
Abstract
Severe acute respiratory syndrome (SARS) is a newly emerging infectious disease, and an effective vaccine is not available. In this study, we compared the immunogenicity and protection efficacy of recombinant proteins corresponding to different domains of the SARS-coronavirus spike protein. Trimeric recombinant proteins were created by fusing the foldon domain derived from T4 bacteriophage to the carboxy-termini of individual domains of the spike protein. While the full-length ectodomain (S) of the spike protein, the full-length ectodomain fused to foldon (S-foldon), the S1 domain (S1), S1-foldon, and the S2 domain(S2) antigens all elicited comparable antibody titers as measured by ELISA, S-foldon induced a significantly higher titer of neutralizing antibody and S2 protein did not elicit virus neutralizing antibodies. When tested in a mouse virus replication model, all the mice vaccinated with the S1, S1-foldon, S, or S-foldon were completely protected.
Introduction
Different vaccine strategies have been explored for SARS, including inactivated whole-virus, DNA, recombinant live vectors, attenuated virus, and subunit vaccines (1,2), and they all have been shown to induce neutralizing and protective responses. However, safety issues relating to either vaccine production or use in humans have made subunit vaccines a favored strategy.
The genome of SARS-CoV encodes four structural proteins, including spike (S), membrane (M), envelope (E), and nucleocapsid (N), and some nonstructural proteins (3,4). The S protein is a 150–180 kDa transmembrane protein and is responsible for receptor binding via its receptor-binding domain located in the S1 region, as well as virus-membrane fusion and tissue tropism involving its S2 region (5). The S protein is the main component of the virus that induces neutralizing antibodies against the virus and is thus considered a main target for SARS vaccines (6). In the present study, we compared the full-length ectodomain of S protein and its fragments (S1 and S2 domains) with respect to immunogenicity and protection against viral infection in mice. In its native state, S protein is a trimer; however, when its ectodomain is expressed as a recombinant protein in eukaryotic systems, the protein exists predominantly in a monomeric form (7). To make trimeric recombinant spike proteins, we exploited a 27-amino acid sequence, called the foldon domain, which was identified in the bacteriophage T4 fibritin protein (8). We compared the immunogenicity and protective efficacy of monomeric full-length spike ectodomain (S) and monomeric S1 domain with trimerized forms of S and S1 generated by fusion of the foldon domain to the carboxy termini of the proteins. Our results showed that recombinant S2 does not elicit neutralizing antibodies and that the full-length ectodomain with a C-terminal foldon (S-foldon) induced higher levels of neutralizing antibodies than S, S1, or S1-foldon constructs. Nevertheless, S, S-foldon, S1, and S1-foldon all protected mice from viral challenge.
Materials and Methods
Cell lines and expression vectors
Sf9 insect cells were maintained in a serum-free insect culture medium (Allele™ Biotechnology). High Five cells were cultured with Ex-Cell™ 405 medium (SAFC Biosciences). VeroE6 cells were cultured at 37°C, 5% CO2, in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% FBS (Invitrogen), 100 U/mL penicillin, 100 μg/mL streptomycin. pAcGP67 baculovirus transfer vectors were from BD Biosciences and linearized baculovirus DNA were from AB Vector (ProEasy™) or Sigma (Diamond Bac™).
Construction of recombinant viruses and production of recombinant proteins
DNA encoding a thrombin cleavage site and a linker followed by a bacteriophage T4 foldon domain plus 9 histidines, NKLLVPRGSPGSG
Recombinant baculoviruses were made by co-transfecting the above plasmids with linearized baculovirus DNA into SF9 cells according to the manufacturers' instructions. For protein production, the supernatants of the cultures of High Five insect cells infected with the recombinant viruses were harvested, concentrated 10-fold, and buffer-exchanged to phosphate-buffered saline (PBS, pH 7.4) with Pellicon XL devices (cut-off size 10 kilodaltons) on a Labscale TFF System (Millipore), and then subjected to chromatography purification with Ni-NTA columns on a BioLogic LP chromatography system (BioRad). Proteins eluted with a linear gradient of imidazole in PBS were dialyzed against PBS and concentrated appropriately by using Amicon Ultra centrifugal filter devices (Millipore). Protein concentration was determined by using a Dc Protein Assay (BioRad) with bovine gamma globulin as the standard. Proteins were stored at −80°C until use.
SDS-PAGE and Western blot analysis
Analysis of proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot were performed with 12% Bis-Tris NuPAGE/MES gels and WesternBreez® kits from Invitrogen, according to the manufacturer's instructions.
Trimer formation analysis
Purified proteins were diluted in 100 μL of 6% (w/vol) sucrose in PBS and added to the top of a 10%–36% sucrose gradient prepared in PBS and centrifuged in a SW55 Ti rotor at 450,000 rpm for 14 h at 4°C. Fractions of 350 μL were collected from the top of the gradient, and 15 μL of each fraction was analyzed by SDS-PAGE and Western blot analysis using mouse monoclonal anti-His tag antibody (Novagen).
Immunization of animals
BALB/c mice 6–8 weeks old (n=5–8 per group) were immunized subcutaneously (s.c.) or intramuscularly (i.m.) with 20 μg each of the purified recombinant proteins formulated with TiterMax Gold Adjuvant or with aluminum hydroxide gel (alum) plus monophosphoryl lipid A (MPL) (all from Sigma) on days 0, 14, and 28, except when indicated. Formulations were prepared according to the manufacturer's instructions. For TiterMax, one volume of protein in PBS was emulsified with one volume of the adjuvant. For Alum/MPL, 100 μL of protein in PBS was mixed with 7.7 μL of alum gel plus 5 μL of MPL on a rotator overnight at 4°C. Each dose was delivered in 100 μL. Animals in the control groups received PBS with the same adjuvants on the same days. Blood samples were collected by tail bleeding at indicated time points and sera were prepared.
Enzyme-linked immunosorbent assay (ELISA)
Immuno MaxiSorp plates (Nunc) were coated with 100 μL per well of recombinant protein at 1 μg/mL diluted in PBS at 4°C overnight. The plates were blocked with 120 μL per well of Blocker™ Casein in PBS (Thermo Scientific) for 1 h at room temperature. Mouse serum was first diluted 1:200 in Blocker™ Casein and was further serially 2-fold diluted. 100 μL of each serum dilution was added to each well and incubated at room temperature for 1 h. After washing the plates three times each with 150 μL of PBS containing 0.5% Tween 20 (PBS-T), 100 μL of peroxidase-conjugated goat anti–mouse IgG (Sothern Biotech) diluted 1:4,000 in PBS-T was added to detect bound antibody. Peroxidase substrate solution (KPL) containing 2,2′-azino-di-3-ethylbenzthiazoline-6-sulfonate and hydrogen peroxide was used to develop the plates and absorbance was read at 405 nm with a Precision Microplate Reader (Molecular Devices).
Serum neutralization assay
Mouse serum was treated at 56°C for 30 min to inactivate complement and then was diluted 1:4 with DMEM. Serial 1:2 dilutions of the serum were then made in a 96-well tissue culture plate by adding 125 μL of the 4-fold diluted serum to an equal volume of DMEM; this was carried through a 1:4096 dilution. An equal volume of SARS-CoV Urbani strain (100 TCID50) was added and the mixture was incubated for 1 h at room temperature. The mixture was then transferred onto 10,000 Vero E6 cells. Each serum was tested in four replicates for each dilution. The cell cultures were incubated at 37°C and examined for cytopathic effect (CPE) under a microscope 4 days later. The highest dilution of serum that prevented CPE in 50% of the cell culture wells was reported as the antibody neutralizing titer.
SARS-CoV challenge
The animal studies were conducted in a BSL3 facility (Bioqual, Inc) in accordance with the approval of the Institutional Animal Care and Use Committees of the US Food and Drug Administration and Bioqual. On day 28 following the last immunization, anesthetized mice were intranasally inoculated with 1×105 TCID50 of SARS-CoV (strain Urbani) in a total volume of 20 μL. Mice were euthanized on day 3 following challenge and lungs were harvested and stored at −80°C.
Determination of viral titers in lung tissues
Virus titers were determined on Vero E6 cell monolayers as described previously (10). Briefly, the whole lung tissue from each mouse was homogenized manually in a sterile microcentrifuge tube with a plastic pestle and was mixed with an appropriate volume of Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, USA) to make a 10% suspension. The homogenate was centrifuged in a benchtop centrifuge at top speed for 3 min, and the supernatant was stored in aliquots at −20°C until use. For virus titer determination, 20 μL of the supernatant was added to a well containing a monolayer of Vero E6 cells, which had been seeded in 96-well tissue-culture plates on the previous day. A series of 10-fold dilutions of each supernatant was assayed in four replicate wells per dilution. Results were evaluated 3 and 5 days after the inoculation and viral titers were calculated by the Reed-Muench method (11).
Statistical analysis
A Student's t test was used to analyze differences in mean values between groups and p values smaller than 0.05 were considered statistically significant.
Results
Six recombinant proteins were designed with a 9-histidine tag at the carboxy-terminal ends to allow for metal-chelating affinity purification (Fig. 1A). They were all secreted into the culture medium as confirmed by Western blot analysis (Supplementary Fig. S1; supplementary material is available online at
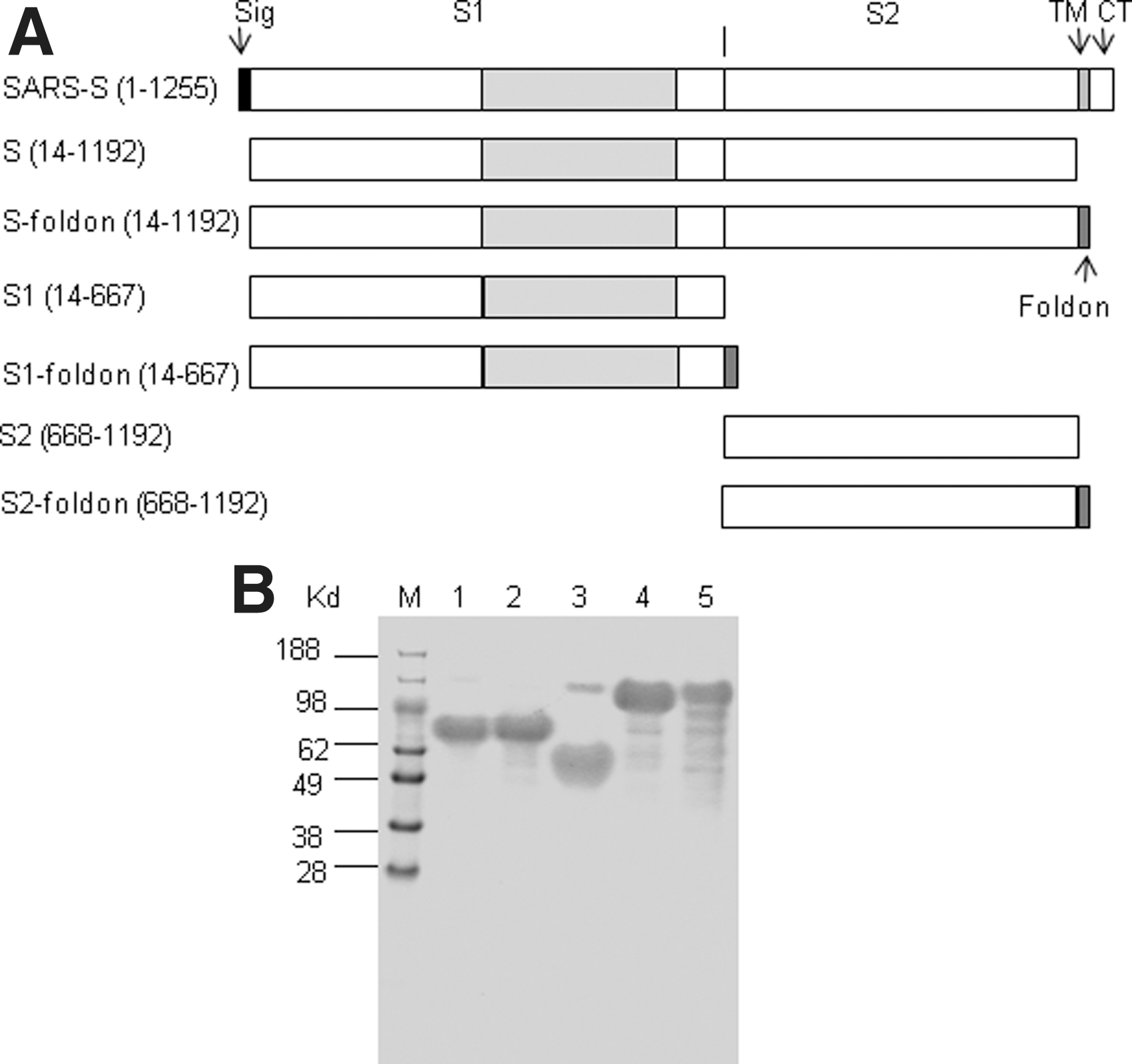
SARS-CoV spike protein and recombinant proteins
To determine if S-foldon forms a trimer, we performed ultracentrifugation in a sucrose gradient and compared the motility of S-foldon with that of S. As shown in Figure 2 upper panel, S-foldon sedimented to a higher concentration of sucrose than the peak for S, indicating that S-foldon formed an oligomer. The position of the S-foldon in the gradient relative to the 660 kD protein marker suggests a trimeric state. Of note, however, a small proportion of S molecules sedimented to where the trimeric S-foldon was located, suggesting that S may also form trimers, although at a much lower efficiency compared to S-foldon. In addition, both S and S-foldon were also found in the bottom fractions, indicating that a fraction of S and S-foldon molecules formed aggregates. It appears that S1-foldon could also form a trimer, but not S1 (Fig. 2, lower panel).
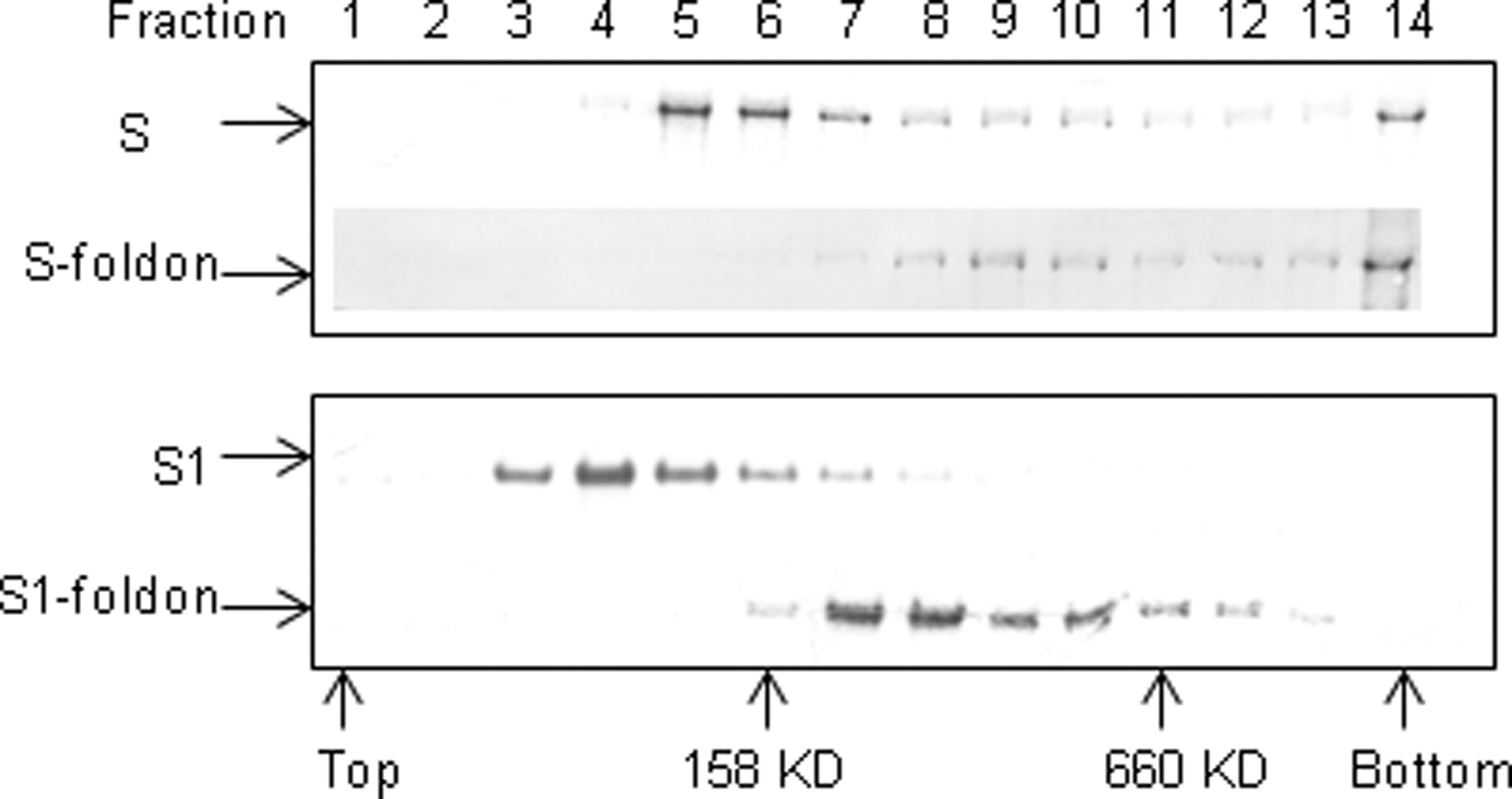
Trimer formation analysis of S-foldon and S1-foldon. Ten micrograms each of S, S-foldon, S1, and S1-foldon were overlaid on a 2% step sucrose gradient of 10%–36% and run in a SW55 rotor at 45,000 rpm, for 14 h at 4°C. Fractions of 350 μL each were collected from the top of the tube and 15 μL of each fraction was subjected to 12% NuPAGE/MES, followed by Western blot analysis with an anti-His Tag antibody. Positions of protein standards (aldolase, 158 kD, and thyroglobulin, 660 kD) are indicated.
The immunogenicity of the recombinant proteins was first tested in groups of BALB/c mice (n=5) administered individual proteins by the subcutaneous (s.c.) route with TiterMax Gold Adjuvant, a water-in-oil adjuvant on day 0, day 14, and day 28. Ten days after the third dose, sera were obtained at the time of euthanasia of the mice and antibody levels were measured by ELISA using cognate proteins as the coating antigens. As shown in Figure 3A,S1, S2, S, and S-foldon elicited geometric mean antibody titers equivalent to or greater than 106, while S1-foldon elicited a geometric mean antibody titer slightly higher than 105. The same sera were then tested for their virus neutralizing activities in vitro (Fig. 3B). Sera from mice immunized with S1, S1foldon, and S, had comparable levels of virus neutralizing activity, with geometric mean neutralizing titers of 171, 90, and 238, respectively. Most interestingly, sera from the mice immunized with S-foldon showed the strongest neutralizing activities, with a geometric mean neutralizing titer of 659.6. This titer is 3.8-, 7.3-, and 2.8-fold higher than that of sera from the mice immunized with S1, S1-foldon, and S, respectively. The differences are all statistically significant (p=0.024, 0.016, and 0.049 for S-foldon versus S1, S1-foldon, and S, respectively). No neutralizing activity was detected in the sera from the mice immunized with S2.

Immunogenicity of recombinant proteins. Serum ELISA antibody titers and virus neutralization titers for mice immunized with recombinant spike proteins adjuvanted with TiterMax. Mice (n=5) were immunized with PBS or recombinant proteins mixed with TiterMax on days 0, 14, and 28, and sera were prepared from sacrificed mice on day 38. End point ELISA titers were determined on plates coated with cognate antigens
We next tested the immunogenicity of these antigens in a formulation that is potentially relevant for human use. Antigens mixed with alum gel and MPL were administered by either the s.c. or intramuscular (i.m.) route to groups of BABL/c mice (n=5 or 8) on day 0, day 14, and day 28. Sera from these mice also have high ELISA titers, although lower than when TiterMax was used (Fig. 3C). In addition, antibody titers were higher in the i.m. group than the s.c. group, and the differences were statistically significant (S1, p=0.006; S1foldon, p=0.000002; S2, p=0.015; S, p=0.00015; S-foldon (p=0.00073) (Fig. 3C).
We then tested the protective efficacy of recombinant proteins in a mouse model. Because S2 did not induce detectable virus neutralization activity, this antigen was not tested for efficacy in mice. Groups of 6 mice were immunized i.m. with S1, S1-foldon, S, S-foldon adjuvanted with alum/MPL or PBS plus adjuvant alone as a control on day 0, day 14, and day 42. Because of a delay in production of live virus for challenge, a fourth dose was given on day 112. The mice were bled on day 133 (3 weeks after the last dose of vaccine) and plasma antibody levels were measured by ELISA on plates coated with cognate antigens. All the mice that received a recombinant spike protein antigen had high antibody titers (Fig. 4A). Insufficient volumes of sera were available for neutralization assays. On day 140, 4 weeks following the last dose, mice were challenged intranasally with 1×105 TCID50 of SARS-CoV. One mouse in the S1 group died during the process of inoculation, likely due to anesthetization. All the other mice appeared normal throughout the challenge experiment. On day 3 post challenge, the mice were sacrificed and their lungs were homogenized for virus load determination. All mice in the group that received PBS plus alum/MPL only had high viral loads in their lungs. In sharp contrast, all mice in the groups receiving recombinant spike protein vaccines had no detectable virus in the lungs (Fig. 4B).
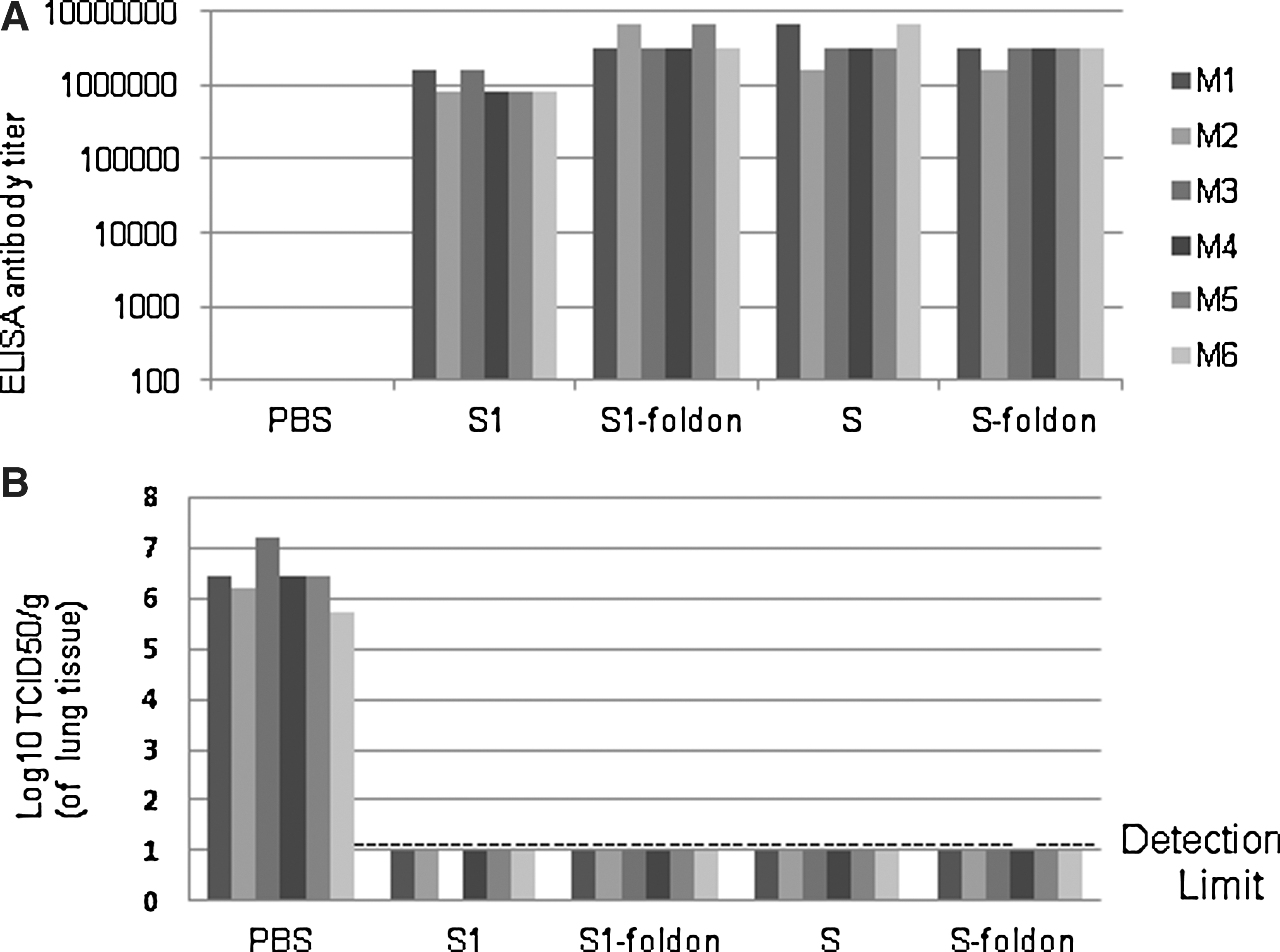
Blood antibody levels and virus load in the lungs of mice challenged with SARS-CoV.
Discussion
Because S protein is present as trimers on the virus (7), it is interesting to examine whether a trimeric S protein is superior to a monomer in terms of immunogenicity and protective efficacy. Recombinant trimeric full-length S proteins have been reported to be highly immunogenic and able to elicit an efficacious protective immune response (12, 13). However, full-length S proteins contain a transmembrane domain, making it impractical in vaccine production. To overcome this problem, we made secreted recombinant proteins by fusing a foldon domain derived from phage T4 fibritin to the ectodomain of S or its domains (S1 and S2). The foldon has been shown to promote trimerization of a number of recombinant proteins such as collagen fibers (14). In our study, both of the recombinant proteins with the foldon (S1-foldon and S-foldon) formed trimers or oligomers. As S1 is predicted not to be directly involved in the formation of a trimeric spike protein, it is likely that the trimeric state of the S1-foldon as seen in the present study was a structure in which three monomers were simply bound together by the foldon. Although S-foldon, similarly to S, tends to form aggregates after freezing-thawing, a major fraction of S-foldon appeared to retain a trimeric state. It is unknown from the present study to what extent the trimeric S-foldon is structurally similar to the native spike protein, however. Nevertheless, while S1-foldon was comparable with S1 in immunogenicity and in the potency of inducing virus neutralizing antibodies, S-foldon showed significantly stronger potency in the induction of neutralizing antibodies than S; sera from mice immunized with S-foldon had a 2.8-fold higher titer compared to mice immunized with S. Indeed, S-foldon was the most potent immunogen among all the five recombinant proteins tested in this study. The principle behind this phenomenon is yet to be elucidated. It is probable that the foldon plays a role in maintaining the native conformation of neutralizing epitopes in the trimeric immunogen. Alternatively, the trimeric protein is more efficiently presented to committed B and T lymphocytes to induce higher levels of antibodies.
A number of different animal models have been described for SARS-CoV but none of them completely recapitulate SARS-CoV pathology in humans (15). Considering cost, availability and ease of use, the virus replication murine model still represents a convenient model for initial proof-of-concept in the evaluation of vaccine candidates. When tested in the virus infection mouse model, the four immunogens, (S1, S1-foldon, S, and S-foldon) fully protected the mice from virus challenge. No difference was observed between the four immunogens tested, perhaps because the mice had been given four doses and they all had very high antibody levels before the challenge. However, given its much stronger potency in inducing neutralizing antibodies than the other constructs, S-foldon might confer an acceptable level of protection with fewer doses than the other constructs. This would be relevant for use in humans and deserves further study.
The S2 domain of SARS-CoV mediates membrane fusion during viral infection and is hence a potential target for protective antibodies. However, reports regarding the presence of neutralizing epitopes in S2 and a protective role for antibodies to S2 have been inconsistent. Several studies have suggested that S2 domain contains neutralizing epitopes. Immunization with Escherichia coli expressed or chemically synthesized S2 peptides elicited neutralizing antibodies (16 –18). Immunization with plasmids encoding the S2 domain was also reported to induce neutralizing antibodies in rabbits (19). On the contrary, there are also reports that showed that recombinant S2 domain could not induce neutralizing antibodies (20,21). Our results demonstrated that recombinant S2 was highly immunogenic but could not induce neutralizing antibodies. The S2 domain of other coronaviruses also failed to induce neutralizing antibodies (22 –25). Taken together, it is likely that the protective epitopes on S2, if such exist, are not immunodominant.
In conclusion, our study corroborates that recombinant proteins based on either the S1 domain or the full-length ectodomain of S protein are suitable vaccine candidates. More importantly, it suggests that the full-length ectodomain engineered to form a secreted trimer may be a superior form of SARS subunit vaccine, and a secreted protein would likely be easier to produce than the native membrane protein.
Footnotes
Acknowledgments
This work was supported by grant RO1-AI064372 (RPV) from the National Institutes of Health. J. Li was supported in part by an appointment to the Research Participation Program at the Center for Biologics Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.