
Abstract
Natural autoantibodies (NAbs) are continually produced throughout life and have an ability to recognize self and altered self, as well as foreign antigens, by recognizing cellular pattern recognition receptors. Sometimes NAb specificity demonstrates overlap between human and pathologic proteomes. This information can be useful in selecting target sequences for screening purposes. In this study we undertook a multi-step bioinformatics search to predict a virus-derived peptide that can be recognized by NAbs in sera of uninfected individuals. We selected protein hepatitis C virus (HCV) NS5A as a target sequence, motivated by the fact that the HCV proteome is characterized by extensive sequence similarities to the human proteome, and because screening for anti-HCV antibodies, including anti-NS5A, is important clinically, particularly in screening of potential blood donors. The virus-specific peptide P1, and the homologous human peptide derived from enzyme-inducible nitric oxide synthase (iNOS), P2, exhibiting not only simple homology, but also complementarities of physicochemical patterns, were synthesized and 80 HCV-negative and 50 HCV-positive blood donor sera were tested by ELISA. These peptides reacted similarly (p < 0.001) with HCV-negative sera, and in several cases the measured reactivity was significantly above the cut-off value of commercial anti-HCV screening assays. In HCV-positive sera, the titers of antibodies reactive with analyzed HCV NS5A peptide were not significantly increased (p < 0.001) compared to host peptide, the implications of which are unclear, but may be consistent with these antibodies being “naturally produced.” Finally, we extended our bioinformatics analyses to the dataset of human self-binding sequences, and propose a general approach for the selection of specific diagnostic and screening antigens for use in immunoassays.
Introduction
Hepatitis C virus is a significant human pathogen infecting about 3% of the world's population (49), and represents a leading cause of cirrhosis and liver cancer. An important characteristic of this pathogen is that a considerable number of extremely widespread and high homologies between the human and HCV proteomes have been observed (23). Consequently, a number of studies have investigated the potential role of molecular mimicry in autoimmune disorders associated with HCV infection (5,6,19). Others, such as Kanduc et al. (21), have focused on the consequences of homologies of HCV and human proteins in healthy individuals, and are concerned with sequence overlapping as an obstacle to vaccine development.
Anti-HCV antibody screening of donor blood is a major step in the prevention of transfusion-associated HCV hepatitis. A major problem with current screening assays is false-positive results (4,11,35). Blood donors comprise a population with low prevalence of infection; thus the sensitivity of screening immunoassays is maintained to be high in order to make sure HCV infection does not go undetected. This high sensitivity results in significant numbers of false-reactive results, each of which requires additional follow-up testing and adds to the cost of these assays.
Immunoassays for the detection of anti-HCV activity include a recombinant antigen from the non-structural 5A (NS5A) protein, in addition to antigens derived from the HCV core protein, NS3 and NS4 proteins, used in previous generations of screening tests (27). Evidence from various studies carried out with NS5-derived antigens have demonstrated that they are not as immunoreactive as the other antigens (3,26), and could be less specific (15).
It has been shown that the electron-ion interaction potential (EIIP) plot of peptide/protein sequences, and its Fourier transform, informational spectrum (IS), may be a good predictor of immunological cross-reactivity (22,43,46). The common frequency components in IS of two proteins are denoted as characteristic frequencies, and represent the determinant of cross-recognition. Here we assumed that the identification of characteristic frequencies will be important for prediction of NAb cross-recognition of HCV NS5A and host peptides.
In this study we applied bioinformatics approaches based on sequence analyses, and identified homologous HCV and human peptides that are afterwards cross-recognized by NAbs in ELISA. Further, this computational analysis was extended to the set of IgG NAbs binding self sequences published by Merbl et al. (33), who performed an investigation with an antigen microarray, and analyzed the natural autoantibody repertoires present in sera from healthy mothers and their newborns. The observed informational characteristics of this dataset are used as the basis for general guidelines for the selection of specific diagnostic and screening antigens for future immunoassays.
Materials and Methods
Human subjects
Serum samples were collected from 80 anti-HCV-negative and 50 anti-HCV-positive informed volunteer blood donors following local ethics committee approval, recruited from the Institute for Blood Transfusion of Serbia.
The sample material used in this study was what remained of samples used for standard donor blood testing. Absence of viremia was confirmed by negative amplification of HCV-RNA by the Roche Amplicor HCV detection methodology. The sera were stored for no longer than 6 months at −80°C, which had no detrimental effect on the results. The presence of viremia in HCV-positive samples was determined by COBAS® AMPLICOR HCV Test, v 2.0.
Protein databases and sequence analysis tools
Human protein sequences were retrieved from UniProtKB/Swiss-Prot. The list of self-binding proteins was made according to findings of Merbl et al. (33). In several instances, antigens of human origin were not tested; so we applied our virtual screening procedure to the human analogues of these sequences. In total we have tested 55 protein/peptide sequences. The homology analysis of HCV NS5A (P26664), and the human protein sequence sub-database created for this research purpose, were performed by using the BLASTP algorithm (1). Multiple sequence alignment was performed by ClustalW (25).
The informational spectrum method
The informational spectrum method (ISM) is a virtual spectroscopy method for structure function analyses of the proteins. The ISM (for reviews see 45,48) encompasses two stages. The first involves the transformation of the amino acid sequence into a numerical sequence. Each amino acid is represented by the value of the EIIP parameter (47), which corresponds to the average energy states of all valence electrons in a particular amino acid.
By using the discrete Fourier transform (DFT), the numerical sequence was transformed into the frequency domain to create an informational spectrum. The absolute value of the complex Fourier transform defines the amplitude spectrum and the phase spectrum. However, in the case of protein analysis, the relevant information is presented in amplitude spectrum. Because the average distance between amino acid residues in a polypeptide chain is 3.8 Å, it is assumed that the points in each derived series are equidistant and the distance is arbitrarily set equal to 1. The maximum frequency in the spectrum is F = 1/2d = 0.5.
In order to identify the common spectral characteristics of two sequences, mathematical filtering is performed by multiplication of the conjugate complex Fourier transform by the Fourier transform of the target signal. The result of the multiplication is the cross-spectral (CS) function for two sequences, or in consensus informational spectrum (CIS) for more than two sequences. The prominent peaks in this function denote the common frequency components of the analyzed proteins. The numerical series that are derived from the analyzed sequences are normalized to zero mean and zero padded to produce a vector that is equal in length to the smallest power of 2 greater than (or equal to) the largest domain in the data set.
Computational peptide mapping
Peptide mapping was used to define linear protein fragments that contribute the most to the amplitude at the characteristic frequency and therefore are responsible for interactions described by the particular spectral characteristic. The entire sequences of NS5A HCV and matching peptide were scanned by the ISM software as 8 and 9 amino acids long, overlapping polypeptides with one residue shift, which led to the identification of the deletions that decrease the most amplitude at the corresponding characteristic frequency.
Hydrophobicity plot
The classic Kyte and Doolittle hydropathy plot of the protein was made by the EMBOSS Pepinfo program (24,39).
RNA isolation and quantitative detection of HCV RNA by real-time PCR
RNA was extracted from 150 μL of serum using a NucleoSpin® RNA Virus kit (Clontech Laboratories, Inc., Mountain View, CA), according to the manufacturer's instructions. The isolated RNA was resuspended in diethylpyrocarbonate (DEPC)-treated water. One-step quantitative real-time PCR was performed on an ABI Prism 7900H machine (Applied Biosystems, Foster City, CA) with the Simplex HCV RNA real-time PCR kit (Cooperative Diagnostics, Greenwood, SC), according to the directions provided by the manufacturer. Briefly, 5 μL of purified nucleic acid were added to 5 μL Simplex HCV RNA Master Mix in a 96-well optical reaction plate (ABI Prism; Applied Biosystems). All of the RNA samples and controls were run in duplicate. Real-time PCR was run 10 min at 55°C, and 20 sec at 95°C, followed by 45 cycles of 1 sec at 95°C, and 20 sec at 55°C, and monitoring fluorescence in the FAM channel during a 55°C annealing/extension step. The results were analyzed with Applied Biosystems SDS 2.3 software. The results indicate the absolute copy number of HCV RNA per milliliter in each tested sample. The lower limit of detection under the conditions used was 1 RNA copy/mL.
Enzyme-linked immunosorbent assay
Polystyrene microtiter plates were incubated overnight at 4°C with 100 μL of peptides (4 μg/well; ProSci Inc., Poway, CA) diluted in carbonate buffer at pH 9.6. The plates were washed with phosphate-buffered saline (PBS) and 0.05% Tween, and non-specific sites were blocked with 200 μL PBS containing 1% BSA for 1 h at 37°C. After two washings with PBS and 0.05% Tween, serum specimens were added to the wells (100 μL/well). Sera were diluted 1:10 in specimen dilution buffer (Ortho HCV ELISA Test Kit, version 3.0; Ortho Clinical Diagnostics, Raritan, NJ). The plates were incubated for 2 h at 37°C. After three washings with PBS plus 0.05% Tween, 100 μL of murine monoclonal anti-human IgG labeled with horseradish peroxidase (Ortho HCV 3.0 ELISA Test Kit), and the plates were incubated for 2 h at 37°C. After four washings with PBS plus 0.05% Tween, 100 μL of substrate (o-phenylene diamine [OPD] dissolved in citrate-phosphate buffer containing 0.02% H2O2) was added, and the absorbance was measured at 490/620 nm after 30 min. Each sample was tested independently twice. The cut-off value was calculated as recommended by the manufacturer, and was equal to an OD value of 0.620. Commercial third-generation ELISA, Ortho HCV Version 3.0, and Architect i2000 were performed according to the manufacturers' instructions.
Statistic analysis
The significance of the differences in OD values was calculated by the Student's t-test and p values <0.001 were considered significant. Calculations were done using STATISTICA software (StatSoft Inc., Tulsa, OK).
Results
Peptide sequence from the human proteome exhibiting several levels of similarities with the HCV NS5A-derived peptide
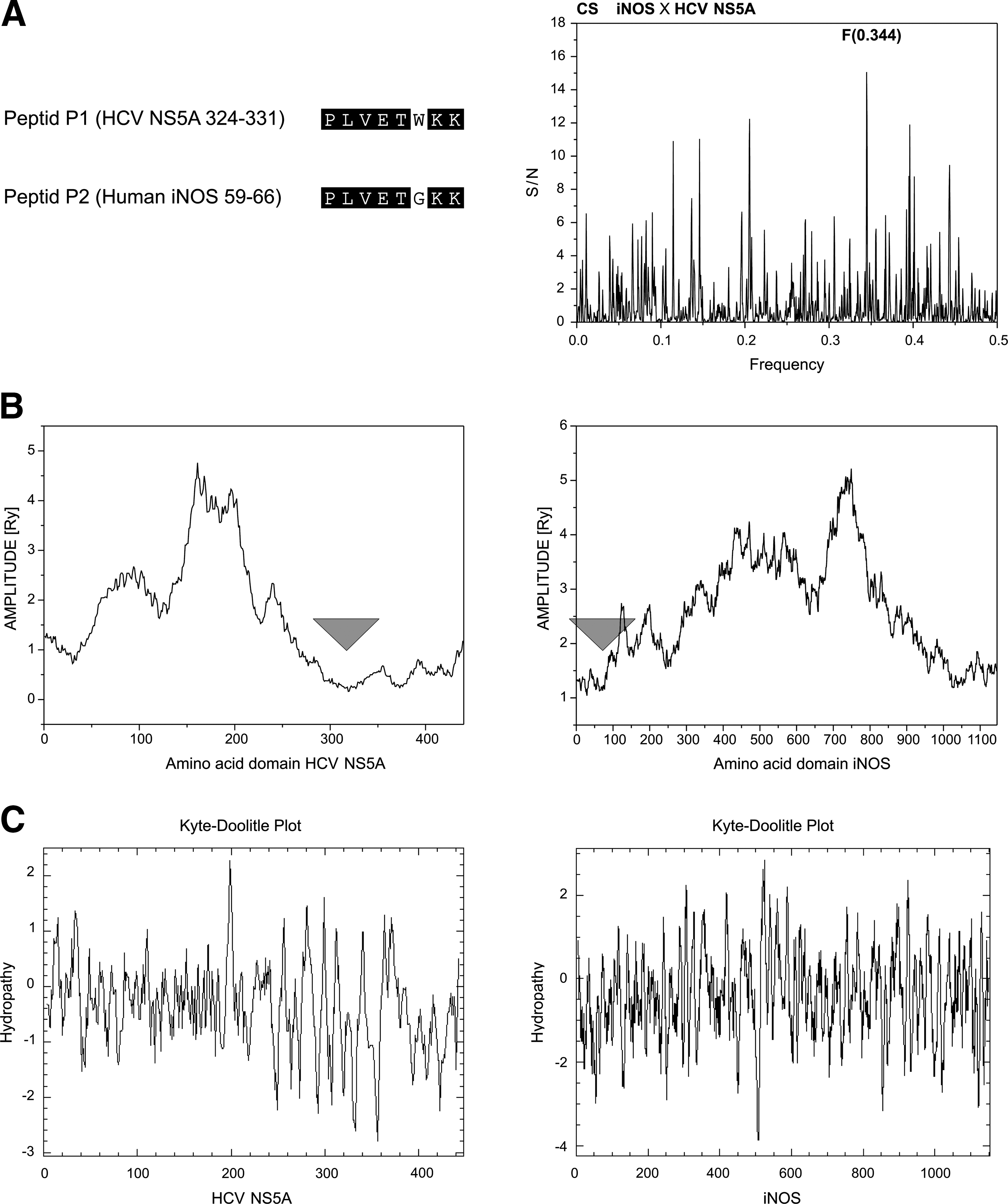
To determine one HCV NS5A domain immunologically cross-reactive with host proteins, we considered three levels of sequence similarities. Generally, local sequence homologies are regarded as a required condition for molecular mimicry between two proteins. Therefore, first we selected the peptides as candidates for further in vitro testing based on the following procedure. The entire sequence of NS5A was divided into sequences 15 amino acids long, for a total of 60 overlapping peptides. The last 8 amino acids of each peptide were overlapped with the first 8 amino acids of a subsequent peptide. In this way, the entire sequence of HCV NS5A was involved in homology analysis of the human protein sequence database, UniProt. After the final step of this process, it was found that the NS5A peptide P1 (aa 324 to 331) shares the highest sequence homology with peptide P2, derived from the N-terminus of inducible nitric oxide synthase (iNOS; P35228) (Fig. 1, panel A). We (43), and others (23), have shown that a peptide sequence homology of 8 amino acids having 4 or 5 amino acid sequence identity is of significant length to draw conclusions about cross-reactivity in ELISA.
(
iNOS is an enzyme that produces high concentrations of the ubiquitous signaling molecule nitric oxide (NO), and which is synthesized by a broad spectrum of cells, such as human bronchial epithelial cells, macrophages, endothelial cells, and vascular smooth muscle cells, and is widespread in the human body. Within cells, iNOS is generally localized in the cytoplasm and membrane fractions (16,18).
In the next step we performed cross-spectral analyses to determine the characteristic frequency present in the informational spectra of HCV NS5A and human iNOS. The peak frequency in cross-spectrum of these two sequences is at Fourier frequency, F(0.344), and represents the main mutual spectral characteristic, which is the ISM determinant of immunological cross-recognition. Computational deletions based on the ISM algorithm (41,45) allowed us to identify peptide sequences contributing the most to the F(0.344). In iNOS, this sequence encompasses amino acid residues from 58 to 66, and in HCV NS5A from 324 to 331 (Fig. 1, panel B), exactly the sequences identified in the previous homology search step. This result corroborates the finding that sequences P1 and P2 may be cross-recognized by HCV antibodies.
Finally, the classical Kyte and Doolittle hydrophobicity plot was used to assure that the peptides were not buried deep within the protein, but were exposed at the surface and accessible for the interaction with antibodies. The Kyte and Doolittle (24) method is a widely applied scale for delineating hydrophilic/hydrophobic characteristics of protein domains. Regions with values below 0 are hydrophilic in character and represent domains exposed on the surface. It is not surprising that P2 is part of a 33-amino-acid-long hydrophilic region, and therefore we can assume that this domain could serve as a self antigen for NAbs. HCV NS5A-derived peptide P1 is also within the rather large hydrophilic region, encompassing amino acids from 315 to 339 (Fig. 1, panel C).
HCV-negative blood donor sera contain antibodies capable of recognizing peptide from the HCV NS5A
In order to compare the immunoassay reactivity we performed an ELISA with synthesized P1 and P2 peptides. Sera for this study were obtained from volunteer blood donors that were routinely tested against other pathogens; therefore, possible cross-reactivity was avoided with antigens of certain other pathogens, including human immunodeficiency virus, hepatitis B virus, and Treponema pallidum. Potential cross-reactivity with antigens of other known pathogens not routinely screened during blood donation, or new, emerging pathogens could not be ruled out. Because of this fact, we used the HCV-positive cohort as a control population to compare against our analysis of the presence of antibodies recognizing peptide derived from the HCV proteome in the HCV-negative, healthy blood donor pool. Thus we tested a greater number of HCV-negative sera in our studies, which minimizes the possibility of false-positive results due to cross-reactivity of our assay with other pathogens.
The statistical analyses of the ELISA results have shown that the reactivity of tested HCV-negative sera with these two peptides is highly similar (p < 0.001; Fig. 2). The titers of IgG NAbs reactive with the host peptide were slightly higher than titers of the HCV peptide (0.120 versus 0.090; Table 1); however, as these differences are not significant, we can assume that they indicate that P2 represents a cellular motif that is recognized as a pathogen-associated molecular pattern. Unfortunately, our attempts to develop a competition experiment were unsuccessful, due to a lack of a significant decrease of reactivity by the P1 peptide ELISA with sera pre-incubated with the P2 peptide (data not shown). We believe that this is due to the low-affinity and poly-reactivity characteristics of natural autoantibodies.
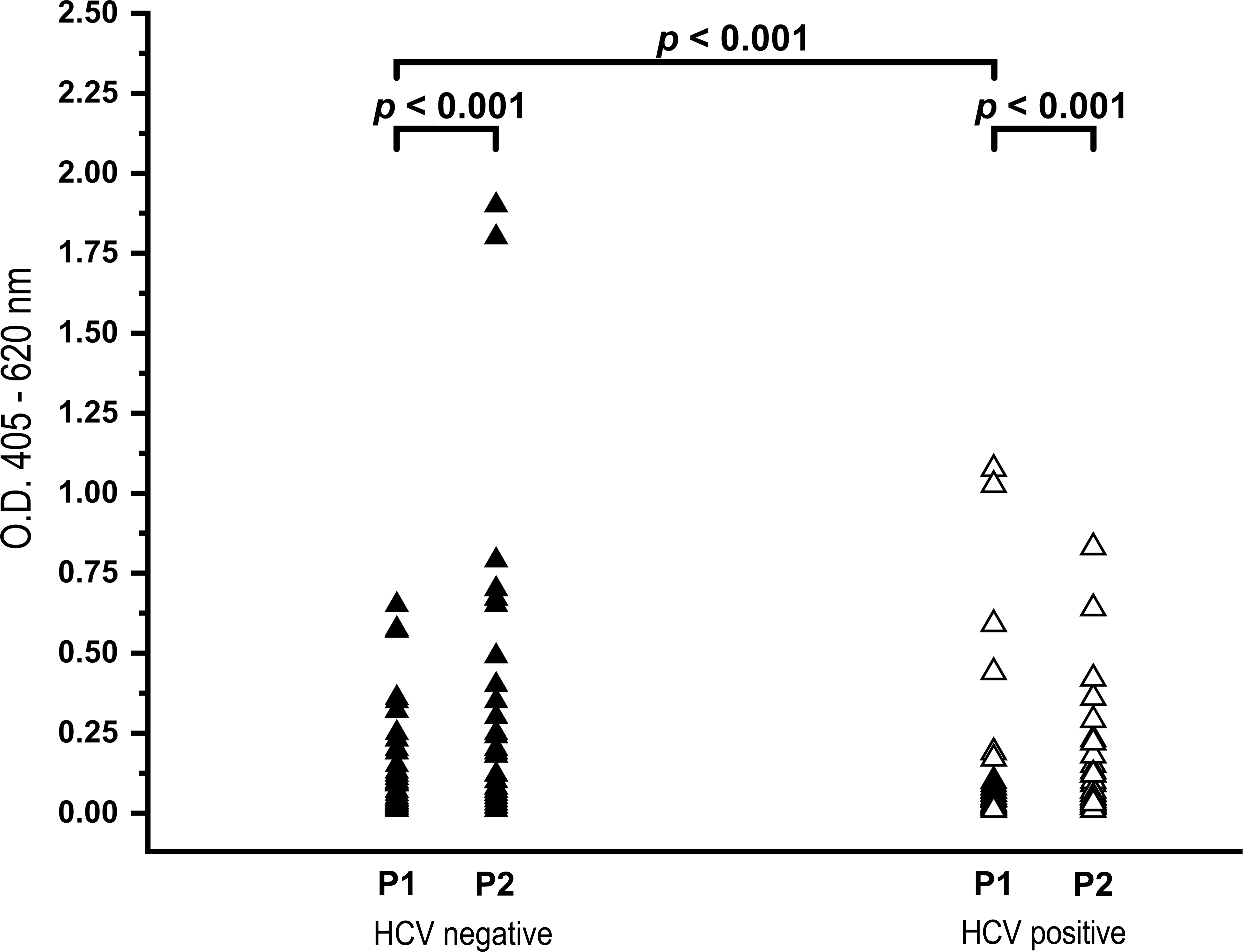
HCV-negative and HCV-positive blood donor sera ELISA reactivity. Titers of antibodies recognizing peptide P1 derived from HCV NS5A and P2 from iNOS are similar in both groups (p < 0.001) with high significance. Titers of antibodies recognizing peptide P1 derived from HCV NS5A in HCV-negative and HCV-positive sera are similar with high significance (p < 0.001; ▴, HCV negative; Δ, HCV positive).
A cut-off value was calculated as recommended by the manufacturer (Ortho HCV 3.0 ELISA), and is equal to an OD value of 0.620.
OD, optical density; HCV, hepatitis C virus; iNOS, inducible nitric oxide synthase; ELISA, enzyme-linked immunosorbent assay.
Four sera exhibiting the highest ELISA activity that recognize P1 peptide in this group also showed the most potent activities with P2 peptide ELISA. Absence of viremia in all sera was confirmed by negative amplification of HCV-RNA by the Roche Amplicor HCV detection methodology. This method of detection is highly sensitive and is state-of-the-art detection for HCV RNA in clinical laboratories. Nevertheless, for the four most potent sera in P1 and P2 ELISA we performed complementary testing with a more sensitive RNA detection method, one-step absolute quantitative real-time PCR (Simplex HCV Real Time PCR; Cooperative Diagnostics), that directly detects viral RNA copies. These results showed zero copies of HCV RNA contained in our cohort, confirming the HCV-negative status of these sera (data not shown).
The four sera that showed the highest OD values by P1 and P2 ELISA gave false-positives with third-generation commercial ELISA tests. They scored positive on Ortho HCV Version 3.0 and on Abbott HCV assay using the Architect i2000 system (Abbott Laboratories, Abbott Park, IL). In our HCV NS5A peptide ELISA, two HCV-negative samples reacted above the cut-off value, and these samples also reacted as false-positive in routine anti-HCV testing. Therefore we can conclude that P1 contributes to false-positive results in routine ELISA testing, but its significance is difficult to estimate precisely because additional viral sequences having differing antigenicity are tested in commercially-available ELISA kits.
The next issue that we addressed was whether the HCV NS5A P1 peptide is immunogenic in humans. The titers of IgG reactive to P1 in tested HCV-positive sera were comparable to a highly significant degree (p < 0.001) to those of the HCV-negative sera (Fig. 2.). Moreover, this reactivity is observed in P2 ELISA with the same group of sera. The finding that the self-binding peptide is not an immune response-eliciting antigen implies that autoantibodies and virus-directed antibodies do not target the same epitopes.
Bioinformatics study of HCV NS5A and IgG NAb self antigens
According to previously described findings, the information about ISM characteristics shared by HCV NS5A and NAb self-binding proteins will be helpful in future immunoassay antigen selection. In order to obtain this information we analyzed the HCV NS5A sequence in cross-spectrum with previously described IgG NAb-binding sequences. This data set was obtained by an antigen microarray device that was used to analyze the natural IgM, IgA, and IgG autoantibody repertoires present in sera from healthy mothers and their newborns (33). While IgA and IgM isotypes were specific for the newborns, maternal and cord IgG autoantibodies showed essentially identical reactivity, because the cord IgG came from the mother by means of active transport across the placenta. Therefore the reported set of IgG NAbs binding self molecules is considered a set of self antigens of adults, although it is gender specific.
The distribution of dominant characteristic frequencies shown in Fig. 3 demonstrates that mutual frequencies of HCV NS5A and host antigens are grouped in a limited number of sub-domains. Most of the common frequencies, 41%, are in the F1(0.25–0.40) domain, while the second domain encompasses F2(0.10–0.15). According to the ISM concept, HCV NS5A domains contributing the most to the frequencies from F1 and F2 will be recognized by NAbs, as we showed here with the HCV NS5A 324–331 domain that contributes the most to the F(0.344), the IS frequency within the F1 domain.
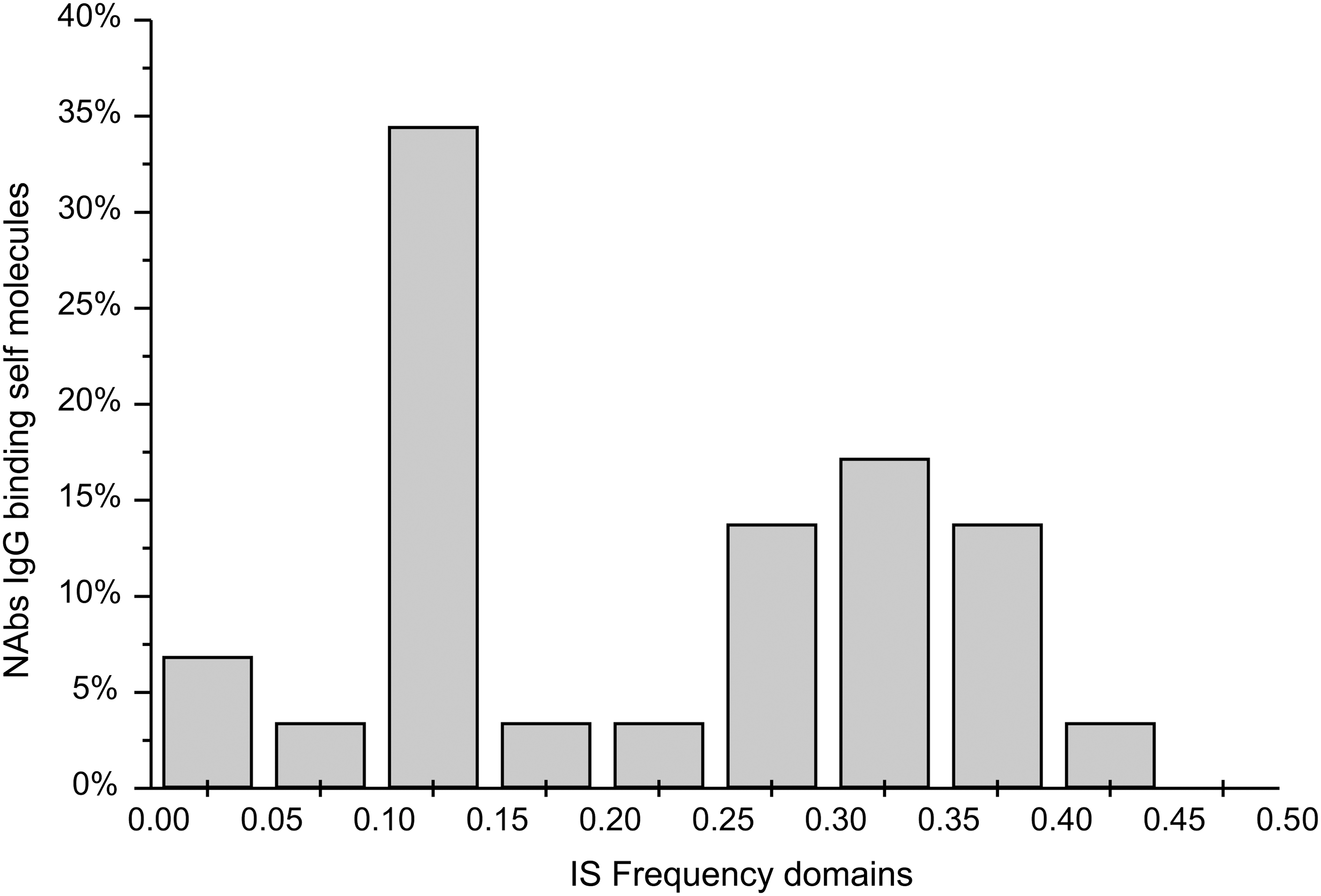
Distribution of characteristic Fourier frequencies for IgG NAb self-binding molecules and HCV NS5A. The x-axis shows the informational spectrum (IS) Fourier frequency domain (0–0.5), and the y-axis the total share of self-binding molecules having characteristic IS frequency in the particular frequency domain.
Discussion
NAbs are polyreactive, low-titer antibodies that could, in some individuals, display high-affinity binding for certain self antigens. The NAb repertoire is genetically determined and stable, representing individual characteristics of a particular person, that react dynamically to changes within the body, fulfilling a role in maintaining homeostasis (9,12,34). We performed this study to show that computational screening allows for identification of pathogen-derived peptides that can be recognized by NAbs in sera of uninfected healthy individuals, and to address the importance of consideration of a rising collection of self-binding sequences in the design of screening immunoassays. Our previous data suggest that the ISM bioinformatics approach is useful in the identification of viral peptide sequences that react with NAbs (14,46). We combined this approach with protein database searches and selected viral-human octapeptide pairs with the potential to be cross-recognized by NAbs. A viral peptide sequence was derived from the human HCV NS5A region, which is included in third-generation commercial ELISAs for anti-HCV screening. The host peptide was from the N-terminus of the human enzyme that generates nitric oxide (NO), iNOS. Besides amino acid sequence homology, these two peptides have similar hydropathic profiles, and share essential determinants of informational spectral similarities. iNOS has important functions in innate immunity and regulation of immune functions (28). Until now this protein has not been recognized as a self antigen, or in any immunological aspect to be associated with HCV. However, it is well documented that interplay of NS5A and iNOS has a role in HCV pathogenesis (13,17,30).
Peptide-based ELISA that was performed with synthesized viral peptide P1 and human peptide P2 against sera of HCV-negative and HCV-positive blood donors showed highly similar reactivity profiles (p < 0.001) for both peptides with both groups of human samples. Not surprisingly, the most active HCV-negative sera as measured by P1 and P2 ELISA gave a false-positive result by commercial ELISA. These findings are particularly interesting, because they corroborate the idea that NS5A is a less-specific antigen in routine blood donor screening tests. Also, antibodies reactive with viral peptide P1 were similarly (p < 0.001) present in infected and uninfected sera. The P1 reactivity with HCV-positive sera shows that this domain of NS5A is not an active T-cell epitope, and NAbs detected by ELISA are not pathogen-related, as is the case with anti-C1q (38) and anti-PCNA autoantibodies (20). This may reflect a certain level of distinction between self- and pathogen-associated epitopes, that has to be further assessed in larger correlative studies. Although great progress in blood safety has been made with effective and highly sensitive serological tests for HCV, the specificity of these assays still requires improvement, given that among referred immunocompetent populations, the proportion of false-positive results averages approximately 35%, and can reach 60% (7,10). False-positive or uncertain EIA results can be clarified using recombinant immunoblot RIBA and RNA-PCR techniques, but these tests are expensive and not always available. In addition, significant efforts are being invested in estimating the relevance and to resolve these problems, as evidenced by over 380 publications that have addressed these issues, as reported in PubMed (36).
The titer of NAbs strongly depends on the genetic profile and the current state of health of an individual (12,44). In order to propose criteria for the discrimination of viral antigen that could be recognized by NAbs and potentially produce false-positive results, we used ISM software and analyzed a dataset of self-binding sequences identified by antigen array. The cross-informational spectrum as a form of representation of the protein sequences allows digital filtering of common frequencies of IgG NAb self-binding molecules and HCV NS5A. The determined two common Fourier frequency domains, F1(0.25–0.40) and F2(0.10–0.15), are determinants of immunological cross-recognition between HCV NS5A and self, as confirmed in our case study with NS5A-derived P1. Namely, the domain P1 that contributes the most to the peak at F(0.344) is recognized by NAbs of HCV-negative sera. The list of NAb-binding sequences revealed in our study is gender-specific and represents part of a growing set of data, and although limited, it is useful, as we have illustrated in this pilot study. Accordingly, we can propose that the domains contributing to the limited common frequency domains can be excluded from the antigen set in order to evade NAb binding. This proposal requires further evaluation with larger numbers of NAb self-binding molecules.
As a corollary, our findings of cross-recognition between HCV and human peptides by natural autoantibodies from healthy, uninfected blood donors supports the idea that the database of natural autoantibody epitopes is useful to analyze for diagnostics by screening antigen selection for effective immunoassay. Increasing knowledge about self-epitopes and application of an antigen chip, as well as informational spectrum analyses, will allow for future accurate characterization of autoantigen sets and efficient choice of specific antigens and biomarkers (32). This will be particularly useful in cases of pathogens that are characterized by a high level of common amino acid motifs shared with human proteins, such as HCV.
Footnotes
Acknowledgments
This work was supported by a grant from the Ministry of Science and Technological Development of the Republic of Serbia (grant no. 173001).
Author Disclosure Statement
No competing financial interests exist.