
Abstract
Secretion of both neutralizing and nonneutralizing virus-specific antibodies by B cells is a key component of immune control of many virus infections and a critical benchmark of successful preventative vaccines. Natural killer (NK) cells also play a vital role in antiviral immune defense via cytolytic elimination of infected cells and production of proinflammatory antiviral cytokines. Accumulating evidence points to multifaceted crosstalk between NK cells and antiviral B cell responses that can determine virus elimination, pathogenesis of infection, and efficacy of vaccine-elicited protection. These outcomes are a result of both positive and negative influences of NK cells on the B cell responses, as well as canonical antiviral killing of infected B cells. On one hand, NK cell-derived cytokines such as interferon-gamma (IFN-γ) may promote B cell activation and enhance immunoglobulin production. In contrast, NK cell immunoregulatory killing of CD4 T cells can limit affinity maturation in germinal centers resulting in weak infection or vaccine induction of antiviral neutralizing antibodies. In this review, we will discuss these and other dueling contributions of NK cells to B cell responses during virus infection or vaccination.
Introduction
Despite the obvious capacity of intracellular pathogens such as viruses to hide from circulating antibodies, humoral immunity frequently contributes to effective antiviral responses (33,124). One clear weakness in viral life cycles exploited by antibodies is the need for virus to exit one cell to infect another. In this regard, development of high-affinity neutralizing antibodies permits capture of virus that has exited the cell to block infection of new cells and thereby constrain viral dissemination. This mechanism is considered the holy grail of many antiviral vaccines, where sufficient titers of circulating neutralizing antibodies can capture virus as it enters the host or effectively contain a localized infection at sites of entry, thereby preventing spread to vital tissues (17). Nonneutralizing antibodies can opsonize virions and shuttle them to professional antigen-presenting cells (3). In addition, some nonneutralizing antibodies can bind virus-derived antigens on viral particles or on the surface of infected cells to attract Fc receptor-expressing effector cells, such as natural killer (NK) cells, which recognize and kill antibody-bound targets via antibody-dependent cellular cytotoxicity (ADCC) (64). These myriad activities highlight the critical role of B cell-derived antibodies in antiviral immune defense.
NK cells are also vital antiviral effectors, particularly against large DNA viruses such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and human papillomavirus (84). Despite shared importance in immune defense against viruses, NK cells lack the somatically rearranged virus-specific antigen receptors that imbue B and T cells with capacity to selectively target specific viruses. Instead, NK cells are activated by virus-induced inflammatory cytokines (e.g., type I interferon), virus-stimulated downregulation of major histocompatibility complex (MHC) proteins ligands for inhibitory NK cell receptors (i.e., missing self), and cell-stress induced ligands for NK cell activating receptors (59). Activated NK cells can kill virus-infected target cells and produce proinflammatory cytokines to constrain infection by stimulating other components of the immune system.
The present review is focused on the functional interactions between innate NK cells and adaptive B cells, which ultimately dictate the success of immune responses against viruses and thereby contribute to viral persistence or immunopathology in different infectious contexts.
NK Cell Activation and Localization During Virus Infection
NK cells are considered innate immune cells owing to their usage of germline-encoded receptors rather than the somatically rearranged antigen receptors characteristic of T and B cells (12). Moreover, NK cells are poised for rapid effector functions that include secretion of interferon-gamma (IFN-γ) and release of cytolytic secretory granules (85). Therefore, while rare virus-reactive T and B cells would need to clonally expand and functionally differentiate to effectively combat a virus infection, preexisting and relatively more abundant populations of NK cells expressing specific combinations of activating and inhibitory receptors favoring stimulation of NK cell activation can rapidly exert antiviral effector functions at early times of infection. As expected, absence of NK cells in humans or mice compromises antiviral immunity against certain viruses (84).
In contrast to the innate characteristics commonly ascribed to NK cells, the discovery of virus-reactive activating receptors that demarcate populations of NK cells that proliferatively expand during virus infection suggests that these cells have adaptive immune functions as well (19,42,60,87). In mice, the receptor Ly49H interacts with a mouse cytomegalovirus (mCMV) gene product, prompting selective proliferation and generation of a long-lived, functionally enhanced population of antigen-experienced cells (82). Likewise, human CMV gene products trigger accumulation of a related population of NK cells expressing NKG2C (32,67). Nevertheless, many different subpopulations of NK cells lacking these receptors are activated and contribute to antiviral responses owing to infection-associated loss of class I MHC ligands for inhibitory receptors and release of NK cell stimulatory antiviral cytokines. Thus, many signals associated with virus infection can stimulate activation of NK cells.
NK cells can be broadly separated into phenotypic grouping based on their variegated expression of defined maturation markers, characteristics of tissue residency, and receptors involved in a process known as arming, licensing, or education [Reviewed in Cooper et al. (25)]. Human NK cells are largely delineated into two groups based on expression of CD56, where CD56dim cells are classically considered to be circulating predominately cytolytic effectors and CD56bright cells are potent producers of cytokines that are enriched in lymphoid tissues (90). Analysis of the diversity of NK cell receptor expression on individual cells in the repertoire of circulating human NK cells reveals up to 30,000 phenotypically diverse subsets of NK cells within individuals (44). These results suggest that the NK cell compartment may achieve a diversity of responsiveness to different infections based on heterogeneous combinations of expressed genes.
After developing in the bone marrow, NK cells widely disperse throughout the body to populate the liver, mucosal tissues, and secondary lymphoid tissues (100). During homeostasis, NK cells represent up to 20% of peripheral blood cells in humans and up to 5% of murine splenic cells. The localization of NK cells within tissues can vary dynamically during infection and inflammation, with importance for the functional contributions of these cells. For example, NK cell migration to lymph nodes and production of IFN-γ contributes to instigation of T cell responses (71). Likewise, NK cell localization in T cell rich white pulp regions of the spleen is critical for limiting potentially detrimental disruption of splenic architecture (10,41,97). Recently, NK cell localization in the B cell follicles of lymphoid tissues was revealed as an important determinant of pathogenesis of Simian Immunodeficiency virus infection in natural host species (e.g., African Green Monkeys) (45). Human NK cells also populate B cell rich regions of the spleen, tonsils, and lymph nodes (7). These and other observations are prompting reconsideration of how receptor-ligand signals integrate with temporal and spatial cues to control the functional contributions of NK cells during infection.
NK Cell Synergy with B Cell Responses
Vaccine development is largely focused on induction of protective high-affinity neutralizing antibodies, yet, evidence exists that nonneutralizing antibodies are critical as well. One major function of nonneutralizing antibodies lies in the capacity of these proteins to bind virions or antigens on virus-infected cells and trigger ADCC (113). In this scenario, the conserved Fc domain of certain antibody isotypes engage Fc-binding receptors on effector cells, including NK cells, providing an activating signal via the Fc receptor that instigates release of cytolytic granules resulting in death of the virus-infected cell (Fig. 1, pink NK cell). Nonneutralizing antibodies specific for influenza virus (31,46,112), hepatitis C virus (66), dengue virus (107), herpes simplex virus (28,77), EBV (86), and HIV (2,11,64) antigens are implicated in the pathogenesis of these diverse infections. Several studies have demonstrated marked changes in the NK cell repertoire during human CMV infection characterized by expansion of antibody-reactive subsets of NK cells (62,99,126). Thus, the antibody producing functions of B cells can converge with cytolytic functions of NK cells to mediate control of virus replication in many different infections. Investigation into the effectiveness of nonneutralizing antibodies, in addition to neutralizing antibodies, in inducing ADCC by NK cells may prove an important readout for vaccine-mediated protection.
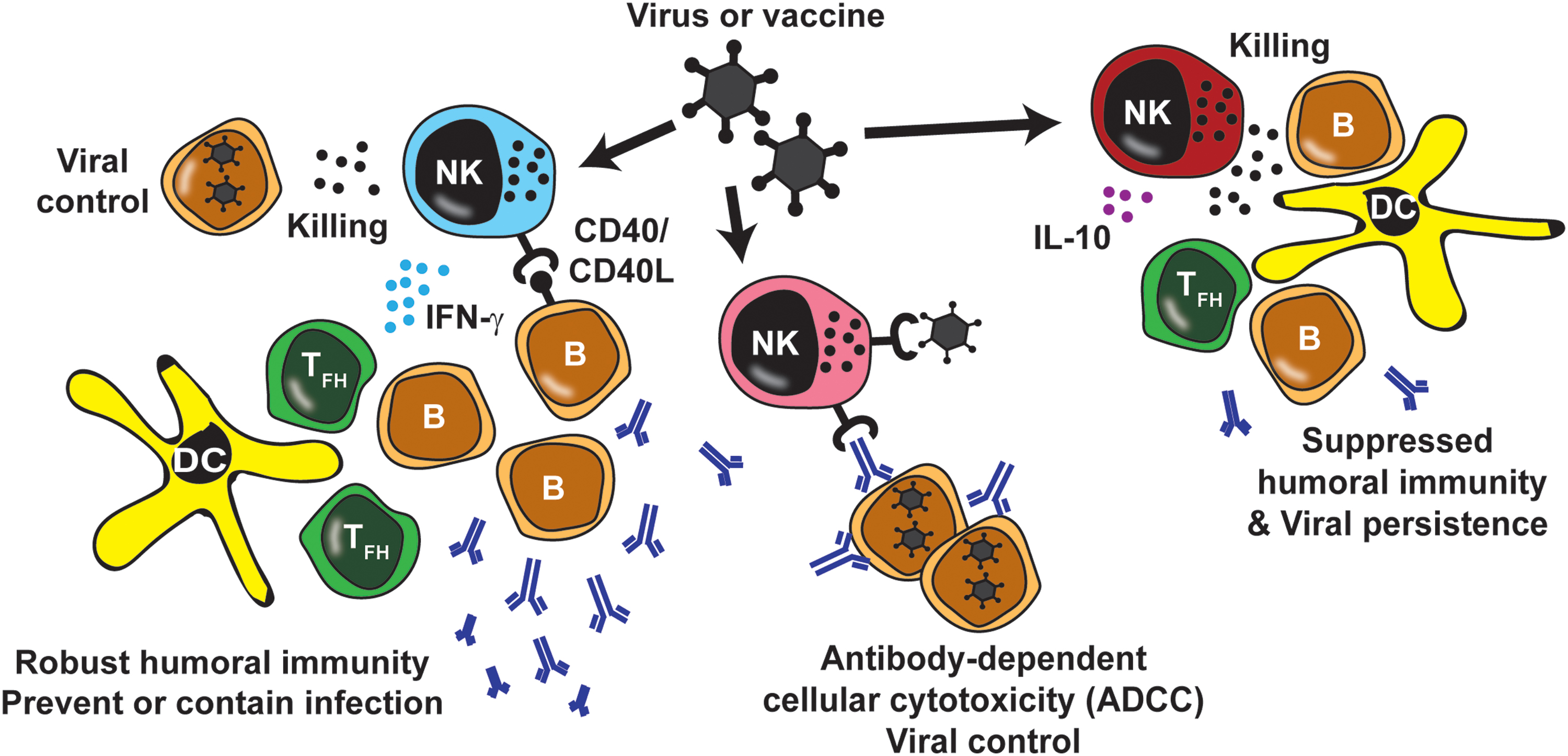
Functional crosstalk between NK cells and B cells during infection or immunization. NK cells (Pink) can synergize with antibodies against viral antigens to promote ADCC-mediated elimination of virus or virus-infected cells. In similarly beneficial capacity, NK cells (blue) can kill infected B cells to promote viral control or exert multiple cytokine- and receptor-mediated functions that promote immunoglobulin production and humoral immunity against viruses. Finally, regulatory functions of NK cells (red) can constrain Tfh and germinal center responses to limit B cell proliferation, differentiation, and affinity maturation. ADCC, antibody-dependent cellular cytotoxicity; NK, natural killer; Tfh, follicular helper T cells. Color images are available online.
Enhancement and Promotion of B Cell Immunity by NK Cells
Key events in generation of antiviral antibodies from the peripheral B cell repertoire include activation and proliferation of virus-specific B cells, isotype class switching, affinity maturation of immunoglobulin (Ig) in germinal centers, and terminal differentiation of plasma cells. Human and mouse NK cells are implicated in influencing many of these events in humoral immune responses. Mouse NK cells can activate B cells and trigger Ig production (47,103,104). In vitro coculture experiments demonstrated direct interactions between mouse NK and B cells, where B cells were demonstrated to promote the induction of IFN-γ by NK cells (76), which in turn induces IgG isotype class switching (36,37,118,123). Early IFN-γ production by NK cells can enhance vaccine-elicited humoral immunity by regulating recruitment of dendritic cells and IL-6 production (34). Likewise, human NK cells can activate B cells (114) and promote Ig production in a T cell independent manner (52) involving IFN-γ and tumor necrosis factor (9). Interactions between signaling lymphocyte activation molecule receptors on B cells and NK cells provide a further IFN-γ-independent mechanism of NK cell stimulation of IgG2a expression (35,38). Contact-dependent activation of B cells by NK cells can also involve CD40-CD40 ligand interactions (13). Studies aimed at uncovering other ligands involved in direct NK cell—B cell interactions could set the stage for novel targeted therapies in promoting B cell antibody generation and maturation activities.
NK cells show potential to enhance B cell activation and antibody production in vivo as well. Immunization of mice with antigen-expressing apoptotic cells prompts NK cell-dependent cytotoxicity of these immunizing cells and enhancement of ensuing humoral immune responses (56). Similarly, mice lacking NK cells display reduced antigen-specific IgG2a production in response to immunization with ovalbumin or keyhole limpet hemocyanin adjuvanted with potent NK cell stimulants such as poly I:C or complete Freund's adjuvant (98,102,119,122). Early IFN-γ responses by NK cells induced by new adjuvants such as AS01 appear to be critical for efficient triggering of humoral immune responses against vaccine (i.e., herpes zoster) antigens (21). Thus, NK cells exhibit multiple beneficial functions that potentiate antiviral humoral immunity (Fig. 1, blue NK cell).
Inhibition of B Cell Responses by NK Cells: Direct Effects on B Cells
In contrast to studies suggesting NK cells enhance B cell immunity, numerous reports detail NK cell suppression of humoral immunity during virus infection and vaccination (Fig. 1, red NK cell). In some instances, human and mouse NK cells could kill activated B cells (15,80,108,109,111). In vitro studies revealed NK cell-dependent suppression of B cell antibody responses triggered by pokeweed mitogen (6) that involved T cell-independent suppression of B cell activation and differentiation (72,78). NK cells also subvert Ig responses against pneumococcal antigens (54) and EBV (57,73), as well as synthesis of IgE (55) and IgA (53). Human NK cells can suppress B cell proliferation (22,50) and transition to plasma cell state (106). Paradoxically, expression of IFN-γ by NK cells is also implicated in the inhibition of B cell proliferation (76). These fascinating and paradoxic mechanisms of NK cell-mediated suppression of B cells clearly needs unraveling to better define the purpose and context of these inhibitory functions.
Inhibition of B Cell Responses by NK Cells: Indirect Modulation of T Cells
NK cells can also suppress the quality and magnitude of B cell responses via effects on helper CD4+ T cells (Th) (1,39,51). This suppression can involve either cytokine secretion or direct cell contact-dependent mechanisms. NK cells can express TGFβ (83) and IL-10 (70,74,89), immunosuppressive cytokines that shape humoral immunity. NK-derived IL-10 can limit the expansion and function of CD4 and CD8 T cells (16,30,63,70,89).
NK cells are also capable of direct lysis of T cells (48,68,81,88,92,101,105). Perforin-dependent in vivo suppression of CD4 T cells during lymphocytic choriomeningitis (LCMV) infection in mice favors viral persistence over fatal immunopathology (115) by modulating T cell exhaustion (24,26,58,115,116,120). Of note, the magnitude of NK cell lysis of activated T cells is controlled, in part, by B cell expressed ligands for NK cell inhibitory receptors (121). The resulting suppression of follicular helper T cells (Tfh) responses by NK cells restricts humoral antiviral immunity during both chronic (23) and acute infection (94). NK cell suppression of Tfh responses also occurs during immunization and constrains germinal center-mediated affinity maturation of vaccine-elicited antibody (95). Disruption of this mechanism during HIV infection facilitates generation of high-affinity broadly neutralizing antibodies (14). Since induction of these antibodies is an ambitious unrealized goal of HIV vaccines, these results suggest that circumventing NK cell suppression of Tfh and B cells may be a viable strategy to enhance vaccine efficacy (96).
Potential Intersections of NK Cells and B Cells
In the context of development of antiviral B cell immunity, we have highlighted multiple points of intersection where NK cells are likely to engage and change size or character of the B cell response. This includes production of cytokines to promote proliferation or class switching of responding B cells, particularly those responding in extrafollicular manner without advantage of receiving these signals from helper T cells in an organized follicle. We also highlighted how NK cells influence SHM and plasma cell differentiation in germinal center responses via modulation of Tfh responses (94,95). As NK cells can enter the bone marrow, it is reasonable to assume that they may interact with developing B cells or long-lived plasma cells, although functional consequences of these unsubstantiated contacts are presently unknown.
In addition to stages of the immune response, there are different subsets of B cells that may differ in their capacity to interact with NK cells. Most of the finding discussed thus far concern follicular B cells, including germinal center B cells. Potential interactions with terminally differentiated plasma cells and memory B cell remain ill-defined, but it is clear that late phenotype B cells are more susceptible to NK cell killing (50). Functional crosstalk between these antigen-experienced cells and NK cells is likely defined, in part, by the isotype of antibody made by the B cell and engagement of Fc receptors on the NK cell. In contrast, NK cells do engage with innate-like B cells, including B-1 cells and marginal zone (MZ) B cells. In vitro coculture of activated NK cells with B-1 or MZ B cells was sufficient to induce Ig release (38,103), suggesting that NK cell support innate B cell compartments needed to promote early T-independent humoral host defense (8,18). However, whether this occurred through direct ligand-receptor interactions or was mediated by cytokine release remains unclear. Intriguingly, a recently identified natural killer-like B (NKB) cell subset seems to drive early IL-12 and IL-18-mediated activation of innate lymphocytes such as NK cells as a means of protecting against microbial infection (117). Collectively these data underscore a potentially large and complex interaction network between these innate populations. Dissection of that interaction network may uncover ways to target and harness these early antibody producers during vaccination (Table 1).
Remaining Questions and Future Directions for Studying NK and B Cell Relationship
NK, natural killer.
Therapeutic Implications of NK–B Cell Interactions
The ability of NK cells to selectively kill target cells and promote beneficial inflammation is a major reason for the increasing popularity of harnessing NK cells in therapy of cancer or virus infection. The ADCC functionality of NK cells is suspected to be critical in the clinical success of rituximab, a monoclonal therapeutic antibody against CD20 used to deplete B cell in leukemia, lymphoma, and autoimmune disease (93). However, similar to T cells, there is a wealth of emerging data suggesting that NK cells can undergo functional exhaustion or be suppressed by persistent antigen and anti-inflammatory properties of the tumor environment (27,40,125). This highlights the potential for checkpoint inhibitors to rescue both T and NK cell function. Two such checkpoints that stand out in the context of the present review are lymphocyte activation gene-3 (LAG-3) and NKG2A. LAG-3 is a CD4 homolog and inhibitory receptor expressed by NK cells that putatively interacts with class II MHC that is abundantly expressed on B cells (5). Therefore, newly developed therapies targeting LAG-3 have the potential to enhance responsiveness of NK cells in contact with B cells. In the case of NKG2A, the recently developed monalizumab antibody relieves human leukocyte antigen E (HLA-E) inhibitory signal via this receptor to potentiate both NK and T cells responses against tumors (4). Notably, NK cell immunoregulatory killing of T cells is also constrained by NKG2A (61,68,81,110), although via engagement of ligands on B cells (121). However, NKG2A+ NK cells appear to play a dominant role in eliminating EBV-infected B cells (43,49,69) and presentation of HIV-derived peptide via HLA-E renders infected T cell susceptible to NK cell attack (29). The results suggest that multiple checkpoint protein may dictate outcome of NK-B cell interactions that will have to be carefully considered with regard to optimal generation of the desired functional response of NK cells in the clinic (Table 1).
In addition to checkpoint blockade and tumor-targeting antibodies, chimeric antigen receptor (CAR)-engineered T cells targeting receptors on B cells have proven highly efficacious clinically, notwithstanding high costs and deleterious side effects of this approach (91). As such, CAR cell therapy approaches are considered further for a variety of antitumor and antiviral applications, it will be important to consider recent evidence that CAR NK cell may represent a less harmful and more cost-effective off-the-shelf strategy than CAR T cells (65,75). Infusion of allogeneic non-CAR NK cells derived from cord blood or cells is already a popular strategy against hematopoietic neoplasms (19,20,79), including those of B cells. Improved understanding of the precise contributions of NK cells to immunity in different contexts will certainly guide the burgeoning field focused on harnessing in NK cells in therapy of a multitude of human disease conditions (Table 1).
Summary
Crosstalk between innate (NK cells) and adaptive (T and B cells) is a critical component of effective immunity against virus infections and a modulator of disease pathogenesis during infection. The dueling capacity of NK cells to enhance or suppress humoral immunity suggests complicated context-dependent activation of different NK cell functions during vaccination and infection (Fig. 1). Simple estimation of the contributions of NK cells in disparate contexts is challenging, and few generalizable patterns have emerged from existing data (Table 1). Certainly EBV infection of B cells and HIV infection of helper T cells are instances where NK cells would engage pathways involving B cell responses with an eye toward elimination of infected target cells. Yet, large DNA viruses (e.g., herpesvirus) often use numerous mechanisms to evade NK cells, highlighting not only the direct evolutionary antiviral role of NK cells in these scenarios but also how viral evasion may modulate the role of NK cells. Additional studies that reveal detailed molecular mechanisms governing these disparate activities will be critical for establishing translational interventions to control NK cell intersections with B cells, and thereby enhance host defense against virus infection.
Footnotes
Acknowledgments
Our sincere apologies to the colleagues whose contributions were inadvertently omitted or misrepresented during crafting of this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors are supported by a National Institutes of Health (NIH) Pathogenesis and Therapeutic Targeting of Immune Disorders T32 training grant AI118697 (A.A.), the Cincinnati Children's Research Foundation, the National Center for Advancing Translational Sciences of the NIH Award UL1 TR001425, and NIH grants DP1 DA038017, R01 AI148080, and R01 AR073228 (S.N.W.).