
Abstract
Zika virus (ZIKV), a single-strand RNA flavivirus, is transmitted primarily through Aedes aegypti. The recent outbreaks in America and unexpected association between ZIKV infection and birth defects have triggered the global attention. This vouches to understand the molecular mechanisms of ZIKV infection to develop effective drug therapy. A systems-level understanding of biological process affected by ZIKV infection in fetal brain sample led us to identify the candidate genes for pharmaceutical intervention and potential biomarkers for diagnosis. To identify the key genes, transcriptomics data (RNA-Seq) with GSE93385 of ZIKV (Strain: MR766) infected human fetal neural stem cell are analyzed. In total, 1,084 differentially expressed genes (DEGs) are identified, that is, 471 upregulated and 613 downregulated genes. Further analysis such as the gene ontology term suggested that the downregulated genes are mostly enriched in defense response to virus, receptor binding, laminin binding, extracellular matrix, endoplasmic reticulum, and for upregulated DEGs: translation initiation, RNA binding, cytosol, and nucleosome are enriched. And through pathway analysis, systemic lupus erythematosus (SLE) is found to be the most enriched pathway. Protein–protein interaction (PPI) network is constructed to find the hub genes using STRING database. The seven key genes namely cyclin-dependent kinase 1 (CDK1), cyclin B1 (CCNB1), histone cluster 1 H2B family member K, (HIST1H2BK) histone cluster 1 H2B family member O (HIST1H2BO), and histone cluster 1 H2B family member B (HIST1H2BB), polo-like kinase 1 (PLK1), and cell division cycle 20 (CDC20) with highest degree are found to be hub genes using Centiscape, a Cytoscape plugin. The modules of PPI network using Molecular Complex Detection plugin are found significant in structural constituent of ribosome, defense response to virus, nucleosome, SLE, extracellular region, and regulation of gene silencing. Thus, identified key hub genes and pathways shed light on molecular mechanism that may contribute to the discovery of novel therapeutic targets and development of new strategies for the intervention of ZIKV disease.
Introduction
Z
ZIKV infects humans mainly through vectors such as Aedes genus (13) and monkeys (30). Recent studies revealed that the viral transmission is through sexual transmission (2), maternal–fetal, and nonvector mode of transmission such as amniotic fluid, breast milk, seminal fluid, saliva, urine, and blood products (19). Recent outbreak in Brazil caused a pandemic health emergency as it is linked to cause microcephaly (6,52), a brain defect in newborn infants and an autoimmune disease known as Guillain–Barre syndrome (37,39) in adults.
Human neuron progenitor cells, human cerebral organoids, skin explants, fibroblasts, dendritic cells, and peripheral blood mononuclear cells are known targets to be infected by ZIKV (10,15,19). However, the severity of the infection increases due to limited knowledge of molecular pathways, mechanism of viral replication, assembly, and interaction with host. This led to unavailability of antiviral therapeutics and vaccines for ZIKV infection. It is a main challenge to diagnose ZIKV infection at an early stage. For early detection of ZIKV infection and diagnosis, finding the potential key genes associated with the infection may be a way to identify the biomarker.
Using RNA-Seq, deregulation of many genes in response to viral infection from neural progenitors (23), human fibroblasts (20), and embryonic mice with microcephaly (31) has been reported. In bioinformatics, RNA-Seq analyses are challenging due to its voluminosity and complexity. However, many computational methods and techniques have been developed and used for the analysis of transcriptomics data. In addition, network-based approach is extensively used to examine gene expression to analyze and identify the potential candidate genes (36).
This study adopted the network-based approach in ZIKV infection to identify the differentially expressed genes (DEGs) associated for further functional enrichment and pathways analysis. Protein–protein interaction (PPI) network is constructed to find the best candidate genes associated with ZIKV infection.
Materials and Methods
Data collection and quality control
The RNA-Seq samples with gene express omnibus accession, GSE93385 (early infection stage SRS1913269, SRS1913271, and SRS1913274 and control SRS1913267, SRS1913270, and SRS1913268) of KO48 cell line, primary human fetal brain-derived neural stem cells were downloaded from Short Read Archive (SRA) database using R-script in RStudio Version 0.99.49 (40) and converted to FASTQ file format using NCBI SRA toolkit.
FastQC(v 0.11.5) (3) was used to observe and check the quality of data sets. Overexpressed sequences originated from primer or other artifacts along with low-quality bases were removed using Cutadapt (33). Student's t-test was performed.
Identification of DEGs
Galaxy, an open source, web-based platform designed for data intensive biomedical research, was used to analyze the transcriptomic data using the Tuxedo tools suite (16,51). Clean reads were aligned against the Homo Sapiens's RefSeq sequence of UCSC database using short read aligner Bowtie 2 v2.2.5 (27). Human reference genome (GRCh38/hg38) was used as a reference genome. Clean reads were aligned to human reference genome using TopHat v2.0.14 (25) with default parameters. Using Cufflinks v2.2.1 (51), the reads were assembled to transcripts and fragment per kilobase of exon per million fragments mapped to estimate the abundance of transcripts expression. All assembled transcripts were merged to get nonredundant unified set of transcripts by Cuffmerge v2.2.1 (51). Cuffcompare v2.2.1 (51) was used to compare the two assembled transcripts with reference annotation. To test and identify the differentially expressed transcripts, Cuffdiff was performed with default parameters. CummeRbund-R package was used to visualize information and perform statistical analysis (17). Integrative Genomics Viewer visualized the aligned reads to ensure the uniformity in mapping (50).
Gene ontology and pathway enrichment analysis
To understand the biological functions and affected pathways, gene ontology (GO) and pathway analysis were carried out by online tools such as Database for Annotation, Visualization and Integrated Discovery (DAVID) (11), FunRich (38), and WebGestalt (WEB-based Gene SeT AnaLysis Toolkit) (57). The analysis was performed against human reference genome with q-value <0.05 (FDR Benjamini and Hochberg method) and permutation value was set to 1,000.
PPI network construction and visualization
The PPI network was constructed for the DEGs identified using STRING database [v10.5] (53). It is a precomputed database wherein association between the proteins is derived from high-throughput experiments, literature mining, gene fusion, cooccurrence, coexpression analysis, and also computational predictions such as genomic meta-analysis. DEGs of confidence score 0.9 was considered for visualization using Cytoscape v3.4.0 (45).
Network centralities
The PPI network constructed was subjected to analysis based on three centrality parameters that are degree, betweenness, and stress by the plug-in Centiscape of Cytoscape (44). Degree: a number of neighbor nodes are connected to a node. The genes with high degree (number of incident nodes connecting the edges) are considered as hub genes. The stress centrality of a node is number of shortest paths between two other nodes. Another topological metric, betweenness centrality is similar to the stress that reflects the amount of control exerted by one node to another connected in a network (43). It is considered as ratio of the node in the shortest path between two other nodes.
Network module analysis
Using Cytoscape's Molecular Complex Detection (MCODE) plugin (4), the highly dense connected regions or clusters were identified with parameters: degree cutoff = 2, k-core = 5, and
Results
Differentially expressed genes
Out of 4,660 identified DEGs, 2,738 genes were found to be upregulated and 1,922 downregulated (Supplementary Table S1; Supplementary Data are available online at
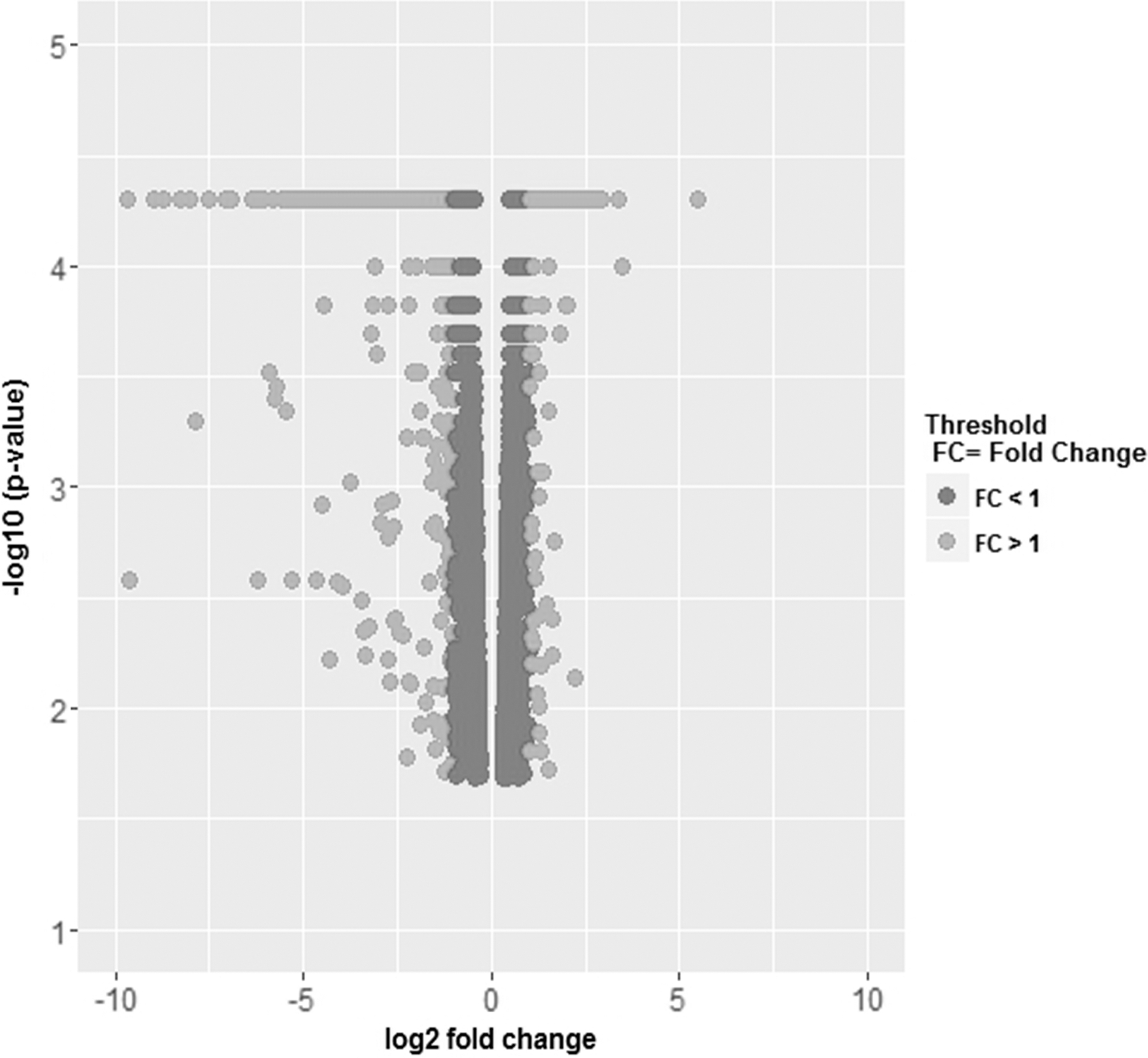
Volcano plot of 4,660 DEGs. DEGs with FC >1 in light gray; DEGs with FC <1 in dark gray. DEGs, differentially expressed genes; FC, fold change.
IFN, interferon; TNF, tumor necrosis factor.
GO and pathway enrichment analysis
The GO and pathway enrichment analysis were performed for 1,084 DEGs. It was found that downregulated DEGs are significantly enriched in the biological process, namely defense response to virus, innate immune responses, and type I interferon (IFN) signaling pathway (Fig. 2A). However, upregulated genes are highly enriched in viral transcription and translation, cell cycle, and chromosome segregation (Fig. 3A). In molecular function, DNA and RNA binding, protein heterodimerization activity, protein binding, and structural constituent of ribosome are significantly enriched in upregulated DEGs, whereas receptor binding, laminin, collagen, and peptide antigen binding are enriched in downregulated DEGs (Figs. 2B and 3B). In terms of cellular component, upregulated DEGs are significantly enriched in nucleosome and cytosol, whereas downregulated DEGs are significantly enriched in plasma membrane and extracellular matrix (Figs. 2C and 3C).
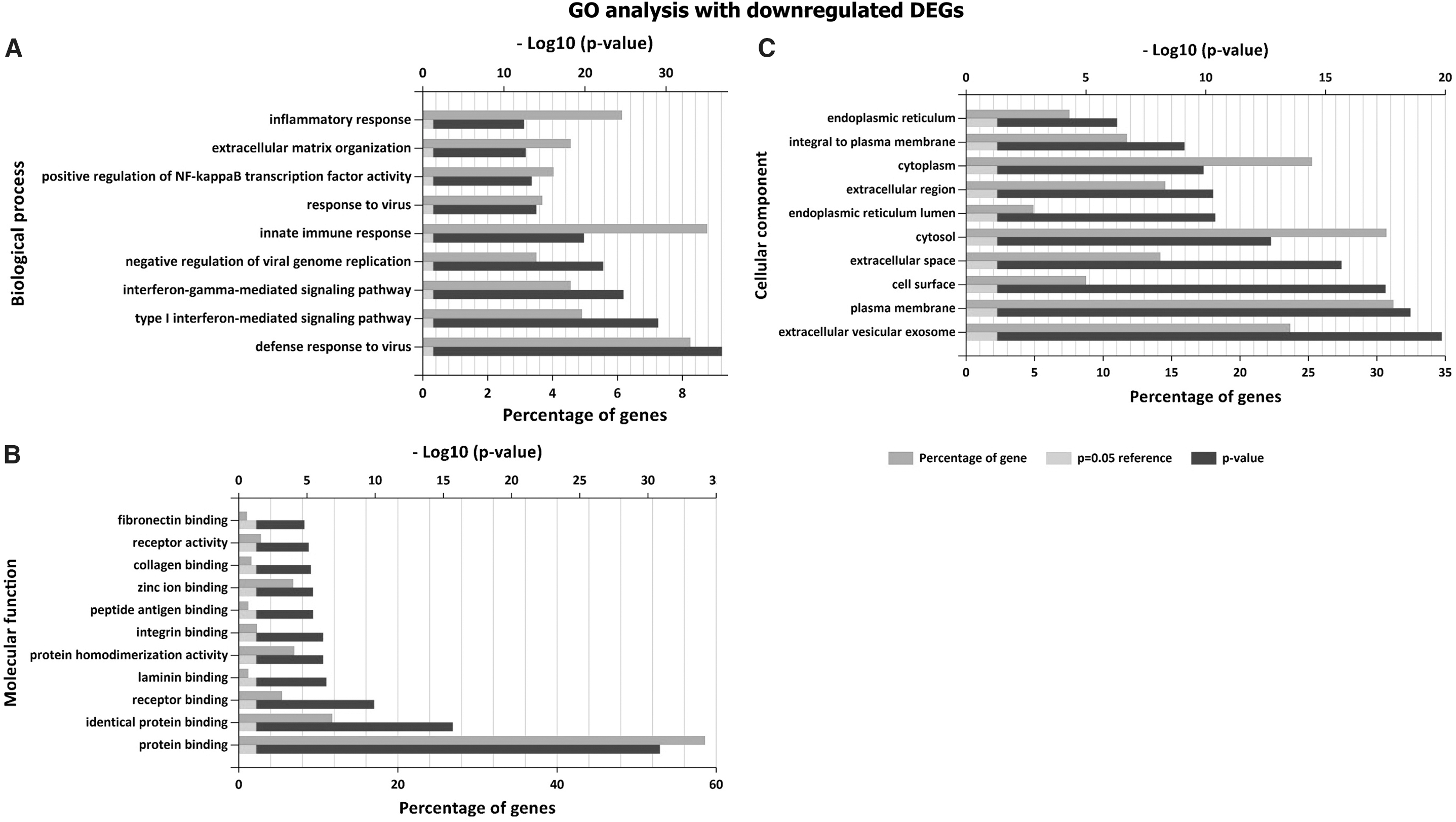
GO analysis result: downregulated DEGs with FC <1.
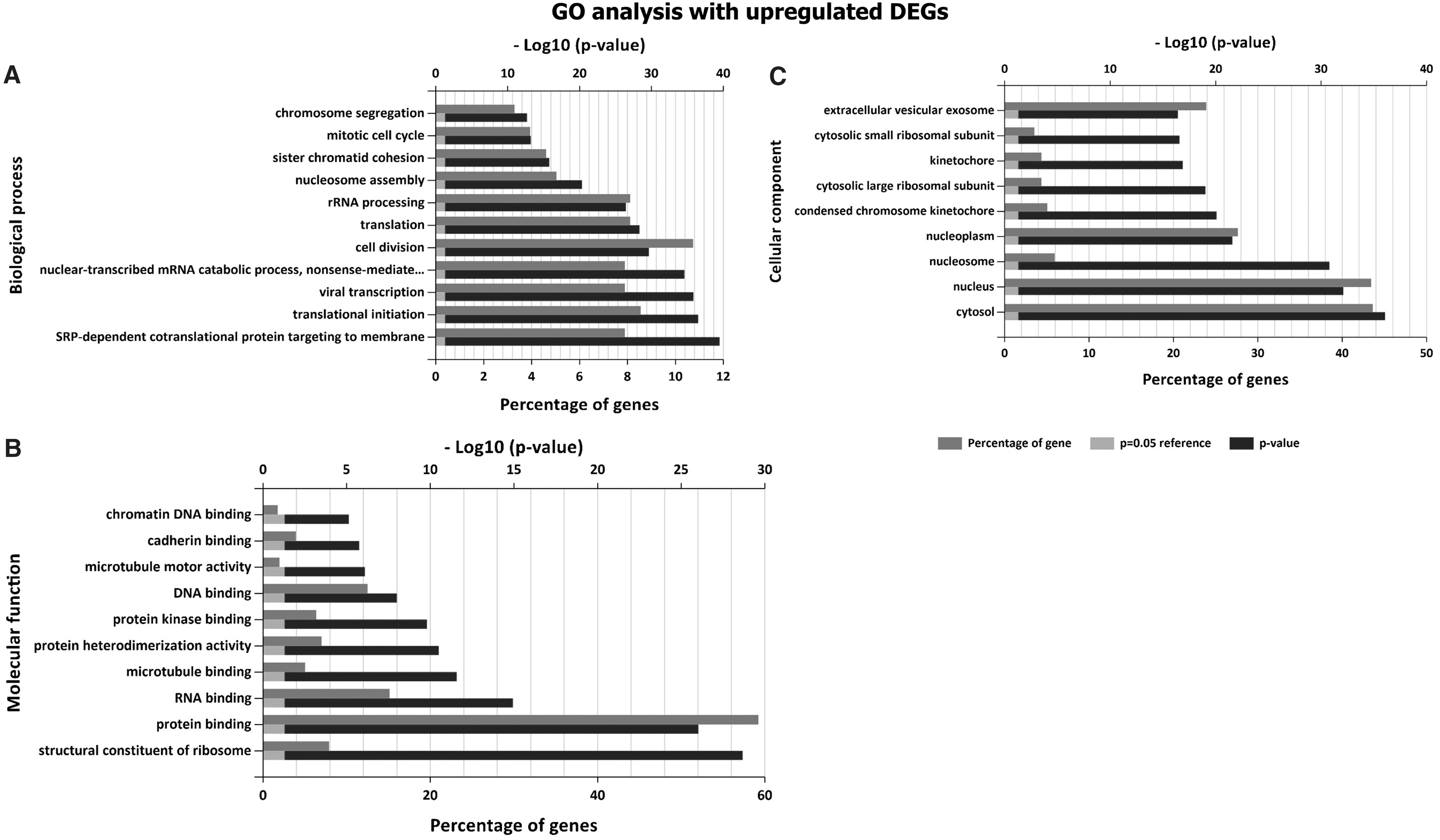
GO analysis result: upregulated DEGs with FC >1.
It is observed from Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, systemic lupus erythematosus (SLE), and alcoholism along with viral carcinogenesis, ribosome, influenza A, and tumor necrosis factor (TNF) signaling pathways are most affected pathways (Table 3).
ECM, extracellular matrix; SLE, systemic lupus erythematosus.
PPI network construction and hub genes
Figure 4 depicts the constructed PPI network and highly connected nodes of degree greater than threshold value (degree ≥50) are considered as potential hub genes. For further study, top seven genes with degree >60 that are cyclin-dependent kinase 1 (CDK1), cyclin B1 (CCNB1, histone cluster 1 H2B family member K (HIST1H2BK), histone cluster 1 H2B family member O (HIST1H2BO), histone cluster 1 H2B family member B (HIST1H2BB), polo-like kinase 1 (PLK1), and cell division cycle 20 (CDC20) are considered to be hub genes. It is noted from network analysis that CDK1 has highest degree, n = 87 followed by CCNB1 with n = 69 (Supplementary Table S3).
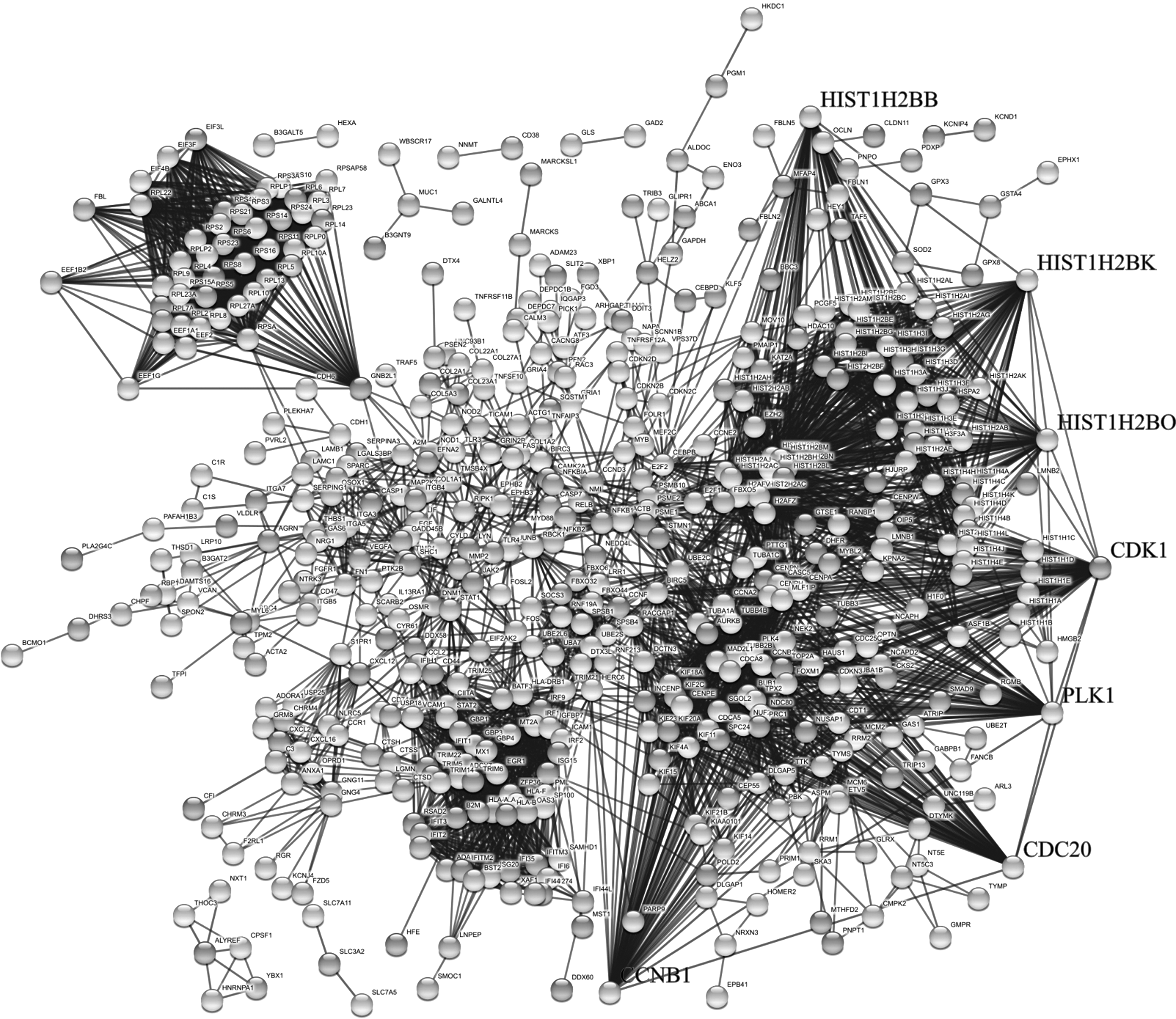
PPI networks. Legends indicate seven hub genes of the DEGs. PPI, protein–protein interaction.
Module identification and enrichment analysis
Modules of PPI network are obtained using Cytoscape plugin MCODE. The pathway enrichment analysis of obtained top five modules/clusters (Fig. 5) shows that they are enriched in SLE pathways, defense responses to virus, structural constituent of ribosome, chromosome, and platelets are all involved in ZIKV progression resulted in microcephaly in newborn.
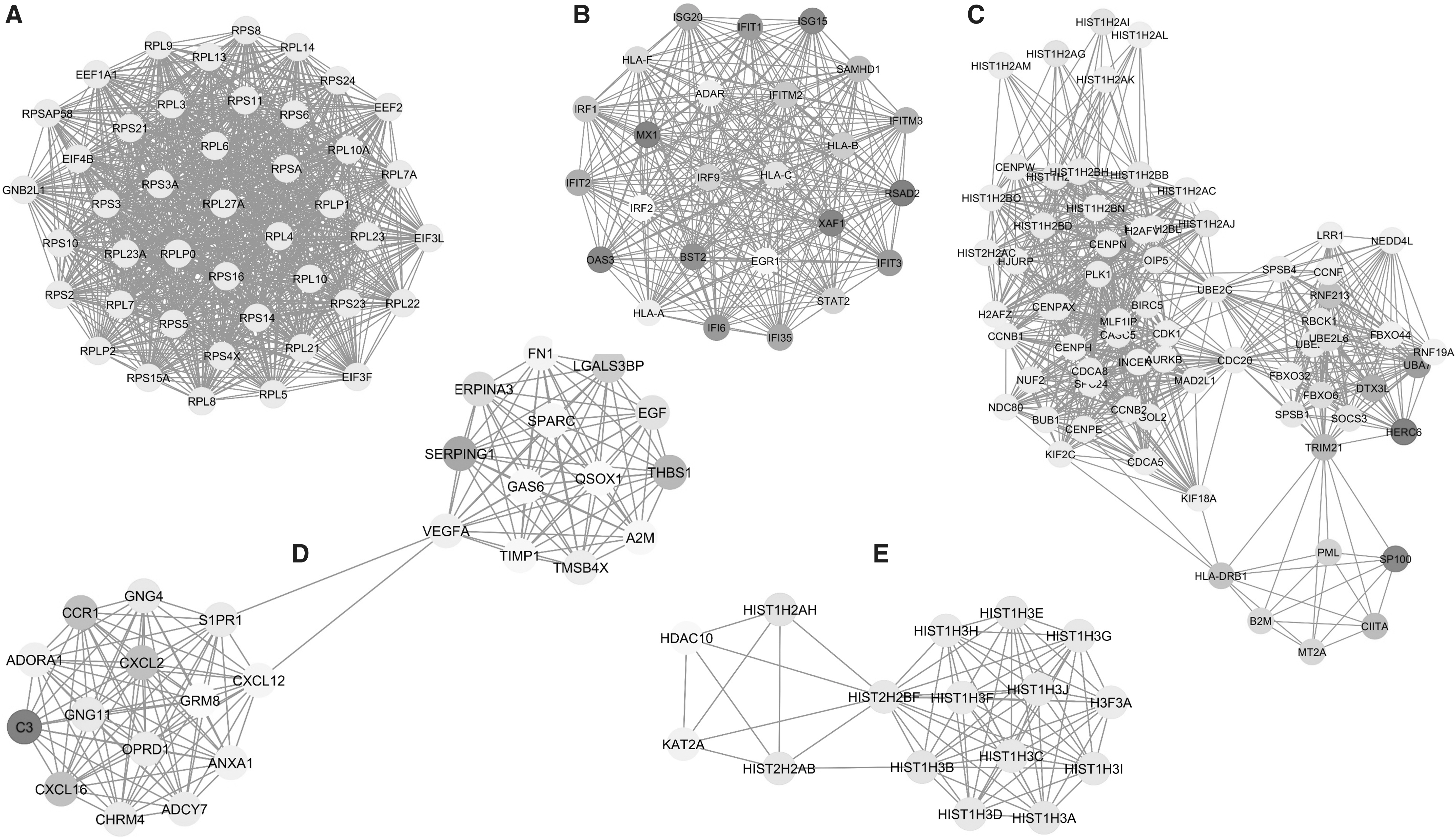
Top five modules of PPI network.
Discussion
As a result of the study, 1,084 DEGs that are 471 upregulated and 613 downregulated genes are identified. GO term enrichment, KEGG pathway analysis, PPI network, and module analysis facilitated to find the highly significant genes and pathways during ZIKV infection.
It is inferred from GO analysis that ZIKV upregulated genes are mostly related to viral translation and progression, whereas downregulated genes are involved in defense response of the host. Similar results are observed in the module analysis also. This can be understood by a recent study that reported that viral NS1, NS4B, and NS2B3 proteins obstruct the induction of IFNs and downstream IFN-stimulated genes (ISGs) by inhibiting TANK-binding kinase 1 level and JAK-STAT signaling pathways (56). However, it is also observed that this protein enhances the viral infection. ZIKV capsid protein induces ribosomal stress and colocalization with the autophagy marker protein LC3 (cytosolic microtubule-associated light chain 3) at autophagocytic vacuoles (20). This restricts the antiviral response and helps in viral replication, also suggesting that ZIKV is efficient to avoid antiviral response. Analysis of molecular function and cellular component shows that the downregulated gene is mostly involved in viral entry such as protein binding, receptor binding, laminin, and collagen binding activity, which are all sublocalized at the outer membrane or extracellular regions. Proteins such as TIM1 and AXL are known to facilitate the entry of ZIKV and also expressed in outer or membrane region (42). Several host factors involved in heparin sulfation (NDST1and EXT1), endocytosis (RAB5C), and transmembrane protein processing and maturation including the endoplasmic reticulum membrane complex and ribosomal proteins (RPL1 and RPL2) also mediating the ZIKV infection during early stage (7,42).
ZIKV infects primary human placental cells, explants-cytotrophoblasts, fibroblasts, etc., that express AXL, TYRO3, and TIM1 (48). Like other RNA viruses, ZIKV hijacks RNA binding proteins for translation. NS5 viral protein has RNA-dependent RNA polymerase activity executed by C terminus and an RNA capping function by the methyltransferase domain of the N-terminus (55). This results in upregulation of molecules associated with ribosome, RNA, and DNA binding activity and microtubule binding in the cytosolic region, which helps in successful invasion of ZIKV into the host and capable of causing inflammatory response.
Through pathway analysis, SLE is the most enriched pathway. Apoptosis, a programmed cell death, leads to ordered destruction before releasing its intracellular contents into the extracellular microenvironment. Dieker et al. (12) reported that inadequate removal of apoptotic cells leads to autoantibodies against chromatin. During SLE, a multigenic autoimmune disorder, apoptosis of peripheral lymphocytes increases, in turn, enhances the leakage possibility of intracellular antigens into the extracellular matrix and triggers an autoimmune response (35). SLE autoantigens bind to toll-like receptors (28), activating NF-κB, IRF5, and IRF7, resulting in the increase of B cells survival and production of cytokines such as TNF-α, IL-10,IL-17, IFN-I, and antibodies, which causes inflammation in various tissues and organs. Histones such as H2BK12 are targeted by autoantibodies for degradation due to apoptosis-induced acetylation of histones during SLE (52).
This study found many histone molecules that are overexpressed, indicating their presence in large amounts in various tissues. Also, complementary proteins such as C2, C3, and C4 are necessary for phagocytosis of autoantigens, neutrophil extracellular traps, etc. (29). However, downregulation of C3 around ninefold is observed. All findings of this study suggested that ZIKV affects the normal phagocytosis activity and stimulates the dendritic cells to secrete IFN-I, auto-antibodies, etc., which causes a permanent damage to tissues. The antagonizing activity of human IFN-I response by STAT2 degradation through ZIKV NS5 protein is seen in mouse models (18). However, NS5 protein is unable to degrade mouse STAT2 (18). SLE and other immune-related diseases such as Aicardi–Goutières syndrome, Singleton–Merten syndrome, and type I interferonopathies are known to be caused by heritable mutations in coding sequences of innate sensing proteins (RIG-I and MDA5). Interestingly, the radiographic characteristics of these syndromes of abnormal immune system overlap considerably with the findings associated with the ZIKV congenital syndrome (26). However, there is no such report on the development of any other autoimmune disorder in immunocompetent mouse model infected by ZIKV. These findings together suggested a crucial role of SLE pathway in ZIKV pathogenesis in host.
The obtained hub genes from PPI network, namely CDK1, CCNB1, PLK1, CDC20, HIST1H2BK, HIST1H2BO, and HIST1H2BB, are found to associate with ZIKV infection. CDK1 exhibits the highest degree followed by CCNB1. The highly connected nodes are known to play an important role in the complex biological systems, which maintain the network integrity and stability. This is shown by a phenomenon known as centrality–lethality rule, which proposes that deletion of a hub protein is more likely to be lethal than deletion of a nonhub protein based on genome-wide studies (21). CDK1 protein, a cyclin family, regulates the eukaryotic cell cycle by modulating the centrosome cycle as well as mitotic onset. Abnormal activation of CDK1 is involved in HIV-1-induced apoptosis and contributes to neuronal apoptosis occurring in neurodegenerative disease (8,49). PLK1 protein belongs to CDC5/polo subfamily with kinase activity and plays an important role in regulation of cell cycle progression, cytokinesis, mitosis, and response to DNA damage. PLK1 is required for proper cortical neurogenesis, which has important implications in the pathogenesis of neurodevelopmental disorder such as microcephaly (41). Both PLK1 and CDK1 also contributed to MCPH1 (the first primary microcephaly gene to be identified) phosphorylation in vivo (32). CCNB1 protein is a cyclin family with serine/threonine kinase activity involved in the regulation of cell cycle. Overexpression of CCNB1 promotes cell proliferation and tumor growth (14). The research conducted by Souza et al. (47) revealed that ZIKV infection induces apoptotic cell death of human neural progenitor cells (hNPCs). This might cause CCBN1 to promote cell growth to retain stability. CDC20 encodes protein that belongs to tryptophan-aspartic acid (single code amino acids). repeat Fizzy family protein with regulatory chromatid separation and nuclear movement before anaphase. Wan et al. (54) reported that apoptosis is suppressed by overexpressed anaphase-promoting complex: CDC20 by targeting BH3-only apoptotic protein Bim. HIST1H2BK, HIST1H2BO, and HIST1H2BB are members of histone 2B family with DNA binding and protein heterodimerization activity and play a role in the stability of nucleosome structure of chromosomal fiber. Phosphorylation of histone 2B is universally observed in quiescent or growing cells during apoptosis, suggestive of apoptosis-specific nucleosomal DNA fragmentation and chromatin condensation (1,24).
Module analyses were performed to explore the biological significance of genes and found that structural constituent of ribosome, defense response to virus, nucleosome, SLE, and extracellular region, and regulation of gene silencing are highly significant. Recent study reveals that capsid protein of ZIKV induces ribosomal stress and proapoptotic activity of Tp53 characterized by structural disruption of the nucleolus (46). ZIKV infection induces several innate immune cell type responses by producing both effector cytokines and cytolytic molecules. The significantly enriched modules in nucleosome and SLE pathway discussed already suggested the critical role in disease progression.
Conclusion
In conclusion, several important hub genes such as CDK1, CCNB1, and H2B family proteins are obtained, which may play a key role in the development of pathophysiology of ZIKV infection. This study has also revealed many critical pathways and submodules for further investigation. Hence, integrative RNA-Seq data analysis and network biology provide some valuable insight into the pathogenesis of ZIKV infection; in turn, it may contribute to the development of novel drug target or diagnosis. However, further validation is required in terms of molecular biology and in-depth studies for better understanding to elucidate the pathogenesis of ZIKV infection to treat more effectively.
Footnotes
Acknowledgments
The authors thank the Centre for Bioinformatics, Pondicherry University, for providing the computer facilities to carry out the work.
Author Disclosure Statement
The authors declare no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.