
Abstract
Programmed death receptor 1 (PD-1) is an important marker of T-cell exhaustion during HIV-1 infection. Natural killer (NK) cells lose their functional capacity during HIV-1 infection, and PD-1 is expressed on NK cells during other chronic viral and bacterial infections. Here, PD-1 expression was increased on NK cells from both viremic and aviremic HIV-1-seropositive individuals, compared to seronegative controls. However, PD-1 was expressed on a small subset of NK cells and at lower frequency than that observed for CD8+ T cells. PD-1 was also induced on a minor fraction of NK cells and CD8+ T cells after long-term culture with IL-15. Raised levels of PD-1 were associated with limited NK cell proliferation, which may have consequences for their maintenance during chronic HIV-1 infection.
Introduction
Programmed death receptor 1 (PD-1) is regarded as an important marker of T-cell dysfunction during chronic HIV-1 infection (2). Expression of PD-1 results in a loss of antigen-specific T-cell proliferation and cytokine production, which can be reversed by blockade of PD-1 or its ligands (5,15). PD-1 can also be induced by cytokine stimulation of lymphocytes in vitro, indicating that antigen-independent signals may be important in controlling the expression of this receptor (9).
PD-1 is significantly upregulated on hepatitis C virus (HCV)-specific CD8+, CD4+ T cells, and NK cells in treatment-naïve patients chronically infected with HCV, compared to healthy controls. The functional significance of this observation was demonstrated in an in vitro model in which PD-1-PDL-1 blockade improved the effector function of HCV-specific CD8+ T cells (6). More recently, an elevated frequency of NK cells expressing PD-1 and its ligands PDL-1 and PDL-2 was observed in the peripheral blood and pleural fluid of individuals infected with Mycobacterium tuberculosis (1). In this study we therefore investigated the impact of chronic HIV-1 infection on the surface expression of PD-1 in NK cells.
Materials and Methods
HIV-1-infected individuals were recruited from the clinics at Chelsea and Westminster Hospital with informed consent and full ethical approval (National Research Ethics committees (U.K.)/Riverside Research Ethics Committee). We recruited 17 HIV-1-positive aviremic individuals receiving highly-active retroviral therapy (HAART; HIV-1 plasma viral loads <50 copies/mL blood, median CD4+ T-cell counts 379 cells/μL blood, range 162–839 cells/μL), and 14 treatment-naïve individuals (median HIV-1 plasma viral load 9901 copies/mL blood, range 174–74,046 copies/mL; CD4+ T-cell counts 403 cells/μL blood, range 259–809 cells/μL). Twenty-two HIV-1-seronegative healthy individuals were recruited as controls.
PD-1 expression was measured by flow cytometry and compared to background staining with an isotype-matched control antibody analyzed using FACS Diva software. For each sample 150,000 events were acquired, gating on CD3−CD56+ NK-cell and CD3+CD8+ T-cell populations, and excluding non-viable cell populations with a green fluorescent amide reactive exclusion dye (GrVid; Invitrogen, Carlsbad, CA). The following monoclonal antibodies were used for flow cytometric analysis: anti-CD3 PerCP, anti-CD56 Pe-Cy7, anti-CD8 APC anti-PD-1 PE, or mouse IgG1 isotype control PE (Becton Dickinson, Oxford, U.K.). Analysis backgrounds for PD-1 staining were set using the isotype-matched control antibody. PD-1-positive NK cells were detected with elevated frequency compared to HIV-1-seronegative controls (median 0.05%, range 0.01–0.6%), in both HIV-1-positive treatment-naïve (median 0.1%, range 0.02–1.7%; p=0.014 by Mann-Whitney U test), and treated individuals (median 0.25%, range 0.02–1.6%; p=0.0015; Fig. 1a). As expected, PD-1 expression was detected with elevated frequency on CD8+ T cells, in both treatment-naïve (median 2.1%, range 0.4–8.1%; p=0.0006), and HAART-treated (median 1.9%, range 0.3–14.6%; p=0.005) individuals, and at a higher frequency than on NK cells (Fig. 1b). Little or no PDL-1 expression was observed on NK cells from HIV-1-positive individuals, and no difference was observed in the expression of this ligand compared to HIV-1-seronegative control individuals (data not shown). PDL-1 expression was, as expected, elevated in CD8+ T cells from treatment-naïve individuals, and was maintained on CD8+ T cells after HAART (p<0.0001, data not shown).
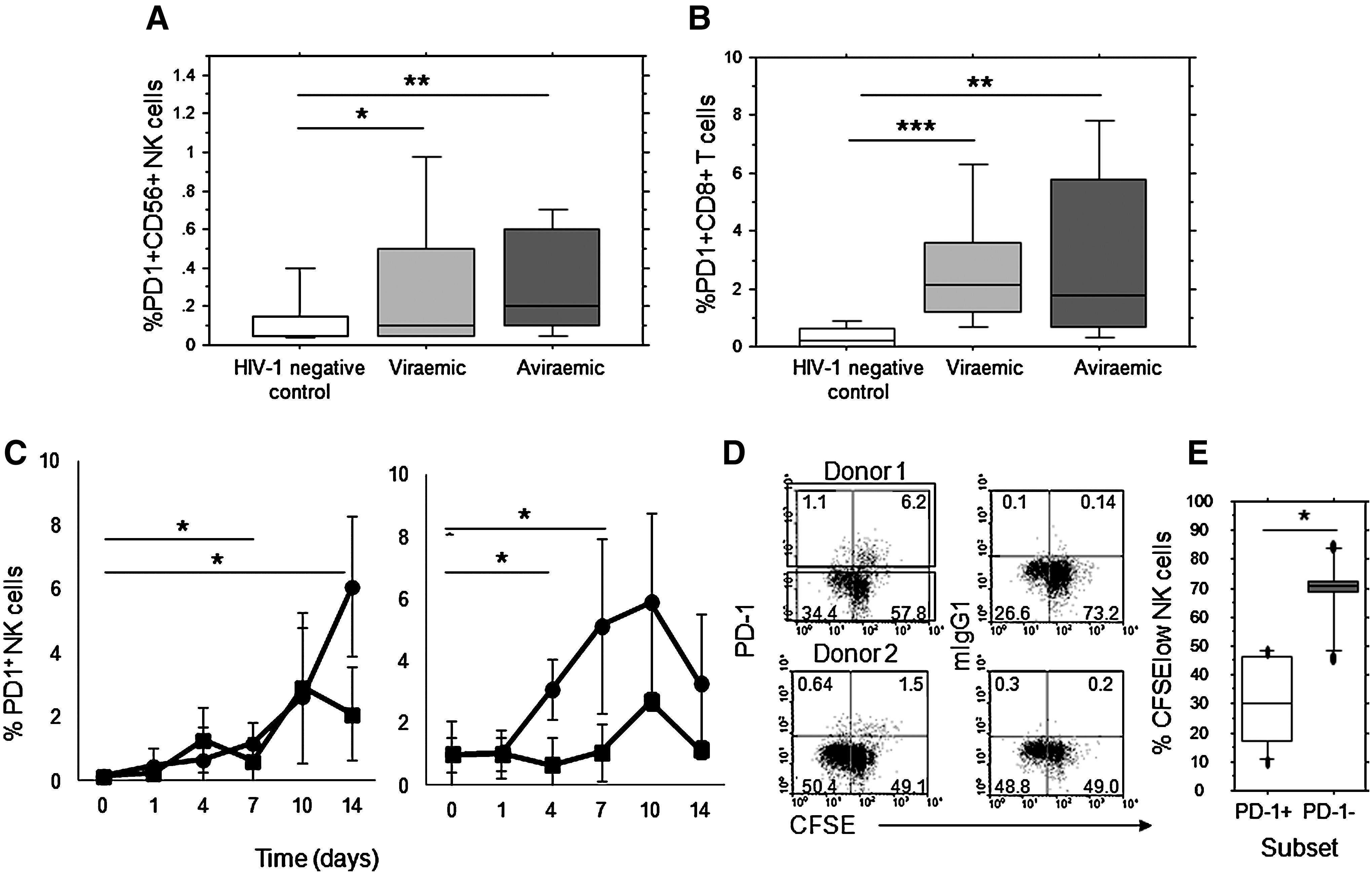
An increased proportion of NK cells expressing PD-1 during HIV-1 infection and induction on NK cells in vitro. Significant increases in PD-1 expression within gated NK cells (
Previous reports have documented the induction of PD-1 expression on CD8+ T cells by antigen-dependent and antigen-independent (cytokine) signals (9,15). We therefore investigated whether NK cell activation played a role in PD-1 expression by in-vitro stimulation with IL-15, as this was demonstrated among IL-2Rγ-chain-utilizing cytokines to be the most potent inducer of PD-1 expression. Additionally, we reasoned that expansion of NK cells will IL-15 would enable detection of sufficient PD-1-positive NK cells to permit analysis of proliferation in this population. Increased expression of PD-1 was observed within 7 d in gated viable CD3−CD56+ NK cells left in medium alone, or stimulated with IL-15 (mean±SD PD-1+NK % day 0=0.14±0.14%; medium day 7=0.61±0.74%, ns; IL-15 day 7=1.7±0.66%, p=0.041; Fig. 1c). The proportion of NK cells expressing PD-1 continued to rise up to 14 d in culture (medium day 14=2.1±1.47%; IL-15 day 14=6.08±2.18%, p=0.0178). PD-1 was also induced on IL-15-activated CD3+CD8+ T cells under the same culture conditions, as previously reported (Fig. 1d) (9).
The proliferative capacity of PD1+ cell populations was assessed by staining PBMCs with CFSE prior to stimulation with IL-15. PD-1 expression was observed on both proliferating (CFSE low, negative) and non-proliferating (CFSE high) NK cells in IL-15-stimulated cultures, indicating that cell proliferation is not a prerequisite for PD-1 expression. However, a significantly lower frequency of proliferating (CFSE negative) NK cells was observed within gated PD-1-positive cells compared to PD-1-negative NK cells (p=0.046 by Wilcoxon signed rank test; Fig. 1d and e).
The frequency of PD-1 expression on NK cells reported here for HIV-1 infection is lower than previously observed during Mycobacterium tuberculosis and hepatitis B infections, and lower than that induced in vitro on CD8+ T cells. These previous studies, however, established frequencies of PD-1 expression in total PBMCs, while we excluded dead cells using amide-reactive dye. It is possible that PD-1 is elevated in apoptotic/dead NK cells, thereby accounting for the difference in observed frequencies. PD-1 expression in CD8+ T cells is known to be associated with advanced maturation, loss of function, and apoptosis (2,12
–14). We have observed that NK cells from HIV-1-infected individuals have increased susceptibility to apoptosis ex vivo, and that PD-1 expression is elevated on annexin V+ cells, indicating that PD-1-expressing cells may also be associated with increased NK cell turnover and apoptosis (data not shown; Supplementary Fig. 1; see online supplementary material at
Our data demonstrate incomplete reversal of PD-1 induction on NK cells in HIV-1-positive aviremic individuals receiving antiretroviral therapy. Such findings are consistent with previous reports showing that CD8+ T-cell expression of PD-1 is maintained in individuals with incomplete immune reconstitution (7). Similar CD4+ cell counts were measured for the groups of treated aviremic individuals and treatment naive viremic individuals studied here, indicating that these may not have fully recovered in the treated group. Whether time to treatment and CD4+ T-cell counts on initiation of antiretroviral therapy influence the maintenance of PD-1 expression on lymphocyte subsets remains to be investigated.
Studies of PD-1 expression during hepatitis C and Mycobacterium tuberculosis infections have detected elevated PD-1 expression on CD56bright NK cells, which are potent in cytokine responsiveness and production, but have limited natural cytotoxicity (1,6). We have also observed an enrichment of PD-1+ NK cells within CD56bright compared to CD56dim cells in HIV-1-infected individuals (CD56bright PD-1+, median 8.8%, range 1.8–23.8%; CD56dim PD-1+, median 1.1%, range 0.1–5.6%; p=0.0044). PD-1 expression within CD56bright cells, which have higher expression of cytokine receptors, may account for the observed reduction in proliferation in this subset in response to cytokines. Blockade of PD-1 on NK cells from individuals with mycobacterial infection was observed to result in partial elevation of NK-cell-derived IFN-γ production, raising the possibility that PD-1 expression on CD56bright NK cells could also influence their IFN-γ production during chronic HIV-1 infection (1). NK cells from HIV-1-infected individuals are partially chronically activated, as demonstrated by elevated expression of the activation markers HLA-DR and CD69 (4,8).
Tissue NK cells have a predominantly CD56bright phenotype, and are partially depleted in the colons of treated and untreated HIV-1-infected individuals (11). PD-1 blockade enhances T-cell immunity in the gastrointestinal tract of macaques chronically infected with SIV, indicating a role in immune regulation in infected tissues. The expression of PD-1 on tissue NK cells has not yet been investigated, but would provide a candidate mechanism for NK-cell depletion in the GI tract during chronic HIV-1 infection. Elevated PD-1 expression on chronically-activated NK cells may therefore lead to reduced proliferation and clonal expansion, and promote apoptosis of NK cells, thereby affecting their turnover during HIV-1 infection.
Footnotes
Acknowledgments
This work was funded by the St. Stephen's AIDS trust, and by the Joint Research Committee of the trustees of Chelsea and Westminster Hospital (to M.R.G.). M.R.G. is also funded by the Campbell Foundation and the Medical Research Council (U.K.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.