
Abstract
Immunopathologic examination of the lungs of mice with experimental influenza virus infection reveals three prominent findings. (i) There is rapidly developing perivasuclar (arterial) and peribronchial infiltration with T-cells and invasion of T-cells into the bronchiolar epithelium, separation of epithelial cells from each other and from the basement membrane, leading to defoliation of the bronchial epithelium. This reaction is analogous to a viral exanthema of the skin, such as measles and smallpox. This previously described but unappreciated reaction most likely is an effective way to eliminate virus-infected cells, but may contribute to acute toxicity and mortality. (ii) After this, there is formation of dense collections of lymphocytes adjacent to bronchi consisting mainly of B-cells, with a scattering of T-cells and macrophages. This is known as induced bronchial-associated lymphoid tissue (iBALT) and correlates with increased interleukin (IL)-17 in the lung. iBALT provides sites for a local immune reaction in the lung to both the original infection and related viral infections (heterologous immunity). (iii) Within the first 2–3 weeks, there is proliferation of type II pneumocytes and/or terminal bronchial epithelial cells extending from the terminal bronchioles into the adjacent alveoli, eventually leading to large zones of the lung filled with tumor-like epithelial cells with squamous metaplasia. The proliferation correlates with IL-17 and IL-22 expression, and the extent of this reaction appears to be determined by the availability of T-regulatory cells.
Introduction
T
Materials and Methods
Methods
Over the course of the last 6 years, the pathologic changes in the lungs of various experimental mouse models in influenza infection studied at the Trudeau Institute were evaluated by immune histology. In brief, hematoxylin and eosin (H&E)-stained slides and paraffin-embedded blocks were obtained from individual investigators at the Trudeau Institute. After review and blinded histologic grading by a board certified pathologist (S. Sell) of each of the H&E slides, six or more serial sections of the tissue blocks were cut at the core histology laboratory at the David Axelrod Institute for immunohistochemistry so that each slide contained two sections.
Immunohistochemistry
Formalin-fixed, paraffin-embedded mouse tissue blocks were sectioned at 5 μm and placed on charged slides. After dewaxing through xylenes and ethanols, sections were brought to water and then subject to antigen retrieval. Five antigen retrieval procedures were used: heating in 0.1 M citrate buffer (pH 6) or 0.1 M Tris HCl buffer (pH 8) for 20 min; proteinase K (25 μg/mL), pepsin (0.5% w/v), or trypsin (0.0025% w/v) digestion (10 min at 37°C). After washes in phosphate-buffered saline (PBS), sections were incubated 1–2 h with serum block (5% serum of the secondary antibody host) in PBS containing 10 mg/mL bovine albumin (Sigma, catalogue no. A-9418). The primary and secondary antibodies used and the conditions for enhancement, as well as others that were tested, but not used for this study, are listed in Tables 1 and 2. Primary antibodies (Table 1) were applied overnight at 4°C. Slides were washed in PBS and then incubated for 15 min in 3% hydrogen peroxide. Secondary antibodies (Table 2) were applied for 1 h at room temperature followed by extravidin peroxidase (Sigma; catalogue no. E2886). Color was developed in diaminobenzidine (Sigma; catalogue no. D8001), and slides were cover slipped with Permount (Fisher Scientific). When amplification was performed, the method of Kerstens et al. was used (20). Briefly, tyramine hydrochloride (Sigma; catalogue no. T2879) was biotinylated with E-Z-Link NHS-LC-Biotin (Thermo Scientific; catalogue no. 21336) following the manufacturer's instructions. Biotinylated tyramine was added to sections for 10 min after the initial extravidin peroxidase step. Sections were washed in PBS and then, the extravidin-peroxidase step was repeated before color development. Staining sections with CD4 and CD8 antibodies was attempted using several different antibodies/clones and all listed antigen retrieval methods, without success.
Antibodies used in final analysis.
Amplified with biotinylated tyramine (see text for details).
LAK, lymphokine-activated killer cells; NK, natural killer cells.
d-Biotinyl-´ɛ-aminocaproic acid-NHS-ester.
These slides were then stained in the following sequence: CD3 (T-cells), Pax5 (B-cells), H&E, F4/80 (tissue macrophages and Kupffer cells), protein C (type II pneumocytes), IAV, and thyroid transcription factor 1 (TTF1) (pulmonary epithelial cells). The controls for CD3, Pax5, and F4/80 clearly show the expected differential staining (Supplementary Fig. S1; Supplementary Data are available online at
Mouse models
The experimental mouse models examined in this article are listed in Table 3. For complete details of the experimental models and “blinded” histologic grading, please see the original papers. All mice were obtained from the animal breeding facility at the Trudeau Institute. Experimental protocols were approved by the Institutional Animal Care and Use Committee of the Trudeau Institute, under Animal Welfare Assurance Number A3075-01 of the National Institutes of Health. Influenza A/PR8 (H1N1) virus was cultured in the allantoic cavity of embryonated hen eggs from stock obtained from St. Jude Children's hospital, and the egg infective dose (EID50) was characterized. The first model compared the immune response in interleukin (IL)-10 knockout (KO) mice with wild-type (WT) mice of the same strain (30). IL-10-deficient mice display increased expression of IL-17 and dramatically increased survival compared with WT mice when challenged with lethal doses of virus (30). BALB/c or BALB/c IL-10 KO at least 8 weeks old were challenged intranasally with 1 LD50 (5,000 EID50) A/PR8. The tissues were taken at 6–8 days after infection. The second and third models involved transfer of naïve memory CD4 T-cells to WT and severe combined immune-deficient (SCID) mice (31). Naive T-cells were obtained from pooled spleen and lymph nodes as previously described (29). IAV–primed memory CD4 T-cells were obtained as previously described (50). 5×106 Th1 polarized memory T-cells were transferred to SCID mice and then, the mice were infected with 2,500 EID50 PR8 virus. SCID mice receiving memory T-cells survived for 4 weeks; SCID mice receiving naive T-cells died after 2 weeks. In the fourth model, CD4 memory T-cells to ovalbumin (OVA) were transferred to WT mice and then challenged intranasally with OVA (3). This model is a control and demonstrated delayed-type hypersensitivity in the lung rather than T-cell cytotoxicity. The fifth model employed Ccr5−/−Cxcr3−/− mice that express increased CD8 T-cell memory (22). WT and Ccr5−/−Cxcr3−/− mice were infected with 3,000 EID50 X31 influenza virus and allowed to rest for 90 days. Mice were then challenged with 50 LD50 PR8 virus. Ccr5−/−Cxcr3−/− cells show a markedly decreased contraction after viral clearance, leading to the establishment of massive numbers of memory CD8+ T-cells. The enhanced potential of virus-specific Ccr5 −/− Cxcr3 −/− cells to progress to memory is caused by their inability to re-encounter antigen in the lung, as the addition of exogenous antigen is sufficient to induce the activation and contraction of virus-specific Ccr5 −/− Cxcr3 −/− T-cells in the lung. Ccr5−/−Cxcr3−/− mice had lower viral titers and increased pulmonary inflammation as graded histologically during the early stages of the immune response (22). In the sixth model (6), Tregs were depleted by injecting an antibody to CD25 (PC61) on days −3 and −1 before secondary infection (12) and then challenged with 60,000 EID50 PR8 influenza virus. In the context of influenza virus infections, memory Tregs control the magnitude of the cellular immune response to secondary viral infection. To examine the antigen-specific contribution of these memory Tregs, NP311–325/IAb Class II tetramers were used to track the antigen-specific CD4 Treg response (8). During secondary influenza virus infection, both tetramer-positive and tetramer-negative Tregs accumulated in the lungs after infection. When memory Tregs were depleted before secondary infection, the magnitude of the antigen-specific memory CD8+ T-cell response was increased as was pulmonary inflammation graded histologically, and airway cytokine/chemokine expression. However, Treg depletion did not alter morbidity, mortality, or virus clearance during the course of the study, but epithelial proliferation was greatly increased when compared with untreated mice (see next section: IL-10-deficient mice). The tissues of at least three mice were examined at each time point.
Increase survival after clearing infection at 2 weeks, but later death from extensive proliferation.
OVA, ovalbumin; SCID, severe combined immune deficient; WT, wild type; 0, no change.
+, ++, +++, and ++++ indicate the extent of pathologic reaction.
Results
IL-10-deficient mice
The lungs of IL-10 KO mice were examined for immunopathologic changes at 6–8 days after challenge with 5,000 EID50 (1 LD50) of influenza PR8 (H1N1). When assayed at these time points, this dose is not sufficient to produce the acute lethal necrotic bronchitis observed classically, but as expected, 50% of the WT mice died at about 10 days, but only 20% of the IL-10-deficient mice died (30). By grading of coded slides (see Table 3), the inflammatory changes were significantly greater in the IL-10-deficient mice than in the WT mice [see McKinstry et al. (30) for details]. Perivascular and peribronchial mononuclear inflammation predominates. There is invasion of the bronchial epithelium by lymphocytes and destruction of epithelial cells with formation of microvesicles (Fig. 1). There is little or no polymorphonuclear infiltration or involvement of distal alveoli; there is no congestion, edema, or hyaline membrane formation. There are perivascular and peribronchial collections of T-cells (Fig. 1E, F, I), and infiltration of the epithelial layer of the bronchus with T-cells associated with separation of epithelial cells. There are very few B-cells in this infiltrate (see smaller arrows in Fig. 1H) and no staining of cells with F4/80 macrophages (not shown). However, there are focal dense collections of peribronchial lymphocytes (BALT, see next section: Transplantation of CD4 T memory cells that consist mainly of B-cells (larger arrow in Fig. 1H), with a few scattered T-cells (Fig. 1I). Epithelial proliferation is not seen at this relatively early time after infection.
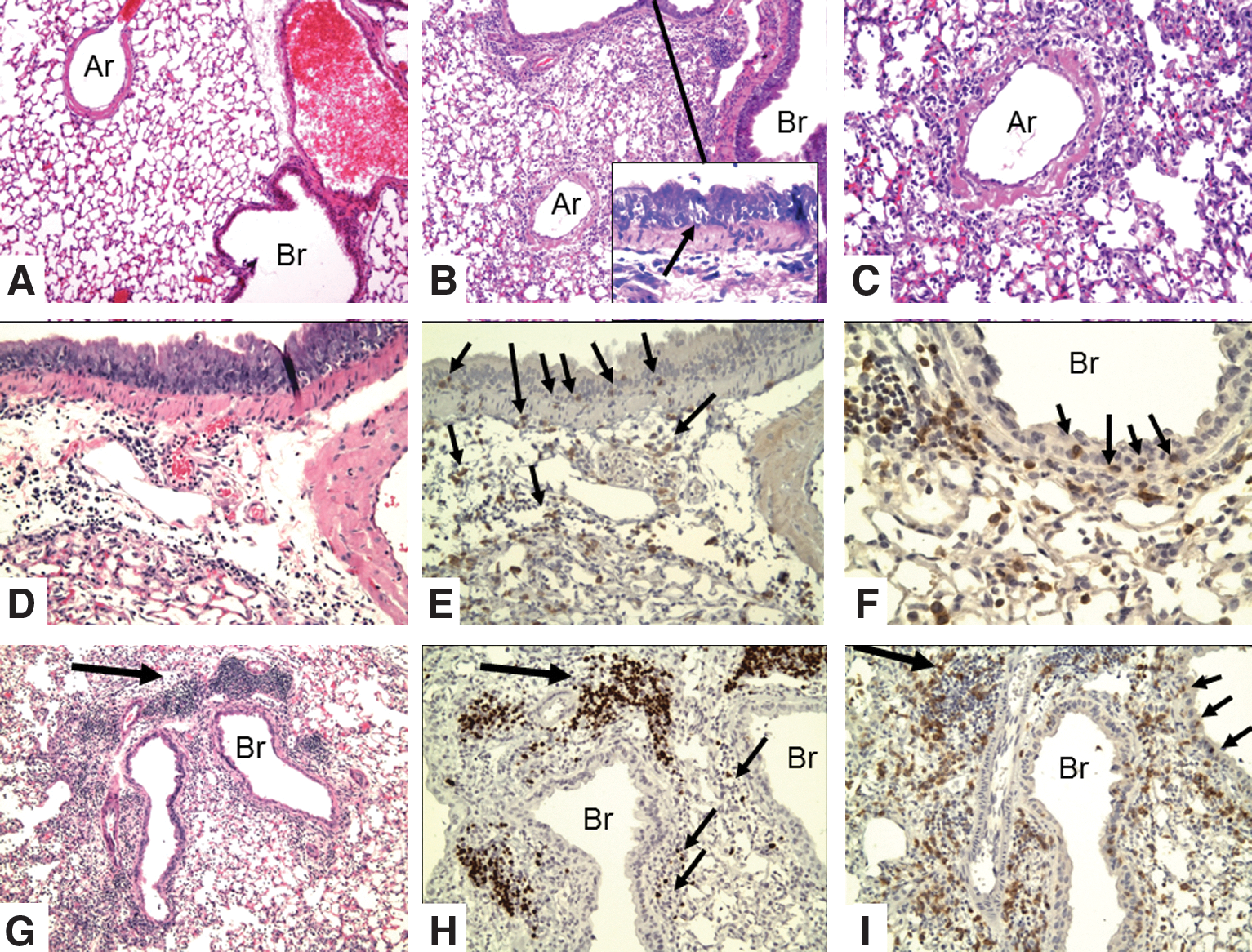
Immunohistologic changes in lungs of IL-10 KO mice at 8 days after intranasal infection with 5,000 EID50 of influenza PR8 (H1N1) virus.
Transplantation of CD4 memory T-cells
Although classically CD8 T-cells are considered cytotoxic cells, there is increasing evidence for CD4 cytotoxic T-cells (7,8,27). Naïve CD4 T-cells do not contribute to viral clearance during primary influenza infection, but memory CD4 T-cells provide potent secondary immunity (8). To better document how protective memory CD4 T-cells contribute to protective immune responses against influenza, and how they might also impact the regulation of repair processes after viral clearance, two sets of experiments were done: transplantation of CD4 memory T-cells to unprimed wild mice and then transplantation to SCID mice.
Transplantation of CD4 memory T-cells to WT mice
Unprimed WT mice received either naïve or memory CD4 T-cells specific for an epitope of the influenza virus before challenge with a lethal dose of 10,000 EID50 (7,8). The slides were prepared 2 days after infection to determine early inflammation driven by memory CD4 T-cells. These studies were inspired by our finding that memory CD4 T-cells drive an enhanced inflammatory cytokine and chemokine response in the lungs that is evident within 48 h of infection and results in a reduction of viral titer evident at 3 days postinfection (27). The mice receiving naïve cells will go on to die (by about day 10 or so), whereas the mice receiving memory CD4 T-cells will survive and clear virus by about day 10. An examination of the lung (Fig. 2) shows slight perivascular and peribronchial infiltration with mononuclear cells after transfer of naïve cells, but a marked increase after transfer of memory CD4 T-cells, including early T-cell invasion and disruption of the basement membrane of bronchial walls and formation of luminal exudate in terminal bronchioles. The degree of cytotoxic T-cell infiltration and reaction with infected epithelial cells (demonstrated by labeling for virus – data not shown) correlates with protection against lethal infection. Changes in the lung were not examined after 3 days.
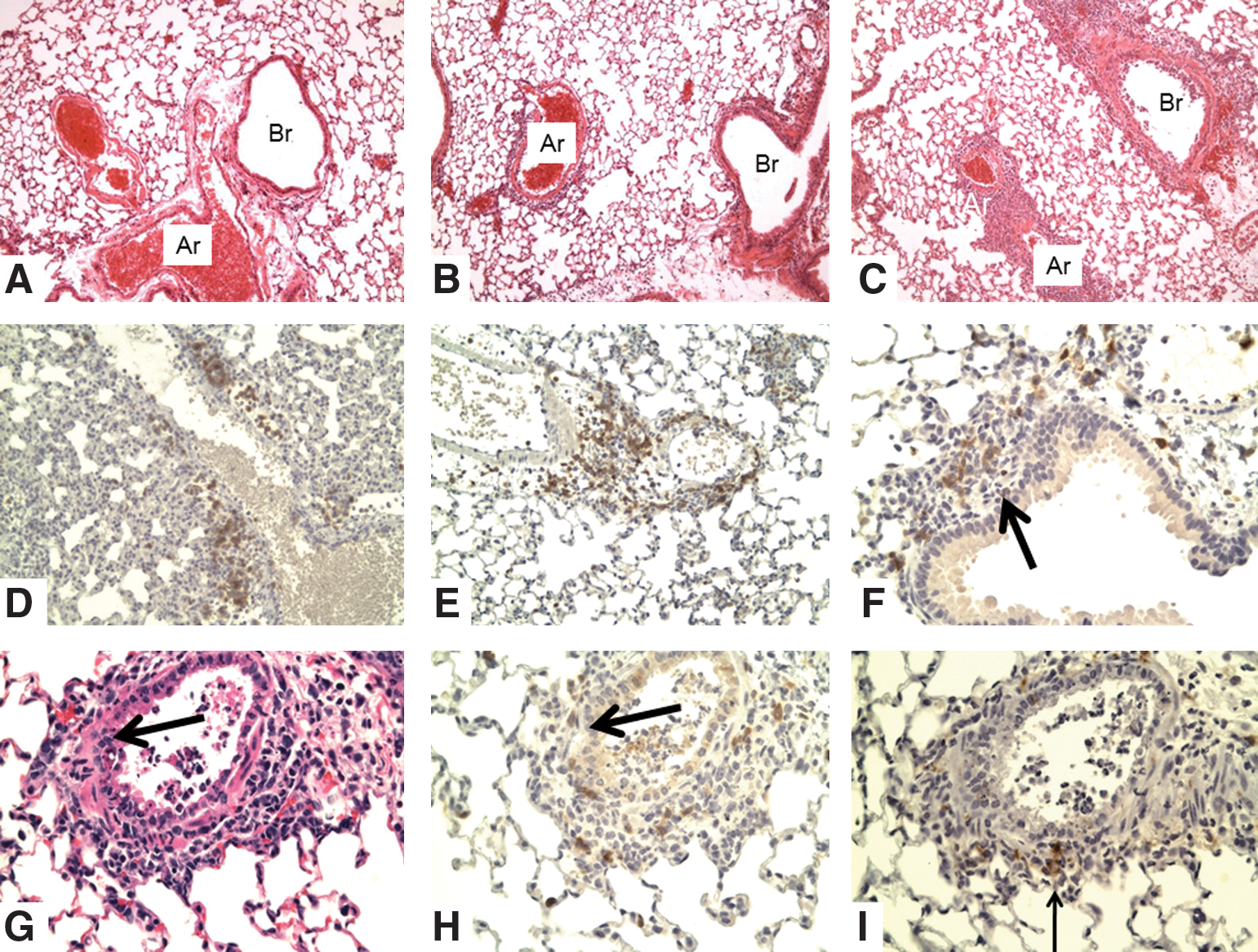
Lungs of WT mice receiving naïve or memory CD4 T-cells at 2 days after intranasal challenge with 10,000 EID50 PR8 influenza virus.
Transplantation of CD4 memory T-cells to SCID mice
SCID mice contain no immune response mechanism and because of this serve as an immunologically blank animal for analysis of function of transplanted cells. Our recent studies showed that memory CD4 T-cells could protect SCID mice against lethal influenza challenge, but that without contributions from CD8 T-cells or neutralizing antibody, the virus is not fully cleared, eventually leading to the death of animals during the third or fourth week postinfection: (27). We investigated the immunopathologic changes mediated by memory CD4 T-cells responding to influenza in SCID mice during the protective phase (weeks 1 and 2) as well as later when memory CD4 T-cell protection begins to fail in SCID mice (weeks 3 and 4). One week after influenza infection of SCID mice bearing transplanted CD4 memory cells for flu virus, there is focal perivascular, marked interstitial, and peribronchial collections of lymphocytes (Fig. 3A, F–H). There is little hyperplasia of the bronchial epithelium at this time (Fig. 3). Most of the periphery of the lung is not involved, and the alveolar sacs are mostly normal. By week 2 (Fig. 3B, I, J), the perivasuclar and peribronchial inflammation is increased, especially around bronchi. The lymphocytic infiltrate, as expected, contains only T-cells (the donor cells) (Fig. 3I, J) and large numbers of T-cells can be seen in the epithelial layer (Fig. 3J). The bronchial epithelium is hyperplastic in some sections, but there is no evidence of continuing injury. By week 3 (Fig. 3C), the most striking finding is focal proliferation of bronchial epithelium extending into the adjacent pulmonary parenchyma, most likely due to proliferation of epithelial cells along terminal air sacs or alveolar walls with squamous metaplasia. By week 4 (Fig. 3D, E, K–O), the degree of focal proliferation of bronchial epithelium, seen as solid collections of epithelial cells resembling small epithelial tumors, has increased with some areas showing early squamous metaplasia. The expanding epithelial cells label for both TTF (stain for bronchial epithelial cells) and protein C (surfactant for type II pneumocytes) (Fig. 3L–O). This most likely represents a hyperplastic reparative response of type II pneumocytes and/or terminal bronchial epithelial cells attempting to restore sloughed epithelium. Some alveoli are filled with loose collections of large mononuclear cells, but these do not stain with F4/80. The proliferative response will be presented in greater detail in later experiments. Although there is a prominent peribronchial lymphocyte infiltration, the organized collections of BALT described next are not seen, most likely because BALT formation requires a major participation of B-cells, which are not present in the SCID mice.
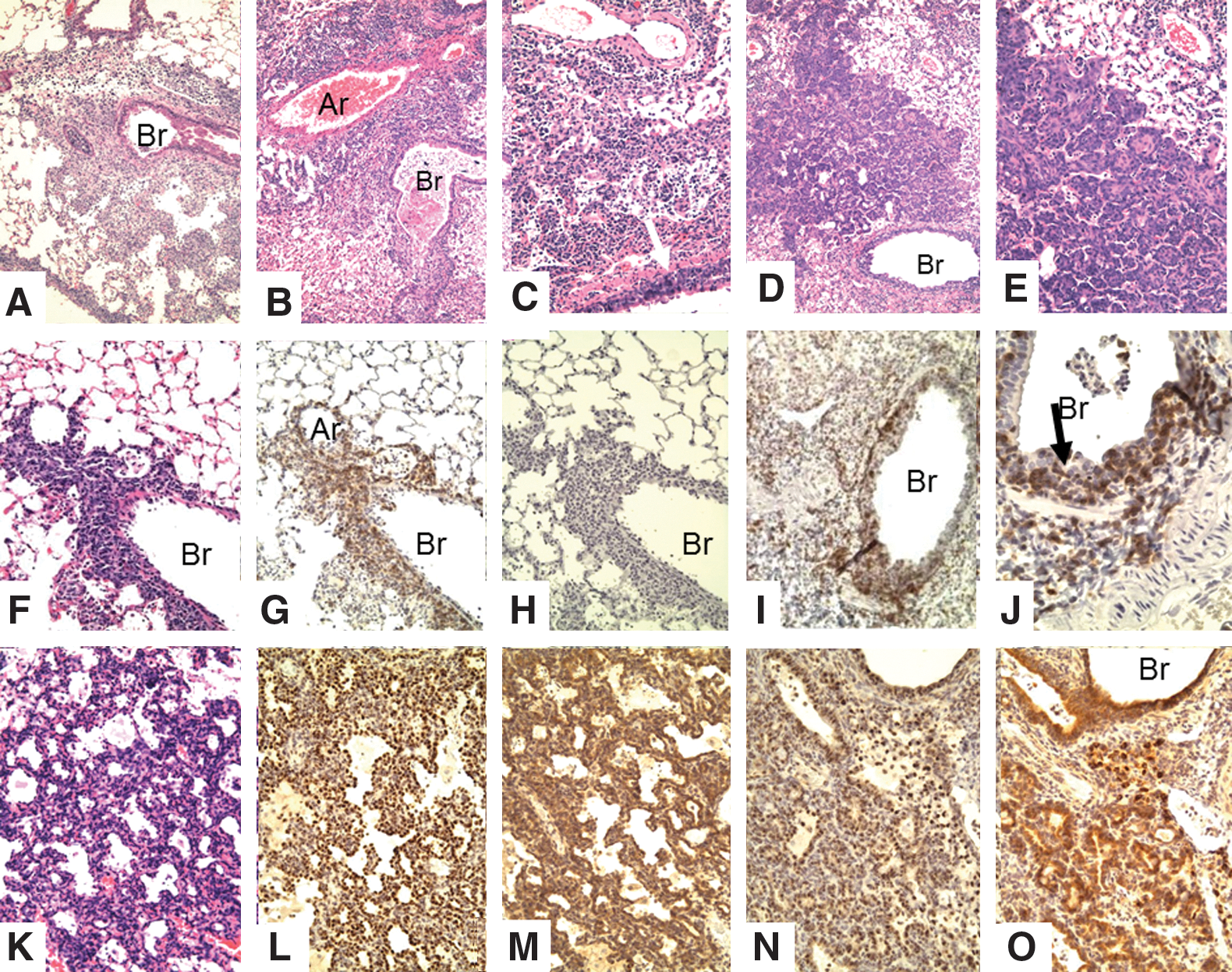
Lungs of SCID recipients of CD4 memory T-cells after challenge with 2,500 EID50 PR8 virus.
Transfer of OVA-specific memory CD4 T-cells to WT mice
The pictures in Supplementary Figure S3 are from WT mice that were given memory CD4 T-cells specific for OVA protein [from OT-II transgenic mice; see Barnden et al. (3)] and then given OVA intranasally. The tissues were taken at 48 h after OVA administration. There is marked perivascular infiltration by lymphocytes with essentially no involvement of the bronchi or bronchioles. This appears to be analogous to a delayed hypersensitivity skin reaction taking place in the lung. Since the eliciting antigen (OVA) diffuses from the bronchus into the lung, the antigen-T-cell reaction takes place in the periarterial space, resulting in the perivascular mononuclear cell reaction observed. Similar to a delayed hypersensitivity skin test, the inflammation peaks at 24–48 h after antigen challenge. In contrast, WT and SCID mice transplanted with IAV-specific CD4+ T-cells before infection with IAV show marked alveolar, peribronchial, and bronchial inflammation due to a reaction of CD4 T-cells with viral antigen located in infected epithelial cells.
Effect of enhancing memory CD8 T-cells
WT and Ccr5 −/− Cxcr3 −/− mice were infected with 3,000 EID50 X31 IAV and allowed to rest for 90 days. Mice were then challenged with 50 LD50 PR8 IAV and sacrificed at various times after challenge. Lung sections from WT and Ccr5 −/− Cxcr3 −/− mice were examined on days 0, 3, 7, and 14 after influenza challenge. Representative sections for WT and Ccr5 −/− Cxcr3 −/− mice are shown in Figure 4. The changes seen were qualitatively similar between the two groups, but were quantitatively much more marked in the Ccr5−/−Cxcr3−/− mice. The two striking findings are a marked mononuclear cell infiltrate around the arteries and bronchi, and extensive proliferation of epithelial cells from the terminal bronchi into the adjacent alveoli (Fig. 5A–E). Again, these epithelial cells stain for both TTF and protein C. The majority of the periarterial and peribronchial lymphocytes were T-cells (CD3+) with few, if any, B-cells or macrophages. The prominent BALT stained heavily for B-cells with a scattering of T-cells (Fig. 4I–K), but the presence of larger unlabeled cells suggests the presence of F4/80-negative dendritic cells (arrows in Fig. 4J, K). The proliferative and BALT changes are even more pronounced in the next model examined in which CD4 T-cells predominate.
Representative changes in lungs of Ccr5−/−Cxcr3−/− mice after secondary intranasal challenge. WT and Ccr5−/−Cxcr3−/− mice were infected with 3,000 EID50 X31 influenza virus and allowed to rest for 90 days. Mice were then challenged with 30,000 EID50 (50 LD50) PR8 virus.
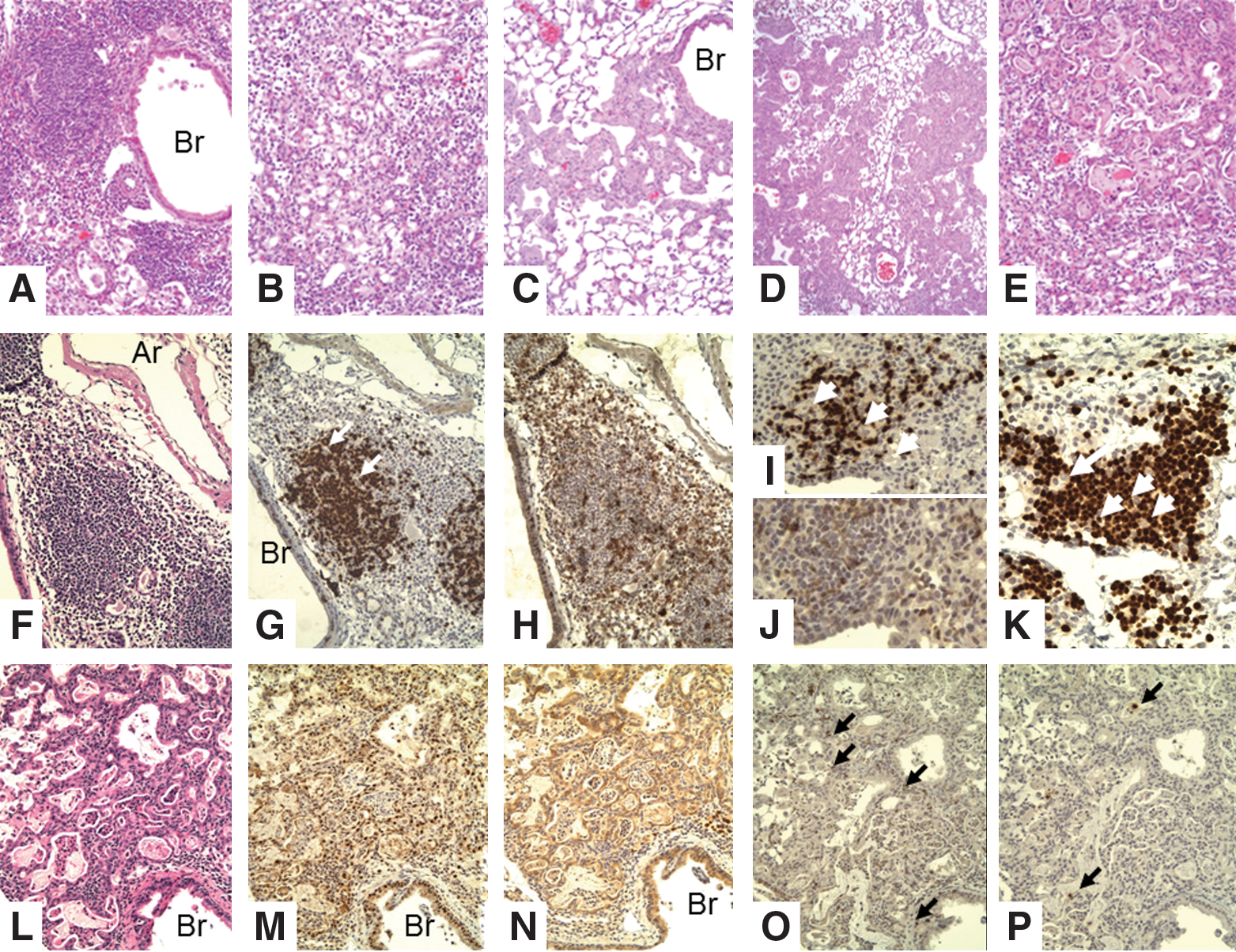
Later lesions in mice depleted of Tregs before secondary virus challenge. FoxP3-GFP mice were infected intranasally under anesthesia with 500 EID50 PR8 virus and allowed to rest for 35 days. Then, they were challenged with 5×104 EID50 PR8 virus and euthanized 4 days later.
Effect of regulatory T-cells (Tregs: CD4+FoxP3+ cells) on extent of inflammation and proliferation after influenza infection
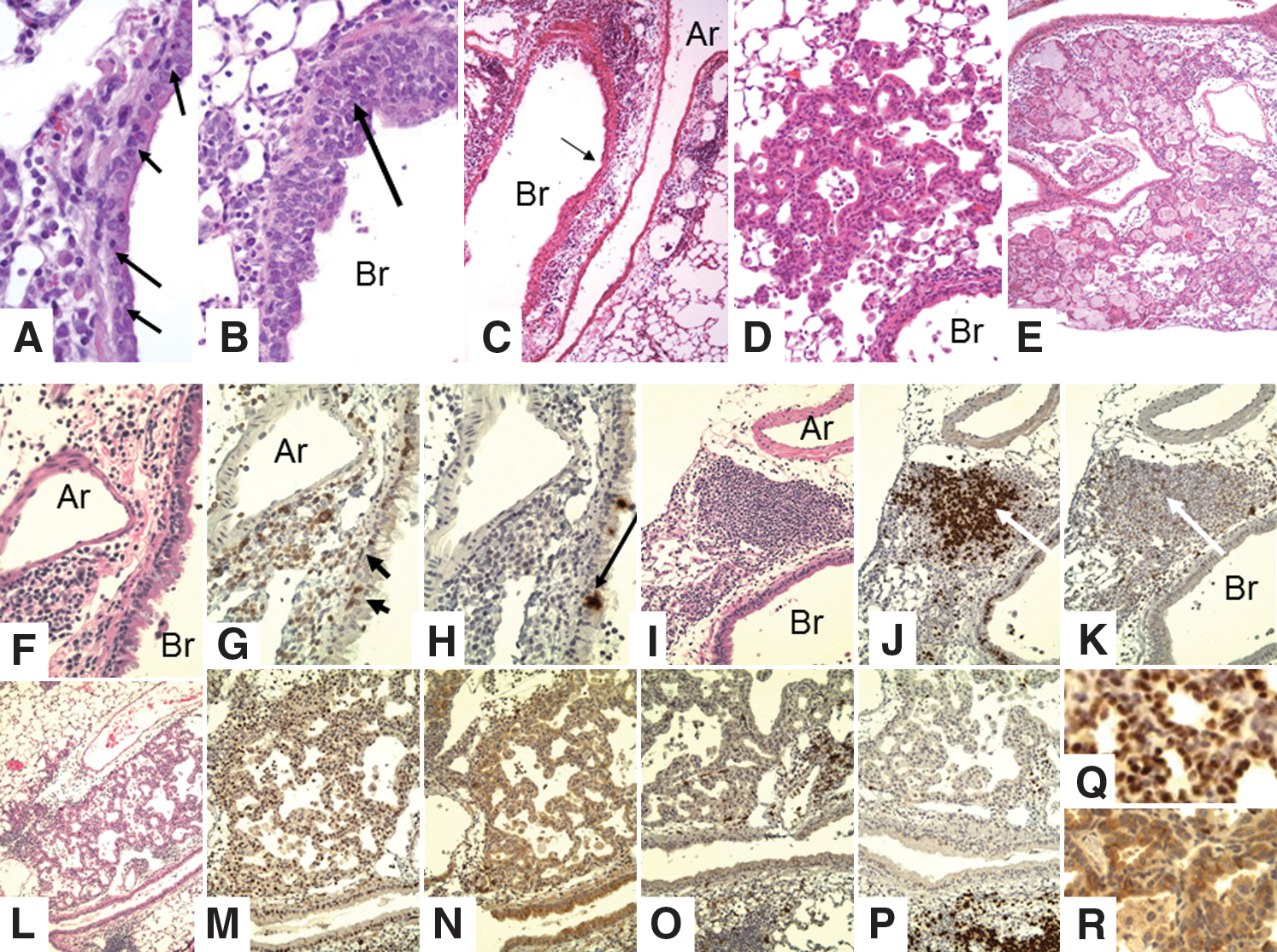
The contribution of Tregs to controlling the immune response was examined in mice treated with anti-CD25 antibody (PC61) that depletes Tregs. Subsequent to depletion, memory-stage mice were challenged with secondary virus infection. Lungs were harvested at 5 days postinfection, fixed, sectioned, and stained with H&E for grading (Fig. 5). Grading of inflammation in this model, as in the other models, is based on both the nature of the lesion and the degree of involvement (see Table 3). Differences between the histology scoring data were determined by the Mann–Whitney U nonparametric test. The mean inflammation index for the PC61-treated mice was 3 and 1.4 for the control, with a p=0.0014. The mean proliferation score for the PC61-treated mice was 1.3 compared with less than 0.3 for the controls with a p=0.008. Thus, depletion of memory Treg before secondary influenza virus infection results in significant increases in both inflammation and epithelial cell proliferation; these changes were accompanied by an increase in antigen-specific memory CD8+ T-cell responses. A broader examination of pulmonary changes revealed a lymphocytic cell infiltrate that varies from small compact foci through large dense areas (BALT, Fig. 5A, 5F–K), loose mononuclear cells in alveolar spaces (Figs. 5B and 5G, 5I, 5K),), and extensive epithelial proliferation (Fig. 5L–P). The conversion of areas of the lung next to bronchi into BALT is much greater in mice depleted of Tregs than in mice with Tregs, which feature loose collections of large monocytes with retention of alveolar structure. In addition to the increased appearance of BALT tissue in the Treg-depleted mice, strikingly large areas of the alveolar spaces are filled with epithelial cells (Fig. 5C–E). Consistent with this observation, the proliferation of the epithelial cells was also much greater in the Treg-depleted mice (Fig. 5L–P). It is well documented in the literature that such proliferation is seen in mice infected with influenza virus beginning with 3–6 days after primary infection, and peaking in the alveoli at about 2 weeks (see Discussion section). The absence of Tregs apparently exacerbates the already robust repair response, as it expands to occupy more than 50% of the lung tissue on the histologic slides. In addition, some of the epithelial cells demonstrated squamous metaplasia (Fig. 5E, P), despite the fact that these mice were sacrificed only 5 days after infection—which is relatively early in the repair response for such extensive proliferation and squamous metaplasia.
Discussion
We will first review the pathology of human influenza, then the pathologic changes described in the literature for WT mice infected with influenza virus, and finally compare these with the observations made on the mouse models in this article.
Pathology of human influenza
The pathologic changes in the lung found in fatal influenza infection have recently been reviewed by Taubenberger and colleagues (24,51). Not only did they extensively review the reported findings of earlier observers, but they also reexamined tissues from the 1918 to 1919 flu pandemic (4). In fatal influenza not complicated by secondary bacterial infection, there is multifocal superficial necrotizing tracheo-bronchitis, alveolar necrosis, and hyaline membrane formation with edema, hyperemia, and mixed mononuclear infiltration of the lamina propria. The necrotizing broncho-alveolitis includes death of type I and type II pneumocytes. Examination of the cases of the 1918 epidemic revealed that the destruction of the bronchial epithelium is followed by extensive proliferation of epithelial cells (2,26,37,54). Mitotic activity can begin as early as 5–14 days after the onset of infection (33) and extends from terminal bronchioles into adjacent alveloli with loss of lung function. The following quote is from Winternitz et al. (54): “It is rare to see such activity on the part of the epithelium as that frequently encountered in influenza. The alveoli may be lined by newly formed cubical cells (Figs. IV, XI, XL VII), and mitotic figures in the injured bronchiolar lining occur in abundance.…… . In a number of cases, epithelial proliferation has been so extensive that it could not be differentiated histologically from an invasive, malignant neoplasm (Figs. XL VIII and XLIX). There is no reason to believe that malignancy might not result from the continuous stimulation of the epithelium to proliferate.” Since these pathologic studies were done on autopsied cases, it is likely that the proliferation contributed to the death of those examined, but this has not been definitively established and the possibility of malignant transformation has not been ruled out. Many cases are complicated by bacterial infection. In these cases, there is greatly increased edema, polymorphonuclear cell infiltrate, necrosis, and fibrin formation. Most of the tissues examined in these studies were from autopsy material of patients who died from the infection. Thus, there is much less known about the pathologic changes that occur early in the course of humans who survive flu infections. Several studies have been done on lung biopsies on patients while still alive less than 2 weeks after infection (56) or within 19 days of infection (35). The acute changes in the lung varied from patchy fibrinous alveolar exudates and hyaline membranes with interstitial edema to severe diffuse alveolar damage and necrosis of the bronchiolar mucosa. Also prominent were reparative changes, including proliferation of epithelial cells, mild interstitial chronic inflammation, and organization within air spaces and in the interstitial tissue. The extensive lung damage in fatal human infections most likely masks the more subtle intraepithelial cytotoxicity seen in experimental mouse models. It is likely that humans with less extensive lung damage that might be cleared by a T-cell cytotoxic response survive without a pathologic examination.
Experimental infection
Swine
Shope (46) first clearly identified a virus as the cause of influenza by infection of swine by intranasal installation of filtrates of infected human tissues or exudates. The principal features of the pathology of swine influenza were an exudative bronchitis accompanied by marked damage of the bronchial epithelium, a peribronchial round cell infiltration, and massive pulmonary atelectasis. The lining epithelium was fragmented, in places partially desquamated, and the cytoplasm of many of the cells appeared vacuolated. In the spaces created by the fragmentation of the lining epithelium, leukocytes, singly or in clumps, were sometimes seen. There was extensive peribronchial round cell infiltration. The observation of lymphocytes in the “fragmented” bronchi is very similar to the T-cell cytotoxicity lesions described in our experiments, but iBALT formation and epithelial proliferation are not described.
Mouse
In 1937, Straub (49) described pulmonary changes in mice infected with the W-S (Wilson–Smith) human flu virus. The classic description of the effects of infection by influenza in mice is as follows: “The terminal and respiratory bronchioles are totally denuded of epithelium. The alveoli are collapsed and partially filled with edematous fluid. The epithelium loses it distinct and regular appearance, the cells stain badly, become low and loosen from the bronchiolar wall; or while attached to it undergo fibrinoid necrosis, forming an adherent layer in which here and there the shadow of a nucleus may be recognized.…The fibrinoid necrotic layer, like the necrobiotic epithelium, desquamates and is then sometimes found loose in the bronchiolar lumen.…Mononuclear cells may be present in the peribronchiolar interstitium. Polymorphonuclear infiltration is either totally absent or present to a very slight extent.….A chronic reparative process in surviving mice has, indeed, been regularly met with. This is not, as so often in man, fibrous but epithelial in nature. The epithelium of the terminal bronchioles proliferates and becomes more or less stratified.…The proliferation does not stop here, but regularly invades the lung tissue. It first enters the respiratory bronchioles and next the alveoli.” This was not fibrous, but epithelial proliferation with squamous metaplasia that invades the lung. These observations were extended by Oliphant and coworkers (34,36), by Taylor (52), by Dubin (13), and by Loosli (25), who noted a slightly later time for the proliferative response to begin and resolve. In some mice, marked hyperplasia and growth of bronchial epithelium into surrounding lung tissue persisted for approximately 3 months, and invasive growth with tumor formation has been described, implying malignant transformation (9).
In a more recent publication, Qiao et al. (38) describe severe interstitial and intra-alveolar fibrosis with thickened alveolar walls, collapsed alveoli, and large fibrotic areas in 8 week-old BALB/c mice at 30 days after intra-nasal inoculation with 1×10 MID50 of A/Chicken/Hebei/108/2002(H5N1) viruses. They state: “The diffuse intra-alveolar fibrosis was a common finding, with an excessive collagen deposition and cell proliferation in air spaces that obliterated the alveolar spaces and severely distorted the structure (Fig. 3E, F).” The finding of fibrosis contrasts with previous observations of epithelial proliferation in mice later after infection and no fibrosis. However, we have carefully examined enlargements of Figure 3E and F of this paper and conclude that these pictures show epithelial proliferation with squamous metaplasia, although fibrosis in other sections cannot be ruled out.
Bronchial intraepithelial lymphocytes in mice infected with attenuated strains
Infection of mice with attenuated strains reported by Bi et al. (5) reveals infiltration of bronchial epithelium by lymphocytes, although this was not recognized by the authors. Bi et al. report on the effects of experimental pulmonary infection of BALB/c mice with 15 representative H9N2 reassorted type A viruses. In particular, we would like to point out the tracheal lesions depicted in their Figure 4 comparing the effect of infection of mice with the highly virulent Ck/SD/WF/98 virus to that of infection with the weakly virulent Ck/SD/03/10 virus. Bi et al. describe “Congestion in the blood vessels of the tracheal submucosa (black triangle) and dropout of the mucous epithelium in the trachea (thick solid arrow) caused by the Ck/SD/WF/98 virus (C and F).….There were no obvious histopathological changes in the respiratory system tissues of the Ck/SD/03/10-infected group (I) and no significant difference compared to the PBS mock-infection group (L).” We agree with the description of the congestion and loss of the mucous epithelium in panel C that is consistent with previous descriptions of the T-cell-mediated destruction of bronchial epithelium after influenza virus infection, but would like to make additional observations on panels F, I, and L. First, panel F shows columnar epithelium separated from the basement membrane by an infiltrate of small round cells, most likely lymphocytes. Second, there is a clear difference between pictures I and L. Panel L shows a normal ciliated columnar epithelium. Panel I shows an epithelium that is invaded and separated from the basement membrane by small round cells. In addition, there is a substantial round cell infiltration of the submucosa. This is consistent with our observations of cell-mediated cytotoxicity of bronchial lining cells. The necrosis of epithelial cells is most likely due to T-cell-mediated cytotoxicity of virus-infected cells, but a role for some components of innate immunity cannot be ruled out (1).
Contact dermatitis/viral exanthema
The infiltration of the bronchial epithelial with lymphocytes associated with destruction of epithelial cells is characteristic of the skin lesions of contact dermatitis and viral exanthema, including smallpox and measles (42). In contact dermatitis such as caused by poison ivy, the antigen, an oleoresin, is lipid soluble. It diffuses into the epidermis and binds to the epithelial cells (hapten). In an individual sensitive to the antigen, cytotoxic T-cells circulating through the skin react with the antigen diffusing into the dermis, and then are attracted into the epidermis most likely because of an antigen gradient. Since most of the lipid-soluble antigen is attached to epidermal cells, in sensitized individuals it takes about 2 days for the reacting cells to invade from the dermis, so that poison ivy reactions usually peak around 48 h. In naïve individuals, it takes about 6–7 days for sensitization to take place and for the cells to enter the epithelial layer. As a result of the epidermal invasion, epidermal cells are separated from each other and destroyed [a form of anoikis (17) also seen in cell mediated autoimmune diseases, such as auto-allergic thyroiditis] (15,42). Small sterile micro-abscesses are formed, eventually leading to vesicle formation. The loss of epidermal cells eventually leads to stimulation of proliferation of the basal stem cells and restoration of the skin. We believe that a similar cytotoxic reaction occurs in bronchial mucosal epithelial cells infected with influenza virus when cytotoxic T-cells are available. This reaction is also responsible for the skin lesions of measles, rubella, erythema infecteosum, and roseola, as well as for the “take” reaction to vaccinia vaccination against smallpox (42). This finding supports the argument that “vaccination” against influenza by intradermal inoculation with living attenuated strains of virus might produce longer-lasting and more effective immunity not only to the immunizing strain, but also to other more virulent strains than currently used vaccines. Intradermal vaccination with recombinant influenza-virus like particles using microneedles has been shown to induce superior protection than intramuscular immunization (39).
The role of other immune mechanisms in reaction to influenza virus infections should also be considered. Classically, four immunopathologic mechanisms were identified by Gell and Coombs (11). This was extended to six in 1972 (43): four antibody-mediated and two cell-mediated reactions (delayed hypersensitivity and T-cell cytotoxicity). There appears to be little or no role for antibody-mediated mechanisms in the production of the lesions of influenza virus infection. Since the virus is intracellular, antibody-mediated mechanisms are not likely to cause tissue damage. However, circulating antibody most likely plays a critical role in preventing re-infection of cells, as such an antibody will block viral receptors for cells, thus limiting infection. The perivascular mononuclear infiltration seen after intra-tracheal OVA challenge of mice bearing OVA reactive CD4 T-cells is a characteristic of a delayed hypersensitivity reaction. In this case, the OVA antigen diffuses across the bronchi and interacts with circulating sensitized CD4 T-cells in the vessel walls. The OVA-stimulated delayed hypersensitivity reaction does not extend to the bronchi. In contrast, with influenza virus infection of the epithelial cells, cytotoxic T-cells migrate across the basement membrane into the infected epithelium where they kill the virus-infected cells.
iBALT formation
iBALT represents an adaptive immune response in the lung and is a site where local immune responses can occur rapidly after exposure to inhaled antigens. BALT is a constitutive mucosal lymphoid tissue in rats and rabbits, but is not usually present in humans or mice. However, BALT is inducible in humans and mice after inflammation in the lung (40,45) (so-called iBALT). iBALT occurs in chronic lung infections in humans (53), as well as in mice with repeated virus infections of the lung (10). Mice lacking spleen, lymph nodes, and Peyer's patches develop extensive BALT in response to influenza challenge, clear influenza infection, and when BALT is present survive higher doses of the virus than do normal mice (32,47). BALT is composed mostly of B-cells with fewer admixed T-cells and dendritic cells and most likely provides a site for a more rapid immune response to subsequent infection. The presence of iBALT in response to one viral infection enhances the immune response to subsequent infection with unrelated viruses (heterologous immunity), by altering cytokine patterns and increasing antibody titers (10). An improved response is seen if the early acute damage mediated by polymorphonuclear cells quickly changes to a lymphocytic response; a worsened response occurs if it changes from a mild lymphocytic response to a severe lymphocytic response depending on the specific viral sequence (10). In any case, BALT may be considered a local site for immune memory that can respond quickly to a second pulmonary infection.
Epithelial proliferation
The epithelial proliferation that occurs after the first week of infection of mice with influenza virus may become extensive, essentially filling up the alveoli of large portions of the lung. Fortunately for the surviving mice, the proliferative lesions appear to be restricted to areas of the lung where the virus infection occurred. Similar to viral exanthema, the infected sites in experimental murine influenza are focal, so that in nonfatal infection, large portions of the lung are not involved. The proliferation appears to extend from the terminal bronchi where it is believed epithelial progenitor cells, including those giving rise to epithelial cancer, may be concentrated (44). The proliferating cells stain for both protein C and TTF, as would be expected for putative lung epithelial progenitor cells (55). In WT mice, this proliferation appears to be self-limiting, but in SCID mice or mice depleted of Tregs, it continues and may result in death. The finding that this proliferation is much greater in mice with impaired Treg function suggests that the proliferation behaves similar to an immune system-sensitive carcinoma. The finding that depletion of Tregs allows progressive epithelial growth raises questions about the recent depletion of Tregs to enhance the immune response in cancer therapy (14).
IL-17 and IL-22
The formation of iBALT and epithelial proliferation are most likely the result of increased IL-17 and IL-22 in influenza-infected mice (23). We have previously shown that increased expression of IL-17 in IL-10 KO mice is associated with increased survival of influenza-infected mice (30). These mice show iBALT formation as early as 6–8 days after influenza infection (see IL-10 Deficient Mice section under Results section). In addition, IL-17-secreting (Th17 cells) as well as IL-22-positive T-cells are increased in mice after challenge with influenza and protect against lethal challenge (19). The major source for IL-17 and IL-22 during acute infection is innate lymphocytes (γδ T-cells and natural killer cells). However, Th17 T-cells arising from maturation of CD4 helper cells contribute to cytokine production during induced immunity (23,28,30). Il-17 is key to iBALT formation (41) with or without the participation of dendritic cells (16). IL-17 acts by increasing expression of chemokines CXCL12 and CXCL13, which are critical for follicle formation. IL-22 promotes epithelial proliferation (18,28), including epithelial cell transformation and tumorigenesis (21), through receptors on epithelial cells involved in STAT3 activation that are not found on immune cells (48). For example, SCID mice have NK cells but do not have other immune components. Thus, SCID mice have epithelial proliferation, but no iBALT formation because they lack B-cells. On the other hand, Th17, produced by innate lymphoid cells or cells derived from the CD4 memory cells transplanted to SCID mice, and IL-22, produced by innate lymphoid cells, could explain the progressive pneumocyte proliferation observed. We conclude that in later stages of experimental flu infections, IL-17 and IL-22 produced by innate lymphoid cells or Th17 cells result in iBALT formation and epithelial proliferation and the effect of both are enhanced in mice with depressed Treg function.
Footnotes
Acknowledgment
Support by the following grants from NIH: P01AI46530 S. Swain and R. W. Dutton; T32 AI049823 D. L. Woodland; and AI083610 J. E. Kohlmeier. Thanks are due to Helen Johnson at the Wadsworth Core Laboratory for cutting tissue sections.
Author Disclosure Statement
The authors have no known conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.