
Abstract
Coxsackievirus B3 (CVB3) is associated with several different acute and chronic forms of human disease, including myocarditis, aseptic meningitis, and pancreatitis. Moreover, CVB3 also infects immune cells like CD19+ B lymphocytes, but the viral uptake mechanism into these cells is not well understood. Therefore, primary murine and human CD19+ B cells were isolated by magnetic-activated cell separation technology and analyzed for virus receptor expression, antibody-dependent enhancement of viral infection, and different cellular surface proteins, that might be involved in mechanisms of viral uptake. Western blot analysis of these cells revealed no significant expression of the coxsackievirus-adenovirus receptor CAR. But incubation of CVB3 with serum dilutions, which exhibited binding but not neutralizing characteristics, increased viral uptake and replication significantly in a dose-dependent manner. Viral entry was reduced when Fc portions of immunoglobulins were blocked by protein A treatment. Moreover, the classical complement system rather than Fc-γ-receptor-mediated mechanisms could be involved in viral uptake. Taken together, these data suggest an antibody-dependent enhancement of CVB3 infection of primary murine and human CD19+ B cells.
Introduction
In addition to solid organs, CVB3 can infect immune cells of different types of cell lines as well (26,36). In situ hybridization techniques also revealed that murine B cells of spleen and lymph nodes were virus-positive after intraperitoneal (IP) infection (23). Moreover, CVB3 is capable of replicating in a certain percentage of murine CD19+ B cells, but only in a limited manner (20). Consequently, B lymphocytes may represent a non-cardiac enteroviral reservoir that may be involved in viral dissemination and maintenance of persistent myocardial infection (1,23,27). However, mRNA expression of the common coxsackievirus-adenovirus receptor CAR is only barely detectable in murine B-cell preparations (27). Therefore, the viral mechanism behind how it infects these lymphocytes so efficiently is not well understood. One strategy to increase CVB3 infectivity could be mediated by antibodies. Thereby, on the one hand neutralizing immunoglobulins (Ig) decrease the ability of CVB3 to infect target cells or organs, but on the other hand virus-specific Ig may enhance virus entry. This mechanism, named antibody-dependent enhancement (ADE) of infection, was first described in 1964 (13), and has been identified for numerous different viruses [reviewed in (18)]. ADE of CVB3 infection has already been studied in vitro and in vivo, demonstrating, for example, that mice with pre-existing low anti-CVB3 antibody titers developed a more severe/lethal myocarditis after a second infection with a highly pathogenic CVB3 strain than did CVB3-seronegative mice (22). In humans, CVB infections are quite common and induce a humoral immune response against at least one of the six CVB serotypes (5). During a second CVB infection, pre-existing group-specific antibodies may worsen the course of the disease. This process may occur not only in patients with cardiomyopathies, but it may also be involved in the onset of type 1 diabetes mellitus (30).
Here, one specific aspect of CVB3-caused pathology, the enhancement of infection of primary murine and human CD19+ B lymphocytes, was studied for the first time with regard to the ADE of viral entry.
Materials and Methods
Cells and virus
Splenic CD19+ B lymphocytes were obtained from male 6- to 8-week-old BALB/c mice. Human CD19+ B cells were isolated from peripheral blood of healthy donors. B cells were purified by magnetic-activated cell separation (MACS) as described previously (20). Briefly, murine single-cell splenocyte suspensions were prepared in a glass tissue homogenizer. Then the cells were centrifuged (5 min, 3600 rpm, 4°C), followed by shock lysis of erythrocytes for 2.5 min in 0.83% NH4Cl at room temperature. RPMI 1614 medium was added, and after an additional centrifugation (5 min, 3600 rpm, 4°C), the cell pellets were resuspended in cold buffer (2 mM EDTA/PBS and 0.5% BSA). Human CD19+ cells of buffy coats from healthy blood donors were obatined by Ficoll separation (Biochrom Corp., Miramar, FL). After isolation, the cells were washed twice with PBS and resuspended in cold buffer (2 mM EDTA/PBS and 0.5% BSA). To avoid cell clumps, the cells were passed through a 30-μm nylon mesh pre-separation filter (Miltenyi Biotec, Gladbach, Germany). Thereafter, the splenocytes were incubated with magnetic beads that were conjugated with anti-mouse or anti-human CD19 antibodies (both from Miltenyi Biotec) for 15 min at 8°C. After washing with MACS buffer (centrifugation at 1000 rpm and 4°C for 10 min), CD19+ B lymphocytes were positively selected by using a VarioMACS separator (Miltenyi Biotec) according to the manufacturer's instructions. Then equal amounts (multiplicity of infection of 10, m.o.i. = 10) of a cDNA-generated CVB3 variant (24) were pre-incubated with murine and human serum (diluted as indicated) for 1 h at 37°C. Thereafter, primary cells were infected with the virus-serum mixture. At the indicated time points, the cells were centrifuged and the supernatants were carefully removed. Lymphocytes were washed three-times with PBS to eliminate all cell-free viruses and stored at −80°C. After three repeated freeze/thaw cycles, cell debris was removed by centrifugation and the supernatants were subjected to sequential 10-fold dilutions in DMEM. Virus titers were determined by TCID50 assays (20). The statistical comparisons were carried out with Microsoft Excel by using Student's t-test. All experiments were performed three times and complied with all federal permissions, guidelines, and international policies.
Serum samples
Murine sera were obtained either from CVB3-infected BALB/c mice at 7 days post-infection (dpi), or from non-infected mice (serum controls). Human sera were obtained from healthy coxsackievirus-seropositive and coxsackievirus-seronegative donors. Neutralizing activity of the serum preparations was analyzed by neutralization assays as described previously (14).
Western blot analysis
Protein detection was performed with non-infected murine or human CD19+ B lymphocytes or Raji cells (ATCC CCL-86™, positive control), or with CVB3/EGFP-infected murine CD19+ B lymphocytes. The recombinant virus variant CVB3/EGFP was constructed by cloning the enhanced green fluorescent protein (EGFP) gene sequence downstream of the CVB3 5′-NTR, together with an artificial 3C protease cleavage site (31). CVB3/EGFP expresses the reporter protein only simultaneously during its normal replication, and thus a clearly visible monitoring of an ongoing CVB3 infection is possible, for example by using Western bolt analysis. Based on the addition of the EGFP gene sequence into the viral genome, the replication kinetics of CVB3/EGFP are substantially prolonged in comparison to wild-type CVB3, without inducing a cytopathic effect at 20 hours post-infection (hpi). Then, virus suspensions were pre-incubated with the murine CVB3-specific serum or the serum control (both diluted 1:100) for 1 h at 37°C. Cell pellets of each sample were prepared for SDS-PAGE by resuspending the cells in NTE buffer (10 mM Tris/HCl [pH 7.4], 100 mM NaCl, and 0.5% NP-40), and vortexing for 30 sec. Cleared supernatants (centrifugation at 14,000 rpm at 4°C for 20 min) were subjected to Bradford protein assays, and equal amounts of protein were mixed with SDS sample buffer and incubated at 95°C for 5 min. The samples were loaded on a 10% or 12.5% gel. After separation by SDS-PAGE, the proteins were transferred to nitrocellulose membranes, probed against an anti-CAR (clone H-300, 1:200; Santa Cruz Biotechnology, Santa Cruz, CA), an anti-GFP (clones 7.1 and 13.1, 1:450; Roche Diagnostics GmbH, Mannheim, Germany), or an anti-β-actin (clone 13E5, 1:1000; Cell Signaling Technology, Inc., Beverly, MA) antibody, and visualized with AP-conjugated secondary antibodies.
Immunocytochemical staining
Viral antigen was detected using the alkaline phosphatase anti-alkaline phosphatase (APAAP) method. First, CVB3-infected African green monkey kidney (GMK) cells were fixed 4 hpi and incubated with different dilutions of the CVB3-specific serum, with the serum control, or remained untreated. Anti-mouse linker-antibodies, AP-conjugated anti-AP-antibodies, and color substrates were applied according to the manufacturers instructions (Dako REAL™ Detection System; Dako Denmark A/S, Glostrup, Denmark). Bright red staining indicates virus-positive cells.
Treatment of murine serum
As previously described, murine serum samples were diluted 1:20–1:1000 using serum-free medium, or 1:100-diluted CVB3-specific serum was treated with different substances. To block the antibody's Fc portions, it was incubated with 200 μg/mL protein A (Sigma-Aldrich Co., St. Louis, MO) for 30 min. Complement inhibition was achieved either by heating for 30 min at 56°C, or by treatment with 10 mM EGTA for 30 min. As control, virus-specific serum remained untreated. Thereafter, equal amounts of CVB3 (m.o.i. = 10) were mixed with these appropriate serum probes for 1 h at 37°C. Then, murine CD19+ B cells were infected with CVB3. At 2 hpi, the supernatants were removed, and the lymphocytes were washed three times with PBS to eliminate all cell-free viruses, and analyzed for the presence of infectious virus by TCID50 assays (20).
Blockade of Fc-γ receptors
In order to block Fc-γ receptors on the cellular surface of murine CD19+ B lymphocytes, cells were incubated either with an anti-Fc-γRII/Fc-γRIII antibody (clones 93, 0.5 μg/106 cells, 90 min at 37°C; eBioscience, San Diego, CA), or with a common mouse FcR-blocking reagent (1:10 diluted; 10 min at 4°C; Miltenyi Biotec). Control cultures remained untreated. After washing with PBS, the cells were infected with CVB3 (m.o.i. = 10), which was pre-incubated with the murine CVB3-specific serum. At 2 hpi, the supernatants were removed, and the lymphocytes were washed three-times with PBS to eliminate all cell-free viruses, and analyzed for the presence of infectious virus by TCID50 assays (20).
Results
CVB3 uptake into primary CD19+ B lymphocytes: Enhancement by virus-positive serum
It was demonstrated previously that primary murine CD19+ B lymphocytes can be infected with CVB3, but only in a limited manner (20). In addition, the transcription of the virus receptor CAR was only slightly detectable in preparations of murine splenic B cells (27). In accordance with this observation, our Western blot analysis revealed no significant expression of the common CVB3 receptor CAR in murine and human primary CD19+ B lymphocytes, since no CAR-specific signal could be detected, whereas Raji cells clearly exhibited CAR expression (see insert in Fig. 1). Therefore, the question remains how the infection of these cells can be enhanced to achieve pathological importance. As shown previously, virus-specific antibodies facilitate coxsackieviral infection of peripheral blood mononuclear cells (PBMCs) (17), and plasmacytoid dendritic cells (pDCs) (37). Therefore, a 100-fold diluted CVB3-specific mouse serum without neutralizing activity and a serum control (1:100 dilution) were used. Comparable conditions were applied to human cells (human CVB3-specific serum and control serum, both 1:200 dilution). Virus was pre-incubated (1 h at 37°C) with these different serum samples and murine (Fig. 1A) and human (Fig. 1B) CD19+ B cells were infected thereafter. During the course of these experiments, pre-incubation with a CVB3-specific but non-neutralizing serum dilution increased viral uptake significantly at 2 hpi. This early event also caused an increase in production of viral progeny in murine samples at 10–11 hpi. A comparable set of experiments with CAR-positive Raji cells revealed no further viral uptake due to ADE mechanisms (data not shown).
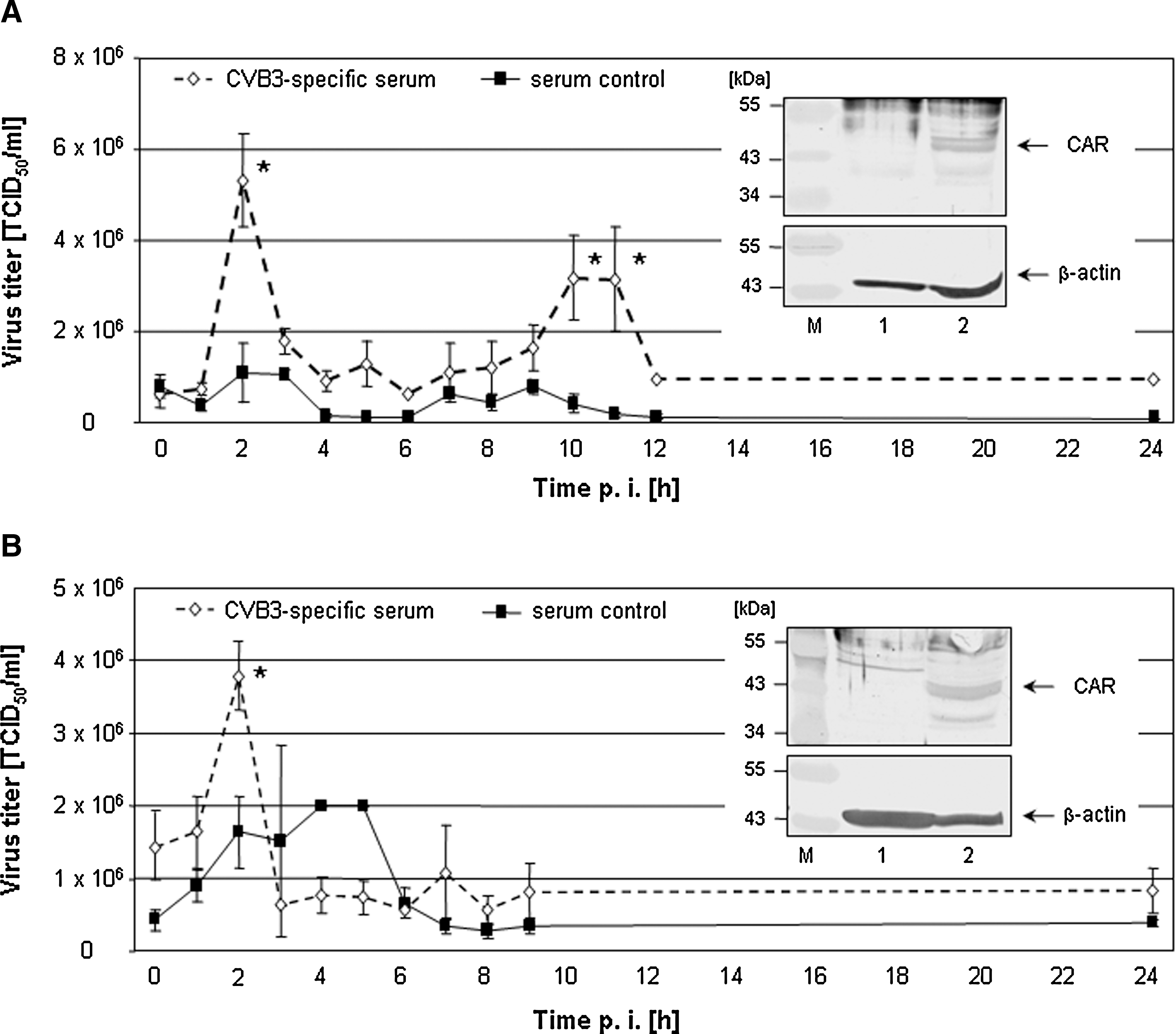
Prior to infection, CVB3 was incubated with virus-specific serum or control serum without CVB3 binding activity (murine sera: 1:100 dilution; human sera: 1:200 dilution). Then, primary murine (
Antibody-dependent increase of intracellular CVB3 replication
In order to demonstrate that increased viral replication occurred due to the ADE of viral uptake, a specific experiment to prove active viral replication was performed. In it, a recombinant CVB3 variant named CVB3/EGFP was applied, which expresses the reporter protein simultaneously only during replication (20). Therefore, if EGFP is detectable in protein samples of CVB3/EGFP-infected cells by Western blotting, this verifies active viral replication. In order to perform these experiments, CVB3/EGFP was incubated with the 100-fold diluted CVB3-specific mouse serum without neutralizing activity, and a serum control (1:100 dilution) for 1 h at 37°C prior to infection. Then freshly isolated murine CD19+ B lymphocytes were infected with these virus-serum mixtures. After isolation of total protein of murine CD19+ B cells and separation on a SDS-PAA gel, an increased virus-based translation of EGFP (27 kDa) was detectable in samples with the CVB3-specific serum by Western blotting using a monoclonal anti-GFP antibody (Fig. 2). EGFP expression was normalized using total β-actin expression. Density values indicate a 3.27-fold increase of EGFP expression in CVB3/EGFP-infecetd murine CD19+ B lymphocytes following incubation with the virus-specific serum at 20 hpi. This long incubation period was used due to the reduced replication activity of the CVB3/EGFP variant. At this time no virus-induced cytotoxicity could be observed (data not shown).
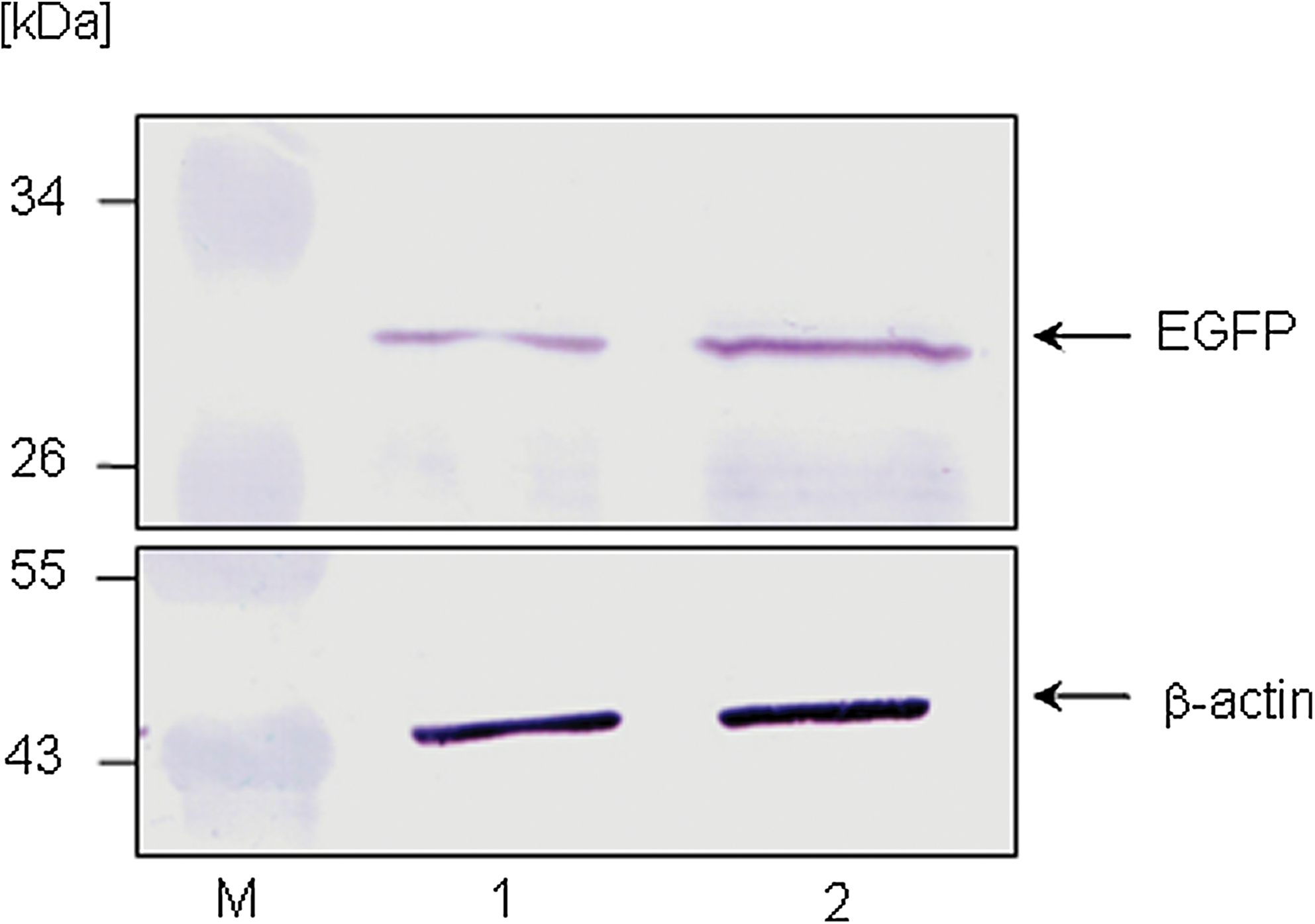
Prior to infection, the recombinant variant CVB3/EGFP was incubated with murine virus-specific serum or control serum without CVB3 binding activity (1:100 dilution). Then primary murine CD19+ B lymphocytes were infected with those serum-treated CVB3/EGFP mixtures, and 20 h later, equal protein amounts of each culture were separated on a 12.5% SDS-PAA gel and probed against an anti-EGFP or anti-β-actin antibody using Western blot technique (M, protein ladder; lane 1:, CVB3/EGFP-positive serum control 1:100; lane 2, CVB3/EGFP-positive CVB3-specific serum 1:100). Color images available online at
Non-neutralizing CVB3-specific serum samples maintain virus-binding properties
Next, equal amounts of CVB3 were incubated with different dilutions of the murine CVB3-specific serum (1:20–1:1000) or the control serum for 1 h at 37°C. Thereafter, remaining infectivity was studied by TCID50 titrations. Fig. 3 shows that the 1:100–1:1000 dilutions of the CVB3-specific serum had no neutralizing effect on CVB3, in contrast to the 1:20 dilution, which caused more than a 90% reduction of viral infectivity. As expected, the serum control had no virus neutralizing effect at all. Interestingly, despite the inability to neutralize CVB3 at a dilution of 1:100, the CVB3-specific serum was capable of detecting CVB3 antigen in infected cells very efficiently. The panels at the bottom of Fig. 3 show CVB3-infected GMK cell cultures, which were analyzed using the APAAP method to detect viral antigen. The 1:100 serum dilution revealed the typical staining pattern of CVB3-infected cells. Serum controls and samples without serum had background activity. The use of the 1:20 dilution caused the strongest coloration, which declined with increasing dilution. These results verify that CVB3-specific serum is able to detect viral antigen in infected cells, even when the neutralizing activity was below neutralizing levels due to high dilution.
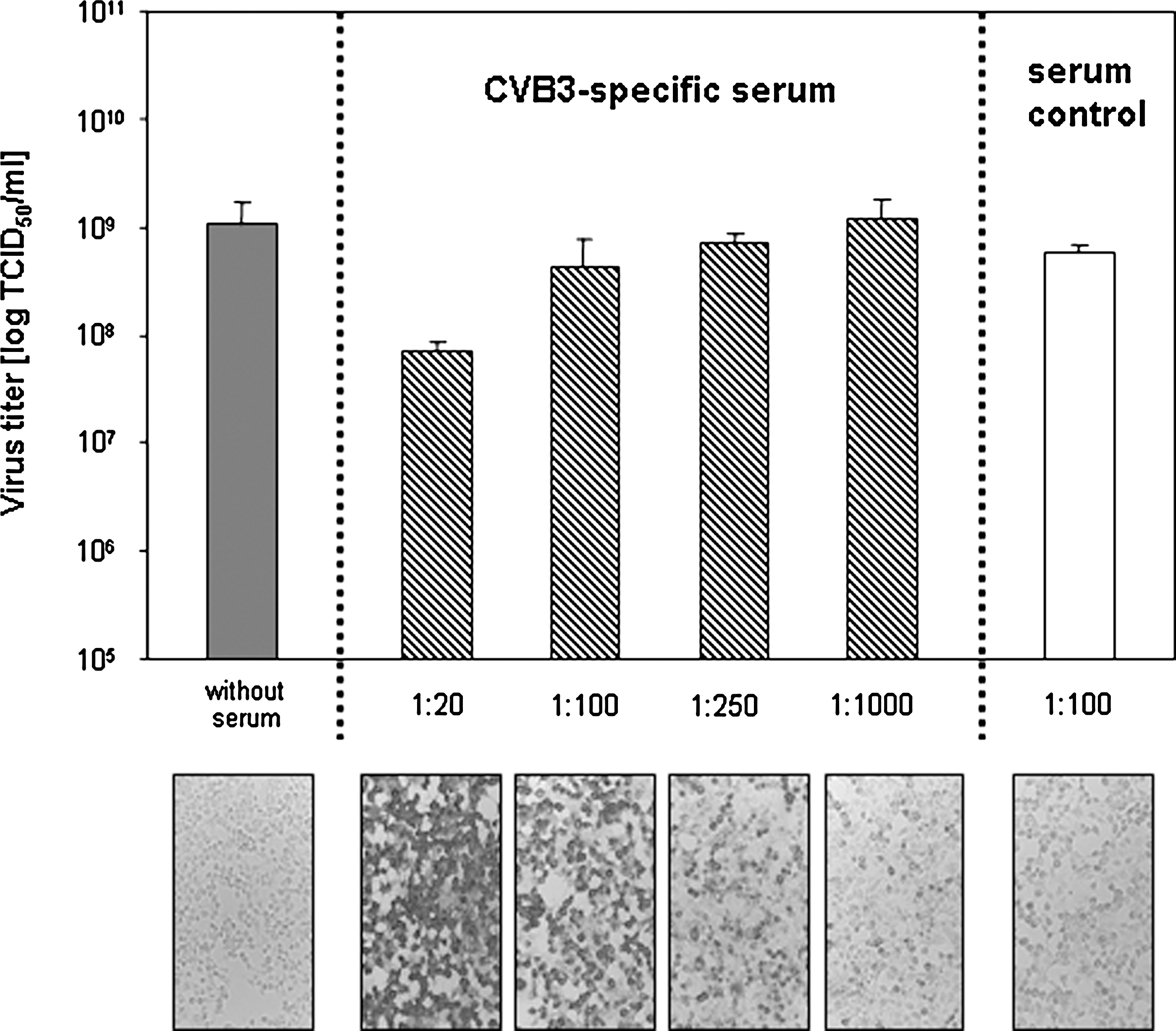
CVB3 was incubated with or without different dilutions of the virus-specific serum or the serum control (1:100 dilution) for 1 h at 37°C. Thereafter, remaining infectivity was analyzed by TCID50 titrations. Only the 1:20 dilution of the CVB3-specific serum decreased viral infectivity. Thereafter, these serum dilutions were used to detect viral antigen in CVB3-infected GMK cells by immunocytochemistry. The APAAP staining of virus-infected GMK cells revealed that the 1:100 dilution of the CVB3-specific serum detected viral antigen efficiently. The application of higher dilutions reduced the staining signal to background levels of the serum control.
CVB3 uptake depends on virus-specific antibody concentration
The concentration of antibodies in CVB3-specific serum samples also influenced the observed ADE (Fig. 4A). Dilutions of 1:250, and especially 1:1000, reduced the enhancing effect of the CVB3-specific serum significantly, until only background activity was achieved. In order to confirm the participation of antibodies in the process, protein A was added to the 1:100 serum dilution. Protein A is a 40- to 60-kDa surface protein originally found in Staphylococcus aureus. It binds with high affinity to the Fc region of human IgG, as well as mouse IgG, through interaction with the IgG heavy chain. This activity prevents the interaction of IgG with its receptors on cellular surfaces. Protein A treatment of the CVB3-specific serum significantly decreased the ADE of CVB3 uptake into murine CD19+ B lymphocytes at 2 hpi (Fig. 4B). This result indicates that due to protein A–based blockade, fewer infectious viruses were internalized by primary B cells. Therefore, the binding of virus-antibody complexes to cellular surface molecules via the Fc region may be involved in the ADE of CVB3 uptake.
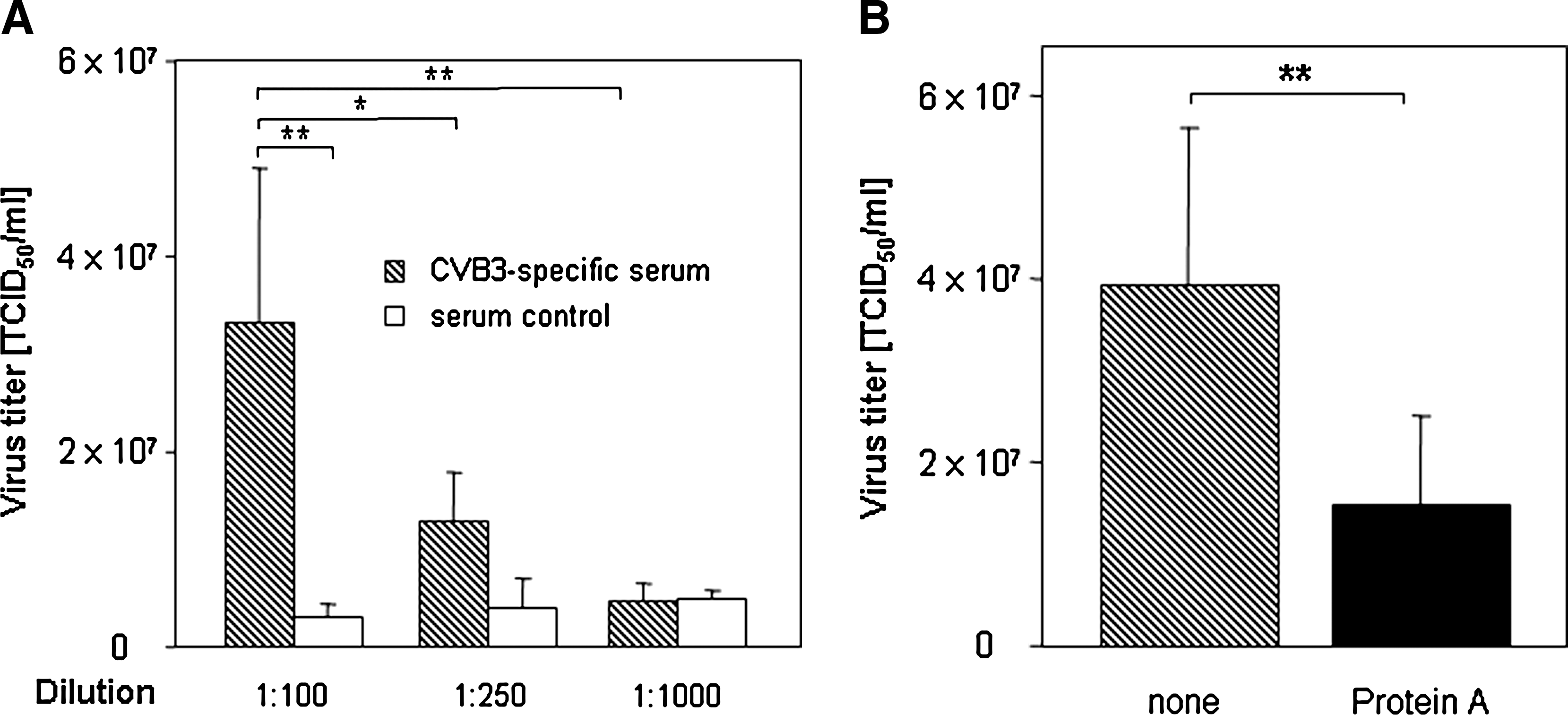
(
Potential role of the complement system
In order to analyze if the cellular Fc receptors (FcR) contribute to the antibody-mediated entry, several experiments were performed. First, the binding capability of anti-Fc-γRII/III antibodies to murine CD19+ B cells was tested using FITC staining. After this binding was confirmed by immunocytochemistry (data not shown), different blocking experiments were performed. In these, primary murine CD19+ B cells were pre-incubated with antibodies directed against common Fc-γRII (CD32) and Fc-γRIII (CD16) molecules on B lymphocytes. At the same time, virus was also treated with the 1:100 dilution of CVB3-specific serum. Then the cells and virus were incubated together for 2 h. Thereafter, samples were obtained and intracellular virus concentrations were determined (Fig. 5). Surprisingly, the anti-Fc-γRII/III blockade on these cells had no inhibitory influence on viral uptake. This result was verified by experiments using a commercially available FcR blocking reagent (Miltenyi Biotech). This reagent efficiently blocks all Fc binding positions on cellular surfaces. Treatment of cells with the FcR-blocking reagent did not influence the ADE-mediated virus uptake either, confirming the data obtained with anti-Fc-γRII/III antibodies. These results indicate that binding of virus-specific antibodies rather than other components to Fc-γR may be involved in CVB3 uptake. The complement system may be involved in this process. For example, in the classical pathway, C1 binds with its C1q subunits to Fc fragments of virus-antibody complexes, which may then increase viral uptake into B lymphocytes. Our initial results support this hypothesis. Conventional heat treatment (56°C for 30 min) of CVB3-specific serum to inactivate complement components prior to virus pre-ncubation significantly reduced the enhancing effects seen in primary murine CD19+ B lymphocytes at 2 hpi. (Fig. 5). Further experiments were performed to confirm this result. Serum samples were treated with 10 mM ethylene glycol tetraacetic acid (EGTA), which is a chelating agent that binds free calcium ions preferentially. Those ions are necessary to activate the classical pathway of the complement system. EGTA treatment of CVB3-specific serum decreased the ADE effect of virus-specific antibodies at 2 hpi, indicating that components of the classical complement system may be responsible for this specific CVB3 uptake mechanism.
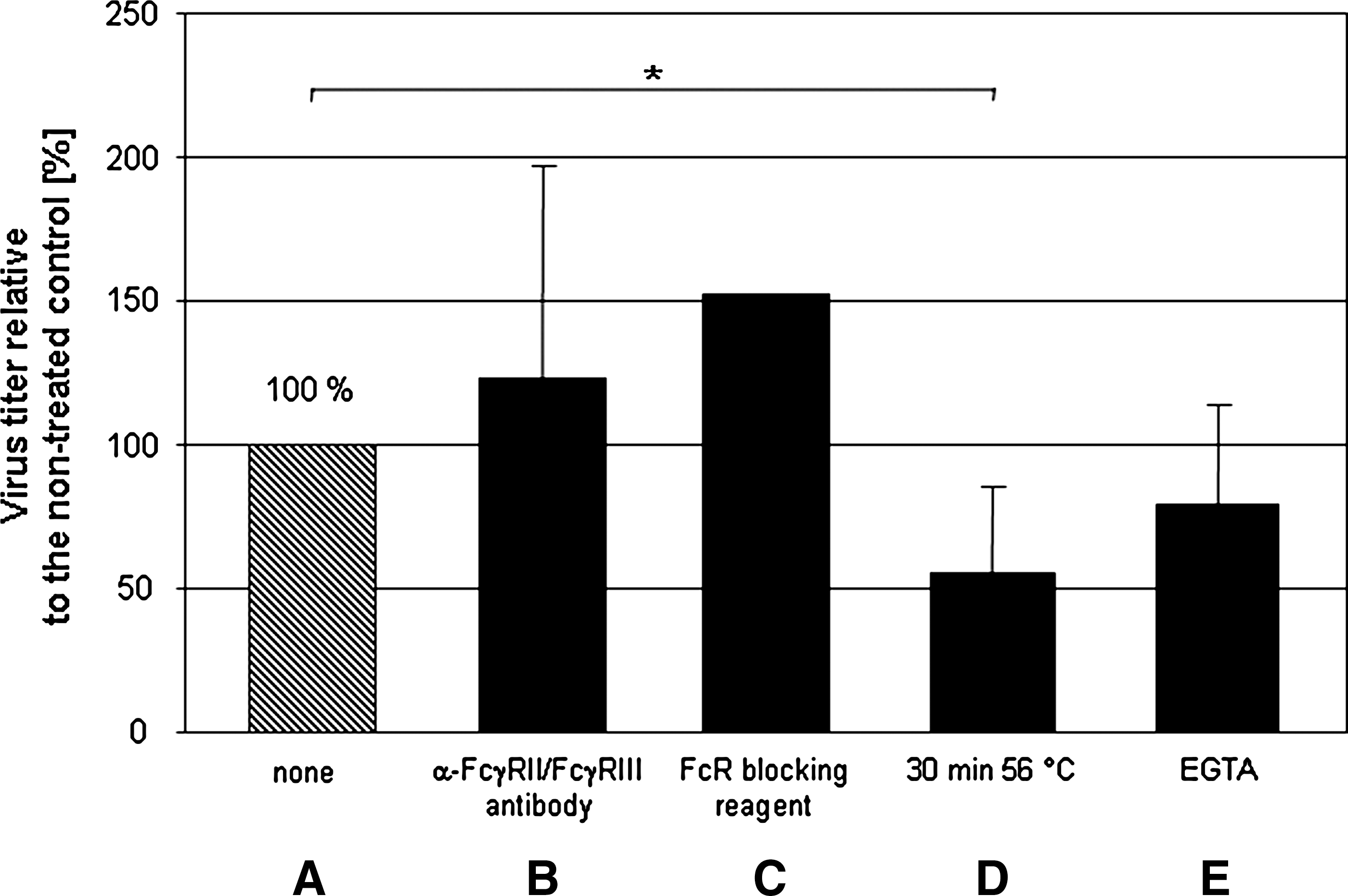
Murine CD19+ B lymphocytes were either incubated with an anti-Fc-γRII/Fc-γRIII antibody (
Discussion
Coxsackievirus infections are a major cause of virus-induced heart disease in humans. However, not only cardiomyocytes, but also different types of immune cells, are susceptible to infection by coxsackieviruses. This has been studied in vitro and in vivo preferentially by using established cell lines (35), human monocytes (16), leukocytes (34), B lymphocytes (20), and PBMCs (3,17). But the viral entry mechanism used to infect these cells is not well understood, because murine B lymphocytes only slightly express CAR (27). This observation was confirmed by our experiments (Fig. 1A), and was extended to primary human CD19+ B cells (Fig. 1B). Therefore, the question remains how CVB3 enhances its ability to infect these cells. One explanation for this process is that virus-specific antibodies may be involved.
Antibody-dependent enhancement of viral uptake is an important mechanism by which viruses infect immune cells. This strategy is responsible for the exacerbation of different diseases, such as AIDS [recently reviewed in (32)] and dengue hemorrhagic fever (18). Moreover, infection of B cells with Epstein-Barr virus may trigger the onset of multiple sclerosis in humans (29).
Here we demonstrated that CVB3 infection of primary CD19+ B lymphocytes was increased in the presence of virus-specific antibodies (Fig. 1A and B and Fig. 2). The results shown in Fig. 2 demonstrate that enhanced viral uptake was accompanied by enhanced viral replication. Using the CVB3/EGFP variant, a clearly increased expression of EGFP was found in murine CD19+ B cells, which were infected with opsonized virus. Here, EGFP expression only occurs if CVB3 actively replicates. Thus far, most of the published data about ADE of coxsackieviral infections are based on results using PBMCs or permanent cell lines [reviewed in (30)]. For example, anti-CVB2 IgGs increased CVB3 infection of J774.1 cells (8). In mice, macrophages may participate in the ADE of viral infectivity in the presence of homotypic or heterotypic virus-specific antibodies, which facilitate viremia and cause more severe myocardial symptoms (8). In human samples, virus-specific antibodies in plasma promoted CVB4 infection of PBMCs (2,17). With the results presented in Figs. 1 and 2, these observations can now be extended to primary B cells as well.
Generally, virus-specific serum contains a mixture of neutralizing and non-neutralizing, but binding, antibodies. As shown in Fig. 3, virus-specific serum dilutions with CVB3-binding, but not CVB3-neutralizing, activity were responsible for the increase in CVB3 uptake in a dose-dependent manner. Clearly, the virus-specific antibody concentration in the 1:100 dilution had no detectable neutralizing activity, but revealed enough binding capacity to detect viral antigen in CVB3-infected GMK cell cultures. Further dilution of the virus-specific serum decreased not only this binding activity, but also the ADE effect (Fig. 4A). A serum dilution of 1:1000 revealed only background binding capacity (like serum controls), and no ADE effect at all. Comparable studies were performed with human 293T cells after infection with a recombinant vesicular stomatitis virus expressing the GP protein of Ebola virus, and in those experiments increasing dilutions of a virus-specific serum also lost the ability to mediate ADE effects (33).
To confirm the involvement of antibodies, the question was raised whether protein A could interfere with the enhancing activity. As shown in Fig. 4B, protein A treatment reduced the virus-antibody complex uptake significantly. A number of different viruses elicit antibodies that enhance infectivity through binding of virus-antibody complexes to FcR on immune cells (such as macrophages) via Fc portions of Ig (6,12,28). This indicates that Fc regions of antibodies are involved in the CVB3 entry strategy, either via FcR or complement factors on the host cell surface. Recently it was demonstrated that CVB3 infection of primary murine and human pDCs required the presence of virus-specific antibodies, whereas FcR mediated the recognition of CVB3-antibody complexes (37). Therefore, several experiments were performed to block FcR on primary B cells. Prior to infection, murine CD19+ B lymphocytes were incubated with anti-Fc-(RII/III antibodies, or a commercially available FcR-blocking reagent (Fig. 5). Despite these treatments, the uptake of the virus-antibody complexes was not reduced, indicating that these FcR were not involved in the ADE effect. One other explanation for ADE could be the participation of the complement system, such as that hypothesized for the antibody-dependent, complement-mediated enhancement of HIV infection (6). As shown in Fig. 5, heat treatment of serum samples decreased the enhancing activity significantly. In addition, EGTA incubation of serum samples to block complement activity reduced CVB3 uptake as well (Fig. 5). These results suggest that components of the classical complement pathway may be involved in the CVB3 strategy to infect primary murine CD19+ B lymphocytes. Further experiments will be performed to analyze the precise mechanisms at work in more detail.
Conclusion
Despite the almost complete absence of the common virus receptor expression, CVB3 has been shown to infect primary murine and human CD19+ B lymphocytes. Diluted virus-specific antibody samples, which revealed binding but not neutralizing characteristics, were identified to increase virus uptake significantly in a dose-dependent manner. Additional experiments demonstrated that components of the classical complement system, and not Fc-γ receptor-mediated mechanisms, may be involved in this viral uptake. These results may be significant for human CVB3-caused disease, because low amounts of already existing virus-specific antibodies may worsen the outcome of a second CVB3 infection, especially in view of the involvement of CD19+ B lymphocytes. Further analysis should be focused on this aspect in patients with acute or chronic myocarditis.
Footnotes
Acknowledgment
This work was supported by the Deutsche Forschungsgemeinschaft grant HE 2910/6-2 (to A.H.).
Author Disclosure Statement
No competing financial interests exist.