ticks (Gray et al. 2016).
Overall, the geographical distribution and abundance of ticks are shaped by the complex interplay of these factors, along with additional considerations such as host availability, habitat fragmentation, and land use patterns (Ostfeld and Levi 2014). Understanding these dynamics is essential for implementing effective tick management strategies and mitigating the public health risks associated with tick-borne diseases.
The Economic Toll of Ticks: Impacts on Agriculture, Public Health, and Tourism
Ticks exert significant economic impacts globally through their direct effects on humans, wildlife, and livestock. The repercussions are extensive, ranging from reduced productivity to increased health care costs and expenditures on control measures. For instance, tick bites on livestock diminish the economic returns for livestock owners and leather industries worldwide by impacting the value of hides and skins (Walker et al, 2003).
Tick-borne diseases represent a substantial global economic burden due to their impact on livestock health and productivity. For example, diseases such as East Coast fever alone impose significant costs on dairy systems globally, with traditional agricultural systems bearing substantial economic burdens (Minjauw and McLeod 2003). These diseases result in direct health care costs and reduce livestock productivity and reproductive success worldwide, thereby amplifying economic losses.
Furthermore, certain tick species inject toxins into animals, inducing paralysis and compromising animal welfare globally. This situation results in economic losses for livestock owners due to decreased productivity and potential mortality (Jongejan and Uilenberg 2004). Additionally, tick-related diseases such as tick toxicosis, such as sweating sickness, affect livestock in various regions globally, leading to economic losses through reduced productivity and increased veterinary expenses for treatment.
Ticks also pose a significant threat to global public health by transmitting diseases known as zoonoses. These diseases incur health care costs and impact productivity and outdoor activities globally. In various regions worldwide, communities may alter their behaviors, such as sleeping outdoors, to avoid indoor-dwelling ticks, highlighting the socioeconomic impact of tick-borne diseases (Talbert et al. 1998).
Addressing the economic impacts of ticks globally requires significant investment in research and the implementation of effective control measures. Funding is essential for research into tick biology, ecology, and control methods, contributing to the economic burden associated with tick management (Soneshine 1991). Moreover, the implementation of tick control measures, such as chemical treatments and management practices, incurs costs for livestock owners worldwide, further adding to the economic impact of ticks on agriculture and society.
In summary, ticks impose substantial economic burdens globally through their effects on livestock production, human health, and associated control measures. Addressing tick infestations and associated diseases is crucial for mitigating these economic impacts and promoting sustainable agricultural practices and global public health initiatives.
Ticks infestation poses significant health risks to livestock, including cattle. Understanding the distribution of ticks on the animal’s body is essential for effective management strategies. Figure 3 illustrates two common sites of ticks infestation in cattle: the udder region (labeled as “A”) and the ear region (labeled as “B”).
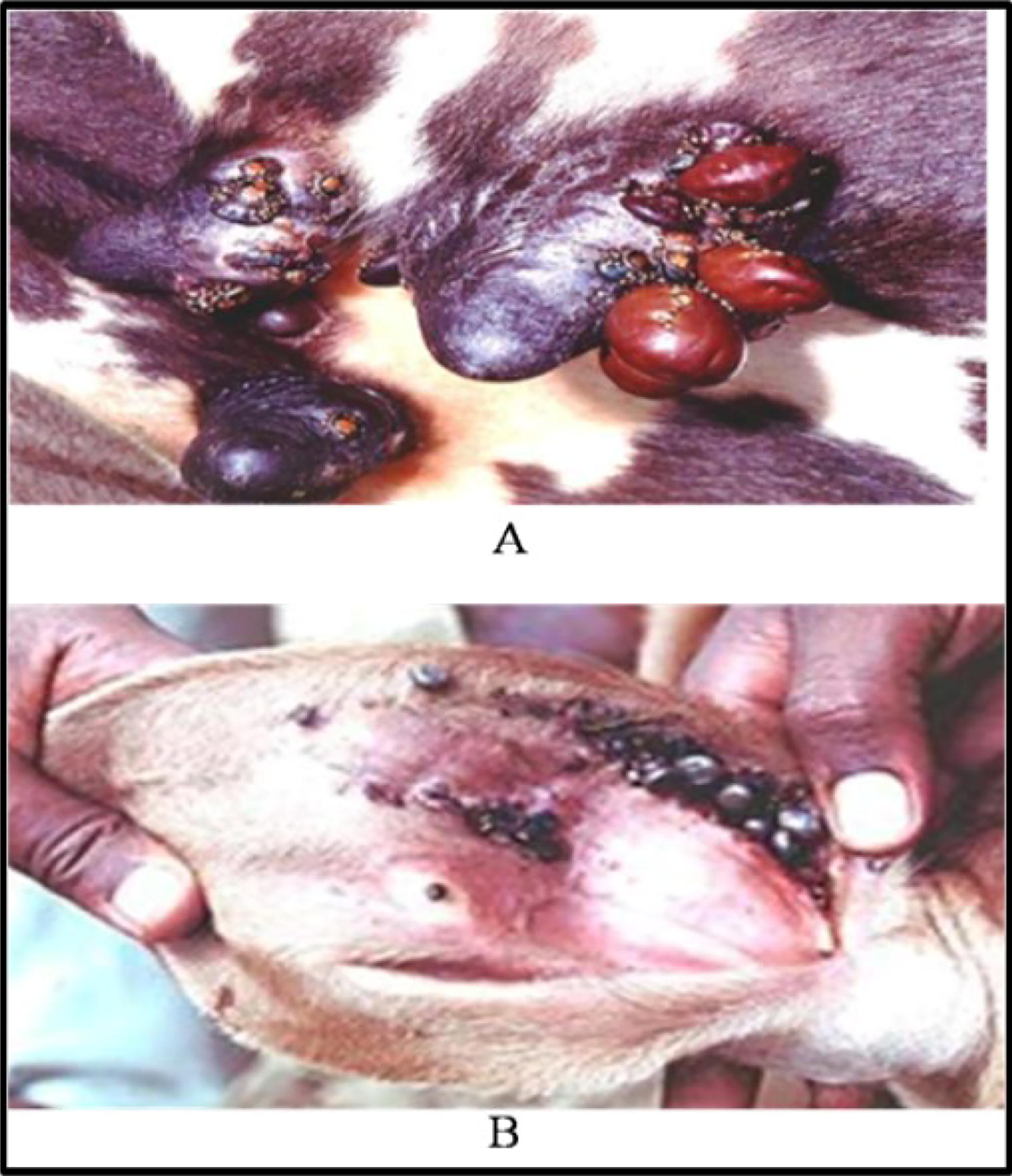
Ticks’ infestation in various animal body parts.
(A) Ticks’ Infestation in the Udder Region: Ticks’ infestation in the udder region of cattle can have detrimental effects on milk production and the overall health of the animal. The udder, being a sensitive and vital part of the cow’s anatomy, is particularly vulnerable to infestations. Ticks in this area can cause irritation, inflammation, and potential transmission of diseases, impacting the quality and quantity of milk produced. Additionally, infestations in the udder region may lead to discomfort for the animal, affecting its well-being and productivity.
(B) Ticks’ Infestation in the Ear Region: Ticks commonly infest the ear region of cattle, posing various challenges for farmers and livestock managers. The ear is a warm and sheltered environment, making it an attractive location for ticks to feed and reproduce. Infestations in this area can result in irritation, inflammation, and the formation of lesions. Furthermore, ticks in the ear region may interfere with the animal’s behavior, causing discomfort and potential complications such as secondary infections. Effective control measures targeting this region are crucial for maintaining the health and welfare of cattle.
Understanding the distribution of ticks on the body of cattle allows for targeted intervention strategies to minimize infestation levels and mitigate associated risks. Regular monitoring, appropriate parasite control measures, and maintaining good herd health practices are essential components of tick management programs in livestock farming.
Tick Species and Associated Tick-Borne Diseases in Sub-Saharan Africa
In sub-Saharan Africa, a region characterized by diverse ecosystems and climates, various tick species act as vectors for transmitting a range of pathogens, posing significant health risks to humans and animals alike. Among the most prominent tick species in the region is Rhipicephalus appendiculatus, commonly known as the Brown ear tick. This species is notorious for transmitting the protozoan parasite Theileria parva, responsible for East Coast fever, a severe and often fatal disease affecting cattle across East and Southern Africa (Nene et al. 2016). Similarly, R. (Boophilus) decoloratus, the Blue tick, plays a crucial role in the transmission of Babesia bigemina, causing bovine babesiosis, which poses a considerable economic burden on cattle production in the region (Mahoney et al. 2019).
Another significant vector species in Sub-Saharan Africa is Amblyomma variegatum, commonly known as the Tropical bont tick. This tick is a known vector for Ehrlichia ruminantium, the causative agent of heartwater, a fatal disease affecting ruminants such as cattle, goats, and sheep (Gubbels et al. 2000). In addition, Amblyomma hebraeum, the South African bont tick, also contributes to the transmission of Ehrlichia ruminantium, further exacerbating the prevalence of heartwater in livestock populations (De Waal et al. 2007).
Beyond livestock diseases, certain tick species in sub-Saharan Africa pose significant threats to human health. For instance, Hyalomma spp., a genus encompassing various tick species, serves as vectors for CCHFV, a severe viral hemorrhagic fever with high mortality rates in humans (Burt et al. 2017). Moreover, A. americanum, commonly known as the Lone Star tick, can transmit pathogens such as Ehrlichia chaffeensis and E. ewingii, which cause human ehrlichiosis, as well as Francisella tularensis, the agent of tularemia (Stromdahl and Hickling 2012).
While these are some of the key tick species and associated diseases in sub-Saharan Africa, it’s essential to recognize the broader impact of tick-borne illnesses on public health and agriculture in the region. Effective control measures, including tick surveillance, vector management, and public awareness campaigns, are crucial for mitigating the risks posed by these vector-borne diseases.
Tick-Borne Disease Transmission Dynamics at the Human–Livestock–Wildlife Interface
The transmission of pathogens from wildlife to humans and domestic animals represents a significant public health concern, often catalyzed by the encroachment of human activities into natural habitats. Viruses with wildlife reservoirs, though traditionally confined to their animal hosts, can breach species barriers and infect humans and domestic animals under certain conditions (Tonetti et al. 2009). This cross-species transmission is influenced by various factors including habitat disturbance, increased human–wildlife interaction, and alterations in land use patterns. Such interactions can facilitate the emergence and spread of infectious diseases, highlighting the importance of understanding the ecological drivers of pathogen transmission.
Arthropods, particularly ticks, play a pivotal role in the transmission of viruses and other pathogens between wildlife, domestic animals, and humans. Ticks serve as efficient vectors, capable of transmitting a wide range of pathogens, thus serving as key players in disease ecology (Weaver and Reisen 2010). Their role in pathogen transmission underscores the necessity of studying their behavior, ecology, and interactions with their hosts to effectively manage and mitigate disease transmission dynamics.
The burgeoning human population, coupled with land use changes, has led to widespread modification of ecosystems worldwide. This alteration of natural landscapes often results in habitat loss, fragmentation, and degradation, leading to a decline in biodiversity (Tonetti et al. 2009). Such changes can create favorable conditions for the proliferation of ticks and their hosts, thereby increasing the risk of pathogen transmission to humans and domestic animals.
Biodiversity loss further exacerbates the dynamics of host-pathogen interactions, as reduced species richness may lead to increased contact between humans, domestic animals, and wildlife. This heightened interaction can facilitate the spillover of pathogens from wildlife reservoirs to novel hosts, including humans, livestock, and companion animals (Tonetti et al. 2009). Consequently, understanding the complex interplay between biodiversity loss and host-pathogen dynamics is essential for predicting and mitigating the risk of disease emergence and transmission.
Wild animals, particularly certain species acting as reservoirs, play a crucial role in the maintenance and transmission of tick-borne pathogens (Allsopp et al. 1999). For instance, African buffalo are reservoirs for diseases such as corridor disease, East Coast fever, and heartwater, while bush pigs and warthogs are implicated in the sylvatic cycle of the African swine fever virus (Thomson 1985). The identification of these reservoir species underscores the need for integrated approaches to disease surveillance and management that encompass both wildlife and domestic animal populations (Mur et al. 2016).
The complex interplay between human activities, biodiversity loss, and wildlife reservoirs significantly influences the transmission dynamics of tick-borne pathogens. Efforts to mitigate the risk of disease emergence and spread require a holistic understanding of these ecological interactions, coupled with proactive measures to preserve biodiversity and mitigate the impacts of habitat modification on disease transmission pathways.
Factors Contributing to the Emergence of Tick-Borne Infectious Diseases
Emerging tick-borne infectious diseases (TBIDs) pose a significant public health concern, with ticks serving as crucial vectors for the transmission of various pathogens. Understanding the factors influencing the evolution and spread of these pathogens is essential for effective disease management and prevention (Brooks and Hoberg 2006).
Host-switching, wherein ticks and their associated pathogens move outside their original habitats and ecosystems, can lead to the adaptation of pathogens to new hosts, potentially resulting in outbreaks in previously unaffected populations (Allsopp et al. 1999). Anthropogenic activities play a central role in the increasing prevalence of TBIDs. Human-induced factors such as biodiversity loss and climate change have accelerated alterations in tick population structures and geographic distributions (Brooks and Hoberg 2006). Biodiversity loss, driven by human activities, not only leads to the extinction of species but also facilitates the introduction of new species carrying novel pathogens, thereby contributing to the emergence of infectious diseases (Allsopp et al. 1999).
Climate change is another critical factor influencing the emergence of TBIDs. Changes in temperature and precipitation patterns directly impact tick populations and their distribution, expanding their geographic range and increasing the risk of disease transmission to humans and animals (Allsopp et al. 1999).
Moreover, ecological changes resulting from agricultural practices, international travel, and microbial adaptation further exacerbate the risk of TBID emergence (Morens and Fauci 2012). These changes alter the dynamics of tick populations and their interactions with hosts, creating favorable conditions for the transmission of pathogens (Brooks and Hoberg 2006). International travel plays a significant role in the spread of ticks and their associated pathogens across borders, introducing these vectors and pathogens to new regions and populations (Brooks and Hoberg, 2006).
Addressing the multifaceted nature of TBID emergence requires a comprehensive approach that considers the interconnectedness of factors such as host-switching, anthropogenic activities, biodiversity loss, climate change, ecological changes, and international travel (Morens and Fauci 2012). By understanding and mitigating these factors through targeted interventions, it may be possible to reduce the burden of emerging TBIDs on public health.
Effective Tick Control Strategies and Management of Acaricide Resistance
Control measures against tick populations encompass a variety of strategies, including the application of single, multiple, and rotational acaricides to limit their numbers and prevent their spread (Chevillon et al. 2007). Complementary approaches such as tick vaccines and selective breeding for resistance in animals also play significant roles in managing infestations and mitigating the impact of tick-borne diseases (Hernandez et al. 2002).
The arsenal of acaricides commonly deployed against ticks includes avermectins, amitraz, pyrethroids, and acetylcholine esterase inhibitors, each targeting specific aspects of tick physiology and behavior (Hernandez et al. 2002).
However, the efficacy of these acaricides is increasingly compromised by the rapid development of resistance in tick populations. The high genetic diversity of ticks, coupled with their pangamy mating structure, facilitates the swift emergence of resistance to multiple acaricides within just a few generations (Cutulle et al. 2009). This resistance is often driven by genetic mutations occurring during DNA replication, a process prone to errors due to the slippage of the replication machinery (Cutulle et al. 2009).
These mutations lead to alterations in gene expression patterns between susceptible and resistant tick strains, rendering acaricides ineffective in targeting their intended sequences (Chevillon et al. 2007). Furthermore, the movement of animals carrying resistant ticks between different geographic areas contributes to the spread of resistance genes via gene flow (Chevillon et al. 2007).
Facilitating this gene flow are various factors such as the sharing of pasture, water sources, and dipping lots among animals, which create opportunities for the dissemination of resistant tick populations and their associated resistance genes (Hernandez et al. 2002).
Although acaricides and other control measures remain essential tools in the management of tick populations, their efficacy is threatened by the rapid evolution of resistance. Understanding the mechanisms driving resistance development and implementing integrated strategies that consider both biological and ecological factors are crucial for sustainable tick control efforts.
Ticks’ Identification
Traditionally, the identification of ticks has relied heavily on morphological characteristics, using phenotypic criteria as the primary basis for differentiation (Soneshine 1991). However, this approach has been found to have several limitations. Studies have indicated drawbacks associated with morphological identification, including its difficulty and susceptibility to misidentification (Anderson et al. 2004). Particularly challenging is the identification of larvae and nymphs of closely related species, which can lead to erroneous classification (Ganajli et al. 2011). Moreover, morphological characters may vary due to factors such as size, age, feeding state, and damage to the tick specimens, further complicating accurate identification (Rumer et al. 2011).
In response to these challenges, efforts have been made to develop alternative characterization methods that offer greater reliability and convenience. Techniques such as protein electrophoresis and cuticular hydrocarbon analysis have been employed in taxonomy studies of various tick species and populations (Rees et al. 2003). However, the most promising approach for characterizing species and populations with precision is DNA sequence analysis (Rees et al. 2003).
DNA sequence analysis provides a direct and accurate method for differentiating subspecies and species of ticks. By analyzing specific genetic markers, researchers can obtain detailed information about the genetic makeup of individual ticks and populations (Rees et al. 2003). This method offers several advantages over traditional morphological identification. First, it eliminates the need for extensive training and experience in morphology and taxonomy, making it more accessible to researchers with diverse backgrounds. Second, DNA sequencing is less susceptible to variations caused by factors such as size, age, and feeding state, ensuring consistent results across different specimens. Additionally, damaged ticks can still be reliably identified through DNA analysis, overcoming a significant limitation of morphological methods (Rees et al. 2003).
Furthermore, DNA sequence analysis allows for the comparison of genetic data across different populations and geographic regions, facilitating the study of tick distribution, evolution, and disease transmission dynamics. By building comprehensive databases of tick DNA sequences, researchers can improve our understanding of tick diversity and ecology, as well as develop more effective strategies for tick-borne disease control and prevention (Rees et al. 2003).
While traditional morphological identification methods have served as the cornerstone of tick taxonomy for many years, they are not without limitations. Alternative techniques such as DNA sequence analysis offer a more reliable and convenient approach for differentiating tick species and populations. By harnessing the power of molecular genetics, researchers can overcome the challenges associated with morphological identification and gain deeper insights into the biology and ecology of ticks.
Unraveling the Genetic Tapestry: Phylogenetic and Genetic Variation Evaluation of Ticks
The investigation of evolutionary relationships among genes or species is a cornerstone of modern biological inquiry, facilitated by phylogenetic studies. As Barker and Murrell (2002) assert, these studies offer invaluable insights into the relatedness of sequences, genes, or species, shedding light on their evolutionary history and divergence. Phylogenies, reconstructed through such studies, serve as hierarchical frameworks for classifying organisms, unraveling the intricate tapestry of evolutionary descent.
Central to phylogenetic analyses is the concept of homology, where genes or sequences share a common ancestry. Black and Piesman (1994) emphasize the importance of this shared ancestry in phylogenetic inference, as it forms the foundation for discerning evolutionary relationships. Homologous sequences, by retaining traces of their common evolutionary origin, furnish rich datasets for comparative analysis, thereby enabling robust phylogenetic reconstructions (Burger et al. 2014).
However, evolutionary forces continually shape genetic sequences, introducing variations such as point mutations, deletions, and substitutions over time. Consequently, sequences diverge from their common ancestors, a process quantified through measures of genetic distance. Norris et al. (1998) elucidate that genetic divergence serves as a proxy for the time elapsed since two sequences shared a common ancestor, offering a nuanced understanding of their evolutionary trajectories.
It’s paramount to recognize that the concept of genetic variation, originally devised for estimating genetic differentiation between populations, transcends disciplinary boundaries. As Norris et al. (1996) underscore, while genetic variation serves as a vital metric in population genetics, its utility extends far beyond, permeating fields such as evolutionary biology and molecular genetics, where it underpins investigations into the origins and relationships of biological entities.
Thus, phylogenetic studies, anchored in the principles of homology and genetic divergence, not only unravel the evolutionary tapestry connecting genes and species but also enrich diverse domains of biological inquiry, underscoring the unity underlying life’s diversity.
Exploring Gene Markers in Tick Molecular Studies: Cellular Locations and Resolution Levels
Molecular analysis stands as a cornerstone in contemporary taxonomy, facilitating the elucidation of genetic relationships among species. At the heart of this methodology lies the pivotal task of selecting an appropriate genomic region, a decision that profoundly shapes the subsequent analytical outcomes. Mangold et al. (1997) emphasize this critical step, highlighting the necessity of choosing regions that strike a delicate balance between conservation and variation.
Regions characterized by near-identical sequences across taxa risk yielding stagnant datasets, wherein the acquisition of invariant data consumes valuable time without contributing substantially to the phylogenetic resolution. Conversely, regions exhibiting considerable divergence among taxa offer promise in generating robust phylogenies, as noted by Norris et al. (1996). However, excessive divergence can render sequence alignments ambiguous, with Norris et al. (1996) suggesting that alignments become problematic when paired sequences differ by more than 30 percent within a given region.
Optimal regions for molecular analysis, as proposed by Norris et al. (1996), typically manifest similarities >70% yet fall short of complete homogeneity. Within the realm of tick species analysis, a myriad of sequences, both coding and noncoding, have been enlisted to unravel genetic relationships. Nuclear DNA sequences such as ITS1 and ITS2, as elucidated by Wesson et al. (1993), offer invaluable insights into intraspecific variations. Meanwhile, mitochondrial markers such as 12S ribosomal RNA (rRNA) and 16S rRNA, alongside nuclear markers such as 18S rDNA and 28S rDNA, have been pivotal in delineating intergeneric and interspecific relationships among ticks (Black and Piesman 1994).
The choice between coding and noncoding sequences introduces additional considerations, reflective of the differential evolutionary dynamics governing these genomic elements (Mangold et al. 1998). Variations in natural selection pressures and mutation rates over time underscore the distinct advantages and limitations associated with each sequence type. Such disparities underscore the need for a nuanced understanding of the biological context and analytical objectives when selecting genomic regions for molecular analysis, particularly when delving into taxonomic inquiries spanning the familial, generic, and specific levels.
The judicious selection of genomic regions stands as a linchpin in molecular analysis, wielding profound implications for the accuracy and interpretability of phylogenetic reconstructions. By integrating insights from diverse sources and heeding methodological guidelines, researchers can navigate the complex landscape of genetic relationships with confidence and precision.
Coding regions
Within the genomic landscape, genes are classified into two distinct categories, each serving pivotal roles in the orchestration of biological processes. The foremost category encompasses structural genes, which harbor the blueprint for synthesizing functional proteins indispensable for an organism’s physiology. Complementing this group are genes relegated to the realms of transfer RNA, rRNA, and small nuclear RNA transcripts, all pivotal players in the intricate machinery orchestrating protein synthesis (Mangold et al. 1997).
In the intricate dance of molecular biology, the integrity of coding sequences, or exons, assumes paramount importance. These segments of genes harbor the precise instructions dictating the amino acid sequence constituting proteins, thereby determining their structure, function, and ultimately, their biological impact. It is within these coding regions that the essence of genetic information is transcribed into tangible molecular entities, underscoring their indispensable role in the fabric of life.
Contrary to the mutational maelstrom often associated with genetic variation, coding regions display a remarkable resilience to mutational onslaught. This robustness stems from the stringent selection pressures acting upon these regions, as any deviation has the potential to disrupt the delicate balance of protein structure and function, with potentially deleterious consequences for the organism (Black and Piesman 1994). However, it is important to note that not all mutations within coding regions lead to catastrophic outcomes. Some mutations may be silent, meaning they do not alter the amino acid sequence due to the redundancy of the genetic code, while others may confer subtle changes that do not critically impair protein function.
The evolutionary trajectory of organisms is intricately intertwined with the conservation of coding sequences. By scrutinizing the rate of sequence conservation within these regions, researchers can glean insights into the evolutionary dynamics shaping genetic diversity across taxa. Furthermore, comparative analyses of coding regions serve as a powerful tool for elucidating the functional constraints imposed on genes throughout evolutionary history, offering invaluable glimpses into the molecular underpinnings of life’s diversity.
The significance of coding regions transcends mere nucleotide sequences; they embody the quintessence of genetic information, shaping the intricate tapestry of life through their orchestration of protein synthesis. Through the lens of evolution, these regions serve as repositories of biological history, bearing witness to the enduring struggle for genetic stability amid the ceaseless tide of mutational flux.
The significance of mitochondrial genes: Insights into species identification
The mitochondrial genome is a treasure trove of genetic information, characterized by its unique inheritance patterns, gene arrangement, and functional diversity. As elucidated by Mangold et al. (1998), mitochondrial genes are inherited exclusively from the maternal lineage, a phenomenon known as maternal inheritance. This feature not only underscores the importance of mitochondria in cellular biology but also facilitates molecular analyses by enabling researchers to trace genetic lineages through maternal ancestry.
The arrangement of genes within the mitochondrial genome is highly conserved across species, albeit with variations. Notably, the cytochrome oxidase subunit 3 (COIII) gene is flanked by cytochrome oxidase subunit 1 (COI) and cytochrome oxidase subunit 2 (COII), with ATP synthase genes intercalated between them, as detailed by Mangold et al. (1998). Furthermore, adjacent to each other lie the 16S and 12S rRNA coding genes, crucial components of the mitochondrial ribosome (Fig. 4).
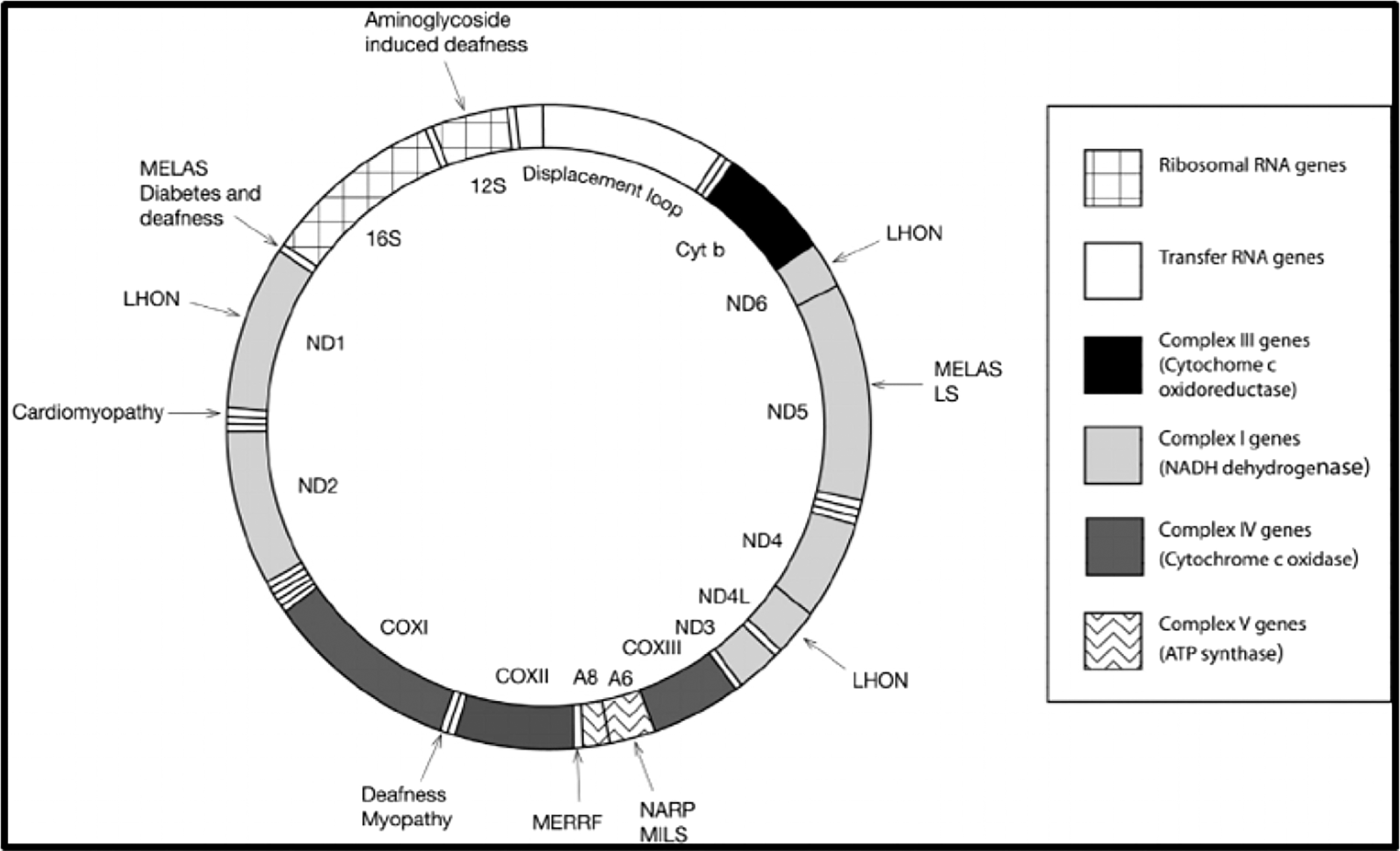
This diagram illustrates the mitochondrial genes commonly utilized in tick molecular analysis, highlighting the positions of the following genes: 16S, 12S, COXI, COXII, and COXIII. Additionally, other relevant genes are depicted within the diagram.
Despite this structural conservation, mitochondrial genomes are subject to high mutation rates and rearrangement events, leading to considerable variability, as observed by Norris et al. (1996). This inherent variability presents a boon for molecular studies, offering insights into evolutionary relationships, population dynamics, and species identification. The detection of such variability among individual ticks, as highlighted by Mangold et al. (1998), holds immense potential for advancing our understanding of tick biology and ecology.
Certain mitochondrial genes have emerged as pivotal targets for molecular analysis in ticks. For instance, the cytochrome c oxidase subunit 1 (COI) gene has been extensively employed for phylogenetic analyses and species identification within genera such as Hyalomma and Rhipicephalus (Black and Piesman 1994). Meanwhile, while COII remains relatively unexplored in this context, COIII has shown promise in quantifying tick species, as noted by Mangold et al. (1998).
The mitochondrial genome serves as a rich reservoir of genetic diversity and evolutionary history in ticks and other organisms. Its distinctive features, including maternal inheritance, gene arrangement, and variability, offer valuable avenues for molecular research, driving advancements in taxonomy, phylogenetics, and population genetics. By leveraging these insights, scientists can unravel the intricate tapestry of tick biology and its implications for human and animal health.
The Significance of Nuclear Ribosomal Genes: Insights into Species Identification
The composition and organization of rRNA genes play a crucial role in molecular analysis, particularly in delineating evolutionary relationships among organisms such as ticks (Takano et al. 2014). Within the nuclear genome, these genes encompass the large subunit (28S) and the small subunit (18S), integral components of ribosomes pivotal for protein translation (Black and Piesman 1994).
Internal transcribed spacers (ITS) delineate these nuclear ribosomal gene clusters, with ITS1 separating the 18S and 5.8S rRNA genes and ITS2 segregating the 5.8S and 28S rRNA genes (Lv et al., 2014). These regions serve as important markers for phylogenetic studies, particularly at family and subfamily levels in ticks, owing to the high conservation of flanking rRNA genes (Lv et al. 2014).
The 18S rRNA gene, extensively studied due to its slow evolutionary rate, is particularly valuable for ancient comparisons (Mangold et al. 1998). Conversely, the 28S rRNA gene exhibits greater variability across its domains, making it a useful tool for exploring evolutionary events spanning the Paleozoic and Mesozoic eras (Mangold et al. 1998).
However, challenges arise with shorter rRNA sequences. The 5.8S rRNA gene, despite its positioning between the ITS regions, proves limited in molecular analyses owing to its low variability across taxa (Mangold et al. 1998). Similarly, the 5S rRNA gene, even shorter than 5.8S, further restricts the utility of such sequences in molecular studies (Norris et al. 1998).
The large subunit (28S) harbors divergent domains or expansion segments, leading to considerable variation in gene size among phyla (Mangold et al. 1998). These divergent domains are valuable for studying recent evolutionary events, though careful selection of regions is imperative, especially when analyzing taxa with recent divergence (Mangold et al. 1998).
Understanding the characteristics and variability of different rRNA genes is paramount for effective molecular analysis, offering insights into the evolutionary history and relationships within tick species and other organisms (Lv et al. 2014).
The Significance of Nuclear Ribosomal Genes: Insights into Species Identification
Understanding the molecular landscape of nuclear protein-coding genes poses a significant challenge in research due to their limited copy numbers. This scarcity complicates the process of amplification, thereby impeding molecular studies (Lv et al. 2014). Moreover, these genes exhibit considerable variability in substitutional and evolutionary rates, further complicating their analysis. In light of these challenges, identifying effective genetic markers becomes paramount for molecular research.
One such marker of particular significance is the adenine nucleotide translocase (ANT) gene. This gene has proven to be instrumental in detecting cryptic species and facilitating molecular analyses within populations of ticks belonging to the same species (Lv et al. 2014). Its utility lies in its ability to provide insights into genetic variation and differentiation among closely related tick populations. By leveraging the information encoded within the ANT gene, researchers can unravel the intricate genetic dynamics within tick populations, aiding in species identification and evolutionary studies.
The use of ANT as a molecular tool underscores the importance of identifying and characterizing genes that offer robust insights despite the challenges posed by low copy numbers. As molecular techniques continue to advance, the exploration of such genes holds promise for enhancing our understanding of diverse biological processes and evolutionary relationships within tick populations. Consequently, ANT serves as a beacon for molecular researchers navigating the complex terrain of tick genetics, offering valuable avenues for further exploration and discovery (Lv et al. 2014).
The Significance of Noncoding Regions: Insights into Species Identification
Noncoding regions play a pivotal role in molecular analysis, especially in the identification and phylogenetic studies of various organisms, including ticks. These regions, devoid of functional transcript coding, exhibit rapid evolution due to minimal selection pressure, rendering them valuable tools for deciphering evolutionary relationships (Black and Piesman 1994).
Among the noncoding regions, the ITS regions stand out for their utility. ITS1, located between the 18S and 5.8S rRNA genes, though less commonly utilized in tick molecular analyses, shares similar evolutionary properties with ITS2. The latter, positioned between the 5.8S and 28S rRNA genes, is particularly favored for molecular analyses due to its propensity to accumulate substitutions rapidly. This characteristic makes ITS2 an invaluable asset in unraveling the intricate genetic makeup of ticks, aiding in species differentiation and phylogenetic reconstructions (Black and Piesman 1994).
Additionally, noncoding regions such as the external transcribed spacer (ETS) and nontranscribed spacer (NTS) contribute significantly to molecular analyses. The ETS, situated upstream of the 18S gene, harbors essential signals for processing rRNA transcripts. Meanwhile, the NTS, also known as the intergenic spacer, contains subrepeating elements that act as enhancers of transcription, effectively separating adjacent copies of the rDNA repeat unit (Wesson et al. 1993).
Despite the immense potential of molecular techniques in tick species identification, regions such as sub-Saharan Africa still rely predominantly on historical morphological records, which inherently suffer from limitations in accuracy and reliability. In Tanzania, for instance, the absence of genetic identification methods and molecular databases for locally collected ticks underscores the urgent need for integration of molecular approaches into tick research and identification practices. Establishing comprehensive molecular databases, particularly focusing on marker genes from indigenous tick populations, could substantially enhance the precision and efficiency of species identification efforts in the region, addressing the inherent challenges associated with morphological methods alone.
Thus, by embracing molecular tools alongside traditional morphological techniques, researchers can navigate the complexities of tick taxonomy more effectively, ultimately advancing our understanding of tick-borne diseases and their ecological dynamics in sub-Saharan Africa and beyond.
Comprises the 18S and 28S genes encoding ribosomal RNA, with the 5.8S gene in between, though not typically utilized in tick diversity analyses. Surrounding these genes are the ITS regions, including ITS1 and ITS2, highly variable segments commonly employed in molecular diversity studies of ticks. Conversely, the ETS and NTS regions, located outside the 18S and 28S genes, are less utilized in tick diversity analyses due to their lower variability compared with the ITS regions (Fig. 5).
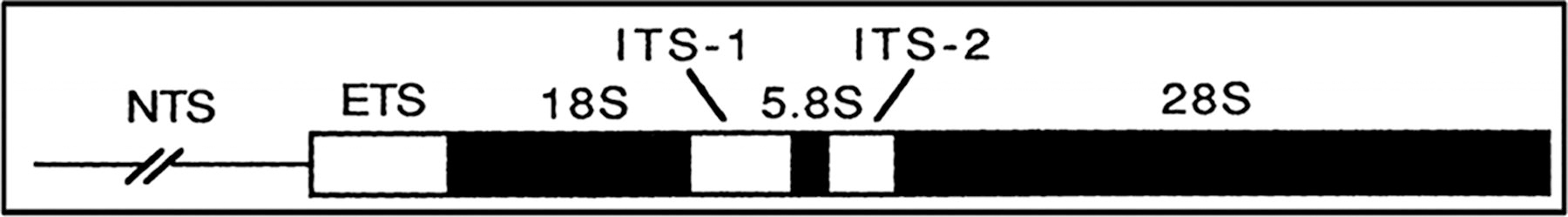
A diagram of nuclear ribosomal DNA (rDNA) gene.
Conclusion
In conclusion, the intricate interplay between infectious diseases and their origins in wildlife, particularly zoonoses, highlights the critical need for a comprehensive understanding of disease transmission dynamics, particularly in regions characterized by extensive interactions between domestic animals and wildlife, such as sub-Saharan Africa. The prevalence of free-range systems in these areas facilitates direct or indirect contact between domestic animals and potentially pathogen-carrying wild animals or vectors, emphasizing the urgency of effective control measures. Ticks, as prominent vectors of disease transmission, impose significant economic burdens and health risks on human populations, livestock, and wildlife alike. The intricate relationship between ticks and their hosts underscores the need for a multifaceted approach to tick control and disease prevention. Despite their pivotal role, our knowledge of tick species in Sub-Saharan Africa remains predominantly reliant on historical morphological records. However, the inherent limitations of morphological identification methods necessitate a paradigm shift towards the adoption of molecular techniques for more precise species characterization. Molecular analysis offers a direct and reliable avenue for discerning the genetic diversity and relationships among tick species and populations. By leveraging molecular insights, we can elucidate key factors influencing tick-borne disease emergence and re-emergence, inform targeted control strategies, and monitor the efficacy of intervention efforts. This review not only provides a comprehensive overview of ticks and their implications but also underscores the imperative of integrating molecular techniques into tick surveillance and control programs. Identifying regions conducive to molecular analysis and harnessing the power of molecular data will be instrumental in refining our understanding of tick ecology, enhancing disease surveillance, and ultimately mitigating the impact of tick-borne diseases on public health and agricultural economies. In essence, by embracing molecular advancements and incorporating them into holistic control strategies, we can forge a more resilient frontline against the scourge of tick-borne diseases, safeguarding the well-being of both human and animal populations in sub-Saharan Africa and beyond.
Footnotes
Author Disclosure Statement
In this review, the authors declare no competing interests, indicating they have no affiliations or financial involvement that could potentially sway their findings.
Funding Information
No funding was received for this article.