
Abstract
Canine parvovirus type 2 (CPV2) is a highly contagious cause of serious and often fatal disease in young dogs. Despite the widespread availability of attenuated vaccines, safer, more stable, and more effective CPV2 vaccine candidates are still under exploration. Vaccinia virus (VV) has already been proved to be a safe, stable, and effective vaccine vector. In this study, we generated a VV-based CPV2 vaccine candidate (VV-CPV-VP2) and then evaluated its immunogenicity in mice and dogs. The exogenous vp2 gene of CPV2, which replaced the major virulence gene hemagglutinin (ha) of VV, expressed efficiently and stably in vitro. Subsequently, intramuscular immunization of mice induced robust and lasting systemic immune responses, including neutralizing antibody against both CPV2a and CPV2b, and CPV2-VP2-specific interferon gamma (IFN-γ) secreting T cell. In addition, administration with a high-dose of VV-CPV-VP2 did not cause significant side effects for mice, thus indicating marked safety of this vaccine candidate. Most importantly, a single-dose vaccination of VV-CPV2-VP2 elicited substantial antibody responses and provided comparable protection for dogs with attenuated CPV2 vaccine. Collectively, this study demonstrated that VV-CPV2-VP2 could be used as a promising vaccine candidate preventing CPV2 from infection for dogs.
Introduction
Canine parvovirus type 2 (CPV2) infection causes highly contagious and acute enteritis in dogs with high morbidity and mortality. CPV2 is a single-stranded negative-sense DNA virus, which belongs to the family Parvoviridae. The virion of CPV2 is a nonenveloped icosahedral particle with a diameter of ∼26 nm, which is composed of three capsid proteins (VP1, VP2, and VP3) and two nonstructural proteins (NS1 and NS2) by alternately splicing the viral mRNAs (34). The VP2 (∼64 kDa), an N-terminally truncated form of VP1 (∼84 kDa), is the major component of the CPV2 capsid. The CPV2-VP2 provides receptor binding, controls the host range (4,30), and elicits neutralizing antibodies (23), thus making VP2 the most attractive target for CPV2 vaccine development.
Currently vaccination is still the most important approach for preventing the spread of CPV2 disease. Since the first neutralizing epitope (2L21) on VP2 was precisely determined (20), the corresponding peptides along with different carriers or adjuvants, such as, keyhole limpet hemocyanin (KLH), poly (DL, lactic-co-glycolic acid) (PLGA), or bovine serum albumin (BSA), have rapidly emerged as new formation of CPV2 vaccine candidates (10,15,20). Vaccination with peptides or peptide-based vaccines induced significant systemic immune responses in mice, guinea pigs, and dogs, with high titer of hemagglutination inhibition (HI) and virus inhibition in vitro of antisera from vaccinated animals (3,8,19). However, the peptides, in general, are expensive and have relatively weak immunogenicity; therefore, peptide-associated vaccines are believed not suitable for wide use in field. In addition, although the conventional attenuated CPV2 vaccines had been extremely successful in reducing the outbreaks of CPV2 disease, the clinical use of CPV2-based attenuated vaccines has still raised several limitations. First, preparation of the attenuated CPV2 vaccine is not efficient currently, likely due to the low replication efficiency of CPV2 in vitro [reference (44) and our unpublished data]. Second, attenuated CPV2 vaccine also raises safety concerns due to the possible virus spread during vaccine preparation. Third, the attenuated CPV2 vaccine has potential risk of virulence reversion under the selective pressure during viral replication (2,9). Therefore, novel types of safe and effective vaccine candidates against CPV2 disease are still in need.
Vaccinia virus (VV) Tiantan strain, which belongs to the smallpox virus, was originally isolated in China and has played a crucial role in the eradication of the domestic smallpox epidemic (13). The VV has been recently exploited as a vaccine vector against influenza virus and HIV-1 (17,18). Our recent research also showed that VV was a versatile vaccine vector when expressing exogenous immunogens, such as HIV-1 env (43,45) and Ebola virus gp (39), with significant humoral and cellular immunity after administration to mice. Based on these findings, we, therefore, wondered whether recombinant VV coding CPV2-vp2 could elicit sufficient immune response preventing CPV2 from infection.
In this study, we developed a recombinant VV harboring CPV2-vp2 gene (VV-CPV2-VP2) and immunized mice with this recombinant virus, followed by evaluating those immune responses against CPV2 in mice and dogs. The results indicated that the recombinant VV induced remarkable systemic immune response against CPV2, thus indicating that VV-CPV2-VP2 could be used as a potential vaccine candidate. The current research also provided a universal approach for generating a VV-based recombinant virus vaccine encoding an exogenous pathogen antigen.
Materials and Methods
Cell lines, antibodies, plasmids, and viruses
The cell lines, including Vero cells (Vero 1008) and F81 cells, grew in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). The parental wild-type VV Tiantan strain was obtained from the Chinese Center for Disease Control and Prevention (China CDC). The VV represents VV Tiantan strain unless otherwise specified. The viral stocks were prepared as previously described (39,43,45). The CPV2-VP2 protein and rabbit antibody against CPV2-VP2 protein were purchased from Abclonal (Wuhan, China). Meanwhile the IgG against VV A34R was obtained with the same process as mentioned. CPV2a-ZD and CPV2b-WH were provided by Dr. Zhang, Huazhong Agricultural University. CPV2 attenuated vaccine (Vanguard plus 5/CV-L) was purchased from Pfizer (NY).
Preparation of recombinant VV
Homologous recombination was used to generate recombinant viruses as previously described (39,43). In brief, an entire expression cassette is composed of a promoter pH5, a CPV2-vp2 ORF, and a BGH pA terminator. The other cassette is composed of pSYN, gfp, and SV40 pA. These two cassettes were both on a single shuttle DNA vector pCDNA3.1_LARA, forming a genetic element, with two flanking partial VV ha gene sequence ∼600 bp in length, which was ready for homologous recombination for VV. The CPV2-vp2 was amplified by primers (5′-GA
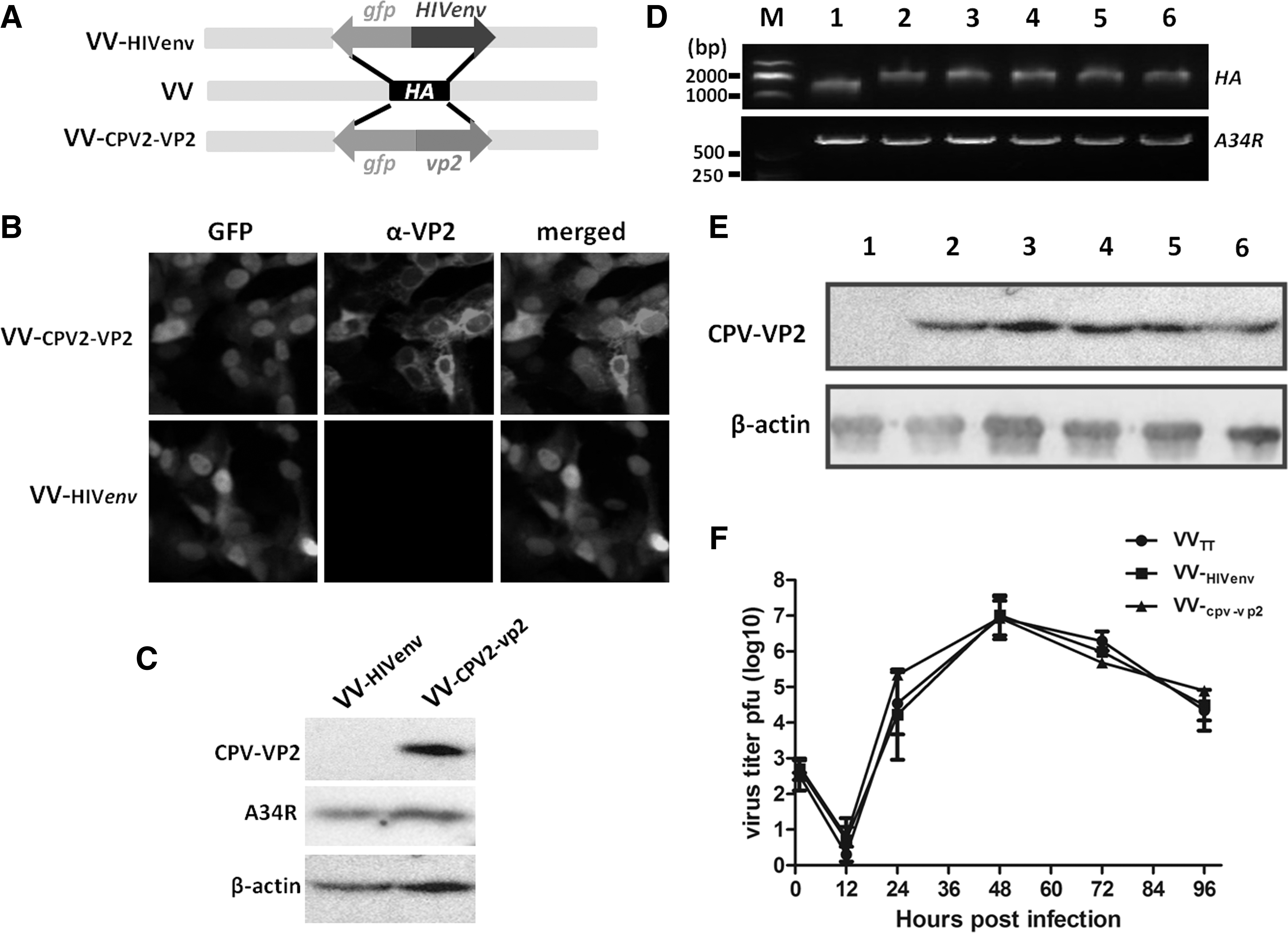
Construction of recombinant VV-CPV2-VP2. Schematic diagram of the construction strategy of VV-CPV2-VP2. Expression cascade harboring gfp and CPV2-vp2 replaced the VV ha by using homologous recombination
Immunofluorescence assay
Vero 1008 cells were seeded in a 100-mm cell plate and infected with VV-HIVenv or VV-CPV2-VP2. At 24 h postinfection, cells were fixed with 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100. After three washes with phosphate-buffered saline (PBS), cells were blocked in PBS containing 5% BSA at 4°C overnight. Thereafter, cells were incubated with rabbit anti-CPV2-VP2 at concentration 1 μg/mL at 37°C for 1 h. After three times of washes with PBST, cells were then incubated with goat antirabbit IgG-Alexa Fluor® 555 (SouthernBiotech, AL) and 4′,6-diamidino-2-phenylindole (DAPI). Finally, cells were washed and subjected to incubate with antifluorescence quenching reagent (Beyotime, China) and observed under a fluorescence microscope (Olympus IX51).
Western blot
Vero cells were infected with VV-CPV2-VP2 or VV-HIVenv. At 48 h postinfection, cells were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (0.45 μm; MerckMillipore, Darmstadt, Germany) followed by blocking with 5% nonfat milk in PBST and probed with rabbit anti-CPV2-VP2 at room temperature (RT) for 2 h. After washing three times with PBST, the membrane was incubated with horseradish peroxidase-conjugated goat antirabbit IgG (1:8,000; SouthernBiotech, AL). The membranes were developed using an enhanced chemiluminescence Western blot detection system (Amersham, Little Chalfont, United Kingdom). In another assay to evaluate the expression of CPV2-vp2 in different passages of VV-CPV2-VP2, virus-infected Vero cells were subjected to SDS-PAGE and Western blot as mentioned.
PCR identification
Viral genomes from Vero cells infected with different passages of recombinant VV were extracted according to the manufacturers' instructions (TaKaRa MiniBEST FFPE DNA Extraction Kit). The ha gene was amplified by primers (5′-GTCGACGATTGTTCATGATGGCA-3′, and 5′-TGAACCAGATGGTCCGCTCA-3′) by using PCR. The genetically recombinant ha should yield a DNA fragment in length of 2,000 bp, whereas the original ha is 1,500 bp in length. The gene A34R with 710 bp in length in the VV genome serves as a control. The resultant DNA fragments were observed by the agarose electrophoresis analysis (SYNGENE, G:BOX).
Mice immunization
Twenty-four 6-week-old female BALB/c mice were randomly divided into four groups (six mice per group). The immunization schedule is indicated in Figure 2A. Mice in groups 1 and 3 were intramuscularly vaccinated with 5 × 106 plaque forming units (PFU) in 100 μL of VV-CPV2-VP2 on days 0 and 21, whereas groups 2 and 4 were vaccinated with same dosage of VV-HIVenv. Mice in groups 1 and 2 were bled from retro-orbital plexus on days 10 and 31 after the first immunization, respectively. Whereas mice in groups 3 and 4 were bled on days 10, 31, and 100 after the first immunization, respectively. The mice in groups 1 and 2 were bled and sacrificed on day 10 after the boost immunization, and then the sera and splenocytes were collected and ready for immune analysis.
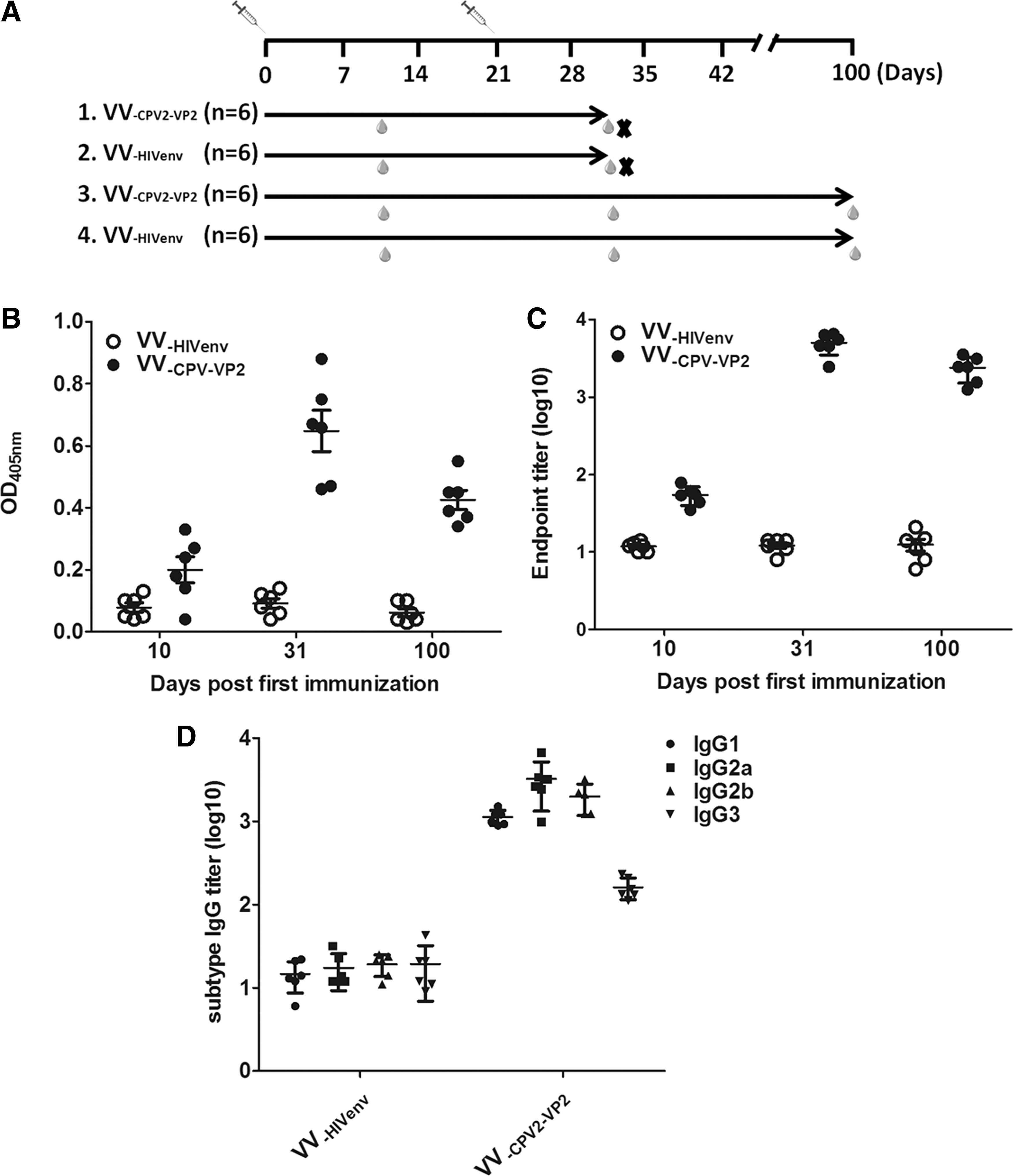
VV-CPV2-VP2 elicited CPV2-VP2-specific binding antibody in mice.
Dog immunization and challenge
Five 2-month old dogs (Chinese rural dogs, CPV2-VP2 antibody free) were recruited in the animal experiment. Two dogs were intramuscularly vaccinated with 1 × 107 PFU in 1 mL of VV-CPV VP2 (group 1), two received 1 mL of commercial CPV2 attenuated vaccine (group 2), and the rest one was mock vaccinated with PBS (placebo) on day 0. And on days 0, 14, 28, 77, and 84 after immunization, sera samples were also achieved and then evaluated by enzyme-linked immunosorbent assay (ELISA). On day 78, all dogs in separate space were challenged with CPV2 on their eyes, noses, and mouths (1 × 107 TCID50/mL, 2.5 mL). The conditions of the dogs, including body weight, temperature, and movement, were monitored daily for 15 days as described by others (20).
Interferon gamma, IL-4, and IL17a ELISpot
The CPV2-VP2-specific T cell immune responses in mice were determined by ELISpot assay. The mice were sacrificed on day 10 after the boost immunization. The individual splenocyte was prepared with Ficoll-Paque (DAKEWE, China). The interferon gamma (IFN-γ)- (DAKEWE, China), IL-4- (U-CyTech, Netherlands), or IL-17a- (R&D systems, MN) secreting T cells were detected by corresponding ELISpot kit according to the manufacturers' instructions. In brief, 5 × 105 splenocytes were added to precoated wells in duplicates and incubated with complete RPMI 1640 containing 10% FBS and 5 μg/mL of protein CPV2-VP2 (Abclonal) for 24 h at 37°C. Spots were visualized by adding 100 μL of the substrates to the wells after their reaction with horseradish peroxidase-labeled avidin and were counted by a MultiSpot Spectrum (AID Diagnostika GmbH, Germany).
Enzyme-linked immunosorbent assay
The binding antibody responses against CPV2-VP2 were determined by ELISA. In general, 96-well ELISA plates were coated with 100 μL of CPV2-VP2 (2 μg/mL) in coating buffer overnight at 4°C. Sera samples from each mouse were serially diluted and then analyzed using AP-labeled goat antimouse IgG (SouthernBiotech), followed by substrate p-nitrophenyl phosphate (PNPP) (Sigma-Aldrich). Finally, the absorbance at 405 nm was measured by a 96-well microplate reader.
Neutralization assay
Neutralizing antibody titers were determined by in vitro inhibition of CPV2 infection on F81 cell line. The mice sera were inactivated at 56°C for 30 min, and then diluted serially (twofold) and mixed with equal volume of 1 × 102 TCID50 CPV2a-ZD or CPV2b-WH in 96-well tissue culture plates. The plates were incubated at 37°C for 1 h and then 100 μL of F81 cell suspension (0.5 × 106 cells/mL) was added. Subsequently the plates were incubated for up to 5 days to assess the cytopathic effect (CPE). The highest dilution of sera showing complete inhibition of CPE was taken as the neutralization titer.
Statistics
Data were analyzed using GraphPad Prism software Version 7.0 (San Diego, CA). All of the data analyses were performed with one-way analysis of variance (ANOVA) or two-way ANOVA. Not significant (NS), p > 0.05; *, p ≤ 0.05; **, p ≤ 0.01.
Results
Preparation of recombinant VV harboring CPV2-vp2
The brief strategy for construction of recombinant VV encoding vp2 of CPV2 was described as indicated in Figure 1A. The target gene CPV2-vp2 replaced the original hemagglutinin gene (ha) of VV by homologous recombination, thus forming a recombinant virus, namely VV-CPV2-VP2. The expression of CPV2-vp2 was assessed by immunofluorescence assay (IFA) and Western blot. Alexa Fluor 555-labeled antirabbit served as the second antibody. As shown in Figure 1B, red signal was observed in VV-CPV2-VP2-infected Vero cells. Whereas the red signal in VV-HIVenv infected Vero cell was not seen. In contrast, the VV-CPV2-VP2-infected cells were subjected to SDS-PAGE and Western blot. As shown in Figure 1C, VV-HIVenv-infected cells yielded no band; however, VV-CPV2-VP2-infected cells exhibited a single band (∼64 kDa) that was consistent with the expectation of VP2 molecule mass. The VV-specific A34R protein serving as a VV vector control could be constitutively detected.
The stability of gene CPV2-vp2 in viral genomes and its expression in Vero cells were then evaluated. The viral genomes from different passages of virus were extracted and used for ha and A34R gene amplification. As shown in Figure 1D, the CPV2-vp2 gene in 1st, 5th, 10th, 20th, and 30th progeny recombinant virus was successfully identified, thus indicating that exogenous CPV2-vp2 was steadily inserted into the genome of VV-CPV2-VP2. Vero cells that were infected with different passages of VV-CPV2-VP2 were subjected to SDS-PAGE and Western blot. As shown in Figure 1E, 64 kDa of bands was observed in 1st, 5th, 10th, 20th, and 30th progeny recombinant virus, thus indicating the efficient expression of CPV2-vp2 gene in different passages of VV-CPV2-VP2. The gene A34R acted as internal reference. Plaque assay was used to evaluate the VV-CPV2-VP2 replication dynamic. As shown in Figure 1F, the replication curve of VV-CPV2-VP2 was basically matched with that of VVTT, thus indicating that the replacement of gene CPV2-vp2 to ha did not reduce the replication capacity of VV-CPV2-VP2. Collectively, the exogenous gene CPV2-vp2 stably inserted into the VV genome and expressed efficiently.
Intramuscular immunization of VV-CPV2-VP2 induced significant humoral immune response against CPV2-VP2 in mice
The CPV2-VP2-specific antibody was considered the most important element preventing CPV2 from infection (5,6). Therefore, we next evaluated the CPV2-VP2-specific antibody level in sera from mice after intramuscular administration. The four groups of mice, two VV-CPV2-VP2 groups and two VV-HIVenv groups, were vaccinated on days 0 and 21. Mice were bled on days 10 and 31 (and day 100 in groups 3 and 4) after first administration (Fig. 2A). Mice antisera taken on days 10, 31, and 100 after the first immunization were diluted 1:100 and then tested. The OD values were calculated and recorded (Fig. 2B). All the antisera from the VV-CPV2-VP2 group reacted strongly with the coating antigen (CPV2-VP2) on day 10 after boost immunization, with a geometric mean titer (GMT) of 5,500 (Fig. 2C). On the contrary, the antisera from the VV-HIVenv group mice did not show any detectable binding activity toward CPV2-VP2. Moreover, we examined the level of binding antibody against CPV2-VP2 on day 100 after the first immunization. As shown in Figure 2C, the binding activity of antibody was GMT of 3,800, indicating a sustainable and high level of CPV2-VP2-specific antibody in mice after twice VV-CPV2-VP2 administration. These results demonstrated that strong and lasting antibody responses against CPV2-VP2 could be induced after immunization with VV-CPV2-VP2. Notably, the ratio of IgG1/IgG2a was close to 0.5 (Fig. 2D), indicating that VV-CPV2-VP2 immunization induced Th1-biased immune responses.
We next investigated the neutralizing capacity of sera derived from VV-CPV2-VP2-immunized mice or VV-HIVenv group. Two homologous CPV2 isolates, CPV2a-ZD and CPV2b-WH, were used to test the cross-neutralizing capacity of antibody. As shown in Figure 3A and B, the control antisera did not show any neutralization effect even at the minimum dilution tested (1:16) and were, therefore, defined as a titer of 8 for GMT calculation. In contrast, antisera from VV-CPV2-VP2-immunized mice effectively neutralized the parent strain CPV2a-ZD, with a GMT of 320 (Fig. 3A). Of note, antisera from VV-CPV2-VP2-immunized mice provided neutralization for homologous CPV2b-WH, with a GMT of 160 (Fig. 3B). Together, VV-CPV-VP2 elicited high level of and sustainable binding and cross-protection antibody against CPV2-VP2 after intramuscular administration for mice.
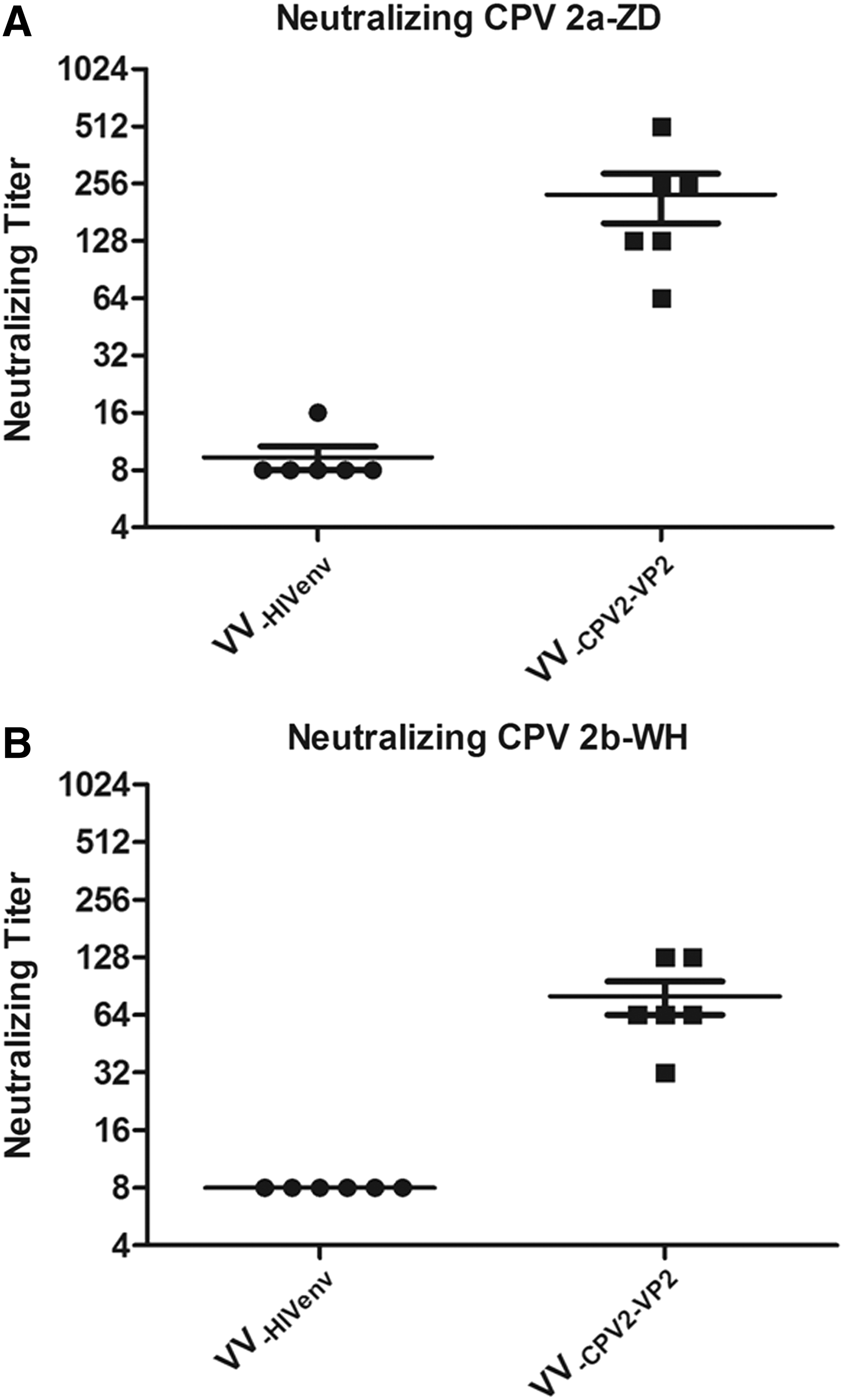
Neutralizing antibody responses after immunization with CPV2-VP2 in mice. Neutralizing titers against CPV2 strains CPV2a-ZD
Cellular immune responses were elicited by VV-CPV2-VP2 in mice
Given the critical roles of cytokines secreted by activated immunocytes in defining the subsequent immune response (42), we evaluated CPV2-VP2-specific Th1-, Th2-, or Th17-like cellular immune responses by measuring the production of Th1-associated (IFN-γ), Th2-associated (IL-4), or Th17-associated (IL17a) cytokines. Splenocytes collected from immunized mice on day 10 postboost were analyzed. As shown in Figure 4B and C, splenocytes from mice in the VV-HIVenv group did not show detectable immunocytes secreting IFN-γ, IL-4, and IL17a, whereas the number of CPV2-VP2-specific IFN-γ-secreting T cells reached an average of 400 spot-forming cells per million splenocytes (Fig. 4A). The number of CPV2-VP2-specific IL4- or IL17a-secreting T cells was ∼20 and 50 (Fig. 4B, C), respectively. Therefore, immunization with VV-CPV2-VP2 seemed to induce a Th1-dominant cellular response, with the number of immunocytes secreting IFN-γ being significantly larger than the other two immunocytes, which was in agreement with the data shown in Figure 2B.
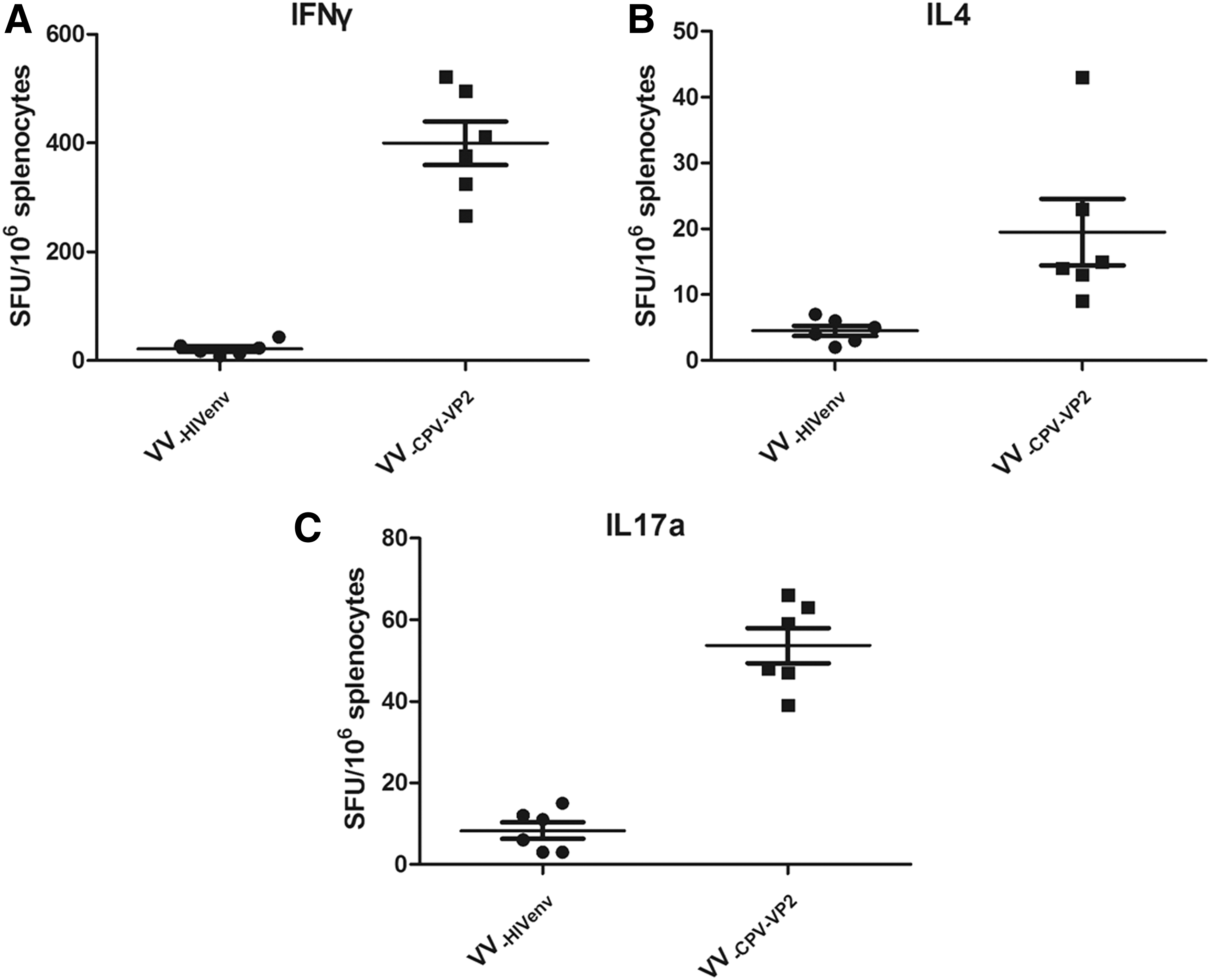
Cellular immune responses in mice. CPV2-VP2-speciFIG IFN-γ-
No significant adverse effects were observed in VV-CPV2-VP2-administrated mice
To investigate whether the VV-CPV2-VP2 causes cytotoxicity in vitro (24), we evaluated the cell death levels of VV-CPV2-VP2-infected F81cells by using MTT assay. As shown in Figure 5A, VV-CPV2-VP2 infection of F81 cells resulted in weaker cytotoxicity than VVTT (VV Tiantan strain) after 36 h of infection (p < 0.05), suggesting a further decreased cytotoxicity, likely due to the replacement of ha by CPV2-vp2 VV-CPV2-VP2.
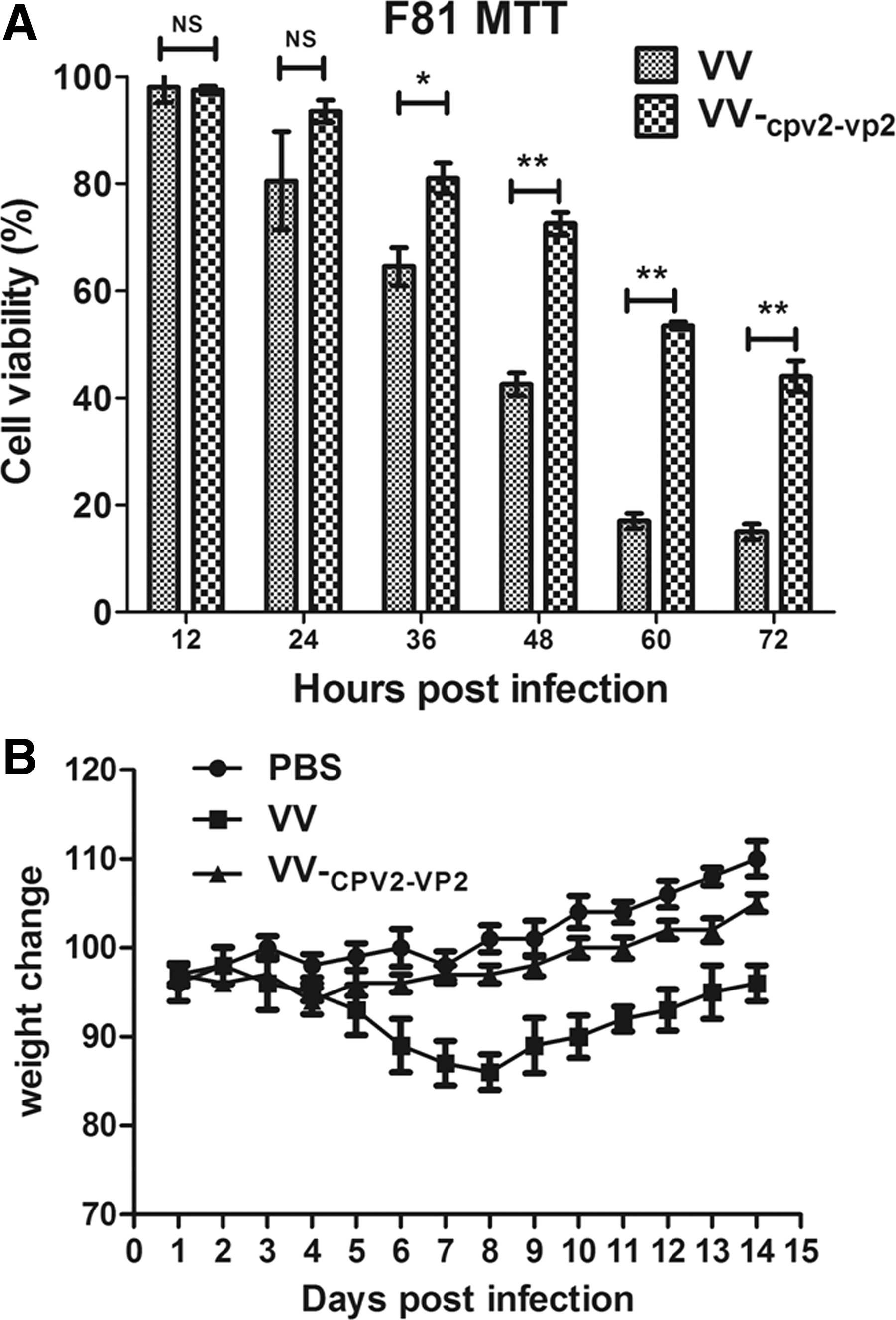
Side effects.
We next evaluated whether intramuscular immunization would lead to side effects to mice with high dosage. Mice were intramuscularly inoculated with PBS or with 2 × 107 PFU of VVTT or VV-CPV2-VP2 in 100 μL, followed by continuous monitoring for body weight of immunized mice. As shown in Figure 5B, body weights of mice in the group with VV-CPV2-VP2 slightly decreased on around day 4 postinfection and then recovered rapidly. However, the body weights in the VVTT-inoculated group decreased significantly compared with the other groups. Thus administration with extraordinarily high dose of VV-CPV2-VP2 did not result in significant body weight loss compared with that with PBS, thus reflecting the marked safety of VV-CPV2-VP2 in mice.
A single-dose vaccination of VV-CPV2-VP2 conferred humoral immune response and provided protection in dogs
We were wondering whether recombinant VV-CPV2-VP2 can induce immune response in dogs. Three groups, VV-CPV2-VP2, commercial attenuated CPV2 vaccine, and PBS, were vaccinated and sampled as mentioned in Materials and Methods section. As shown in Figure 6A, sera achieved before immunization had no binding antibody, whereas after the first vaccination, the level of antibody against CPV VP2 increased rapidly up to ∼500. Whereas the sera from PBS-immunized group always did not show any binding antibody toward CPV VP2. The antiviral activity of sera was also evaluated. As shown in Figure 6B, the neutralizing titer of sera from mice immunized with VV-CPV2-VP2 was ∼180, which was comparable with that of attenuated vaccine. Even on day 77 postimmunization, CPV VP2-specific antibody and its antiviral capacity were also detectable, thus indicating that a single immunization may induce durable antibody immune response in dogs.
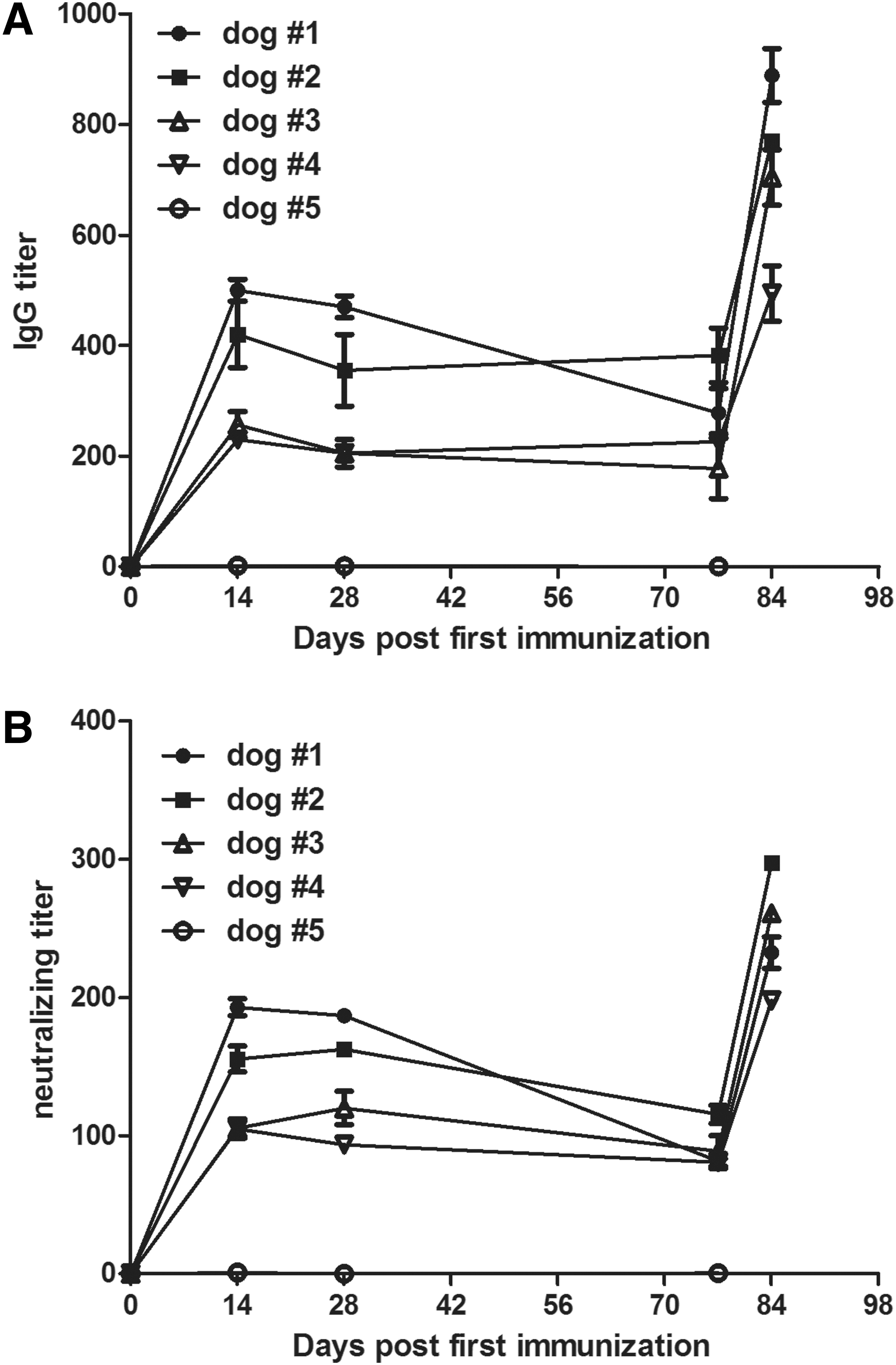
VV-CPV2-VP2 induced protective immune response in dogs. All dogs received vaccines on day 0 and then were challenged with CPV2 on day 78. On days 0, 14, 28, 77, and 84 after immunization, all dogs were subjected to bleed from ear vein.
To evaluate whether vaccination of recombinant VV-CPV2-VP2 provides protection, dogs were challenged with CPV on day 78 postimmunization, and then the clinical symptoms were monitored daily for 15 days. As given in Table 1, vaccinations with VV-CPV2-VP2 or commercial attenuated vaccine provided complete protection from CPV infection. However, the placebo-treated dog (group 3) developed typical CPV2 disease symptoms including vomiting, hemorrhagic diarrhea, depression, and loss of appetite, and was subjected to euthanasia after 7 days' observation. Of note, both binding IgG titer and neutralizing titer for CPV2 increased rapidly to a higher level after CPV2 challenge, likely due to the stimulation of live virus CPV2.
Vaccination and Challenge of Dogs Against Canine Parvovirus
Group 1 and group 2 contained one 2-month-old female and one 2-month-old male dog, respectively, whereas group 3 contained only one 2-month-old male dog.
Vanguard plus 5/CV-L (Pfizer).
Mock injected PBS.
CPV2, canine parvovirus type 2; PBS, phosphate-buffered saline; VV, Vaccinia virus.
Discussion
CPV2 is a highly contagious pathogen resulting in severe clinical syndromes including hemorrhagic enteritis, fever, and vomiting in young dogs. Despite the widespread availability of attenuated vaccines, CPV2 disease still outbreaks locally, thus calling for new formation, safer, and more effective vaccines. In this study, we constructed a stable and safe recombinant VV encoding CPV2-vp2 (VV-CPV2-VP2) that is the major protective capsid protein, and evaluated its immunogenicity in mice. Consequently, recombinant VV-CPV2-VP2 vaccine candidate induced robust systemic immune response against CPV2 in mice and dogs.
VV-CPV2-VP2 is a safe and stable vaccine candidate against CPV2. In the past 5 years, several biosafety issues regarding virulence reversion occurred in attenuated virus vaccines, such as severe acute respiratory syndrome coronavirus (SARS-CoV) (18), influenza virus (46), porcine reproductive and respiratory syndrome virus (PRRSV) (21), and African horse sickness virus (35). The current available CPV2 vaccines are all attenuated formations, thus raising safety concerns of virulence reversion. The VV has been recently developed as a multifunctional vector for vaccines against influenza virus and HIV-1 (14,22,38,47), demonstrating that VV is a superior vaccine candidate with high safety and stability. In addition, VV has been verified to be not permissive for dog infection (29); therefore, it is an another safety evidence for VV-based vaccine development. Furthermore, our findings in this study showed that no mutation and deletion of the exogenous gene CPV2-vp2 occurred after even 30 passages' replication in vitro (Fig. 1D, E). After administration of recombinant myxoma virus that also belongs to the Orthopoxvirus genus for dogs, no detrimental effects of MYXVΔserp2 treatment were observed in any canines. No clinically significant changes were measured (25). Collectively, VV is a safe and stable virus vector when used for developing a VV-CPV2-VP2 vaccine candidate.
Proliferation of VV- CPV2-VP2 is more convenient than CPV2 in vitro. Yu et al. (44) and our unpublished data showed that CPV2 replicates in several cells at low efficiency. In general, after a round of CPV2 proliferation in vitro, the virus titer was only at the level of about 1 × 102 TCID50/mL. However, VV can rapidly replicate in Vero cells, with high yield of virus (often 1 × 107 PFU/mL) after one round of replication. Therefore, it is more cost-efficient for VV-CPV2-VP2 proliferation than for CPV2.
Administration of VV-CPV2-VP2 induced strong CPV2-VP2-specific systemic immune responses in mice, likely due to several aspects given hereunder. First, exogenous gene CPV2-vp2 expresses highly efficiently in multiple cell types. With no specific cell tropism (12), VV can easily infect many cell types. Moreover, the genome replication and viron assembly of VV occur in cytoplasm that is a hallmark property compared with other viruses (12), which enhanced the efficiency of viral replication and increased the number of progeny viruses. In addition, two artificially optimized VV-specific promoters, namely pH5 and pSYN, were inserted in front of the multiple cloning sites of shuttle vector for recombinant virus construction. The promoter (pSYN) strongly directs the transcription and expression of exogenous gene vp2 (37), which significantly elevates transcriptional efficiency of exogenous gene. Second, VV serves as an “adjuvant,” thus elevating the immune response specific for CPV2-VP2. Several components of VV induce strong immune response in animals. Both humoral and CD4+ and CD8+ T cell responses are induced by VV infection in mice (41) and humans (11,27,32). Therefore, the exogenous protein induces both MHC I-mediated T cellular immune response and MHC II-mediated humoral immune response upon the recombinant virus infection. Subsequently, VV infection recruits multiple cytokines and immunocytes toward the compartment of viral infection, then the resulting immunocytes (e.g., macrophage, dendritic cell, and T cell) and many cytokines collaborate together to establish an adaptive immune response against the exogenous antigens. After this process, CPV2-VP2 achieves an enhanced immune response, which benefits from the VV vector. In summary, VV-mediated exogenous gene CPV2-vp2 highly efficient expression may lead to robust and lasting systemic immune response in host.
CPV2 has evolved to CPV2a, 2b, and 2c with several amino acid substitutions in VP2 (26). CPV2a and 2b are currently the predominant strains in China. Several studies claimed that single CPV-derived immunogen did not induce cross-protection between different subtypes of CPVs (17). Whereas Wilson et al. reported that vaccination of dogs with CPV2b induces neutralizing antibody responses to both CPV2a and CPV-2c (36). Our data also suggested that VV-CPV2-VP2 could induce cross-protection antibody against CPV2a and CPV2b infection. The cross-protection likely resulted from specific vectors, unique immunization routes, or certain adjuvants, which warrants further investigation.
The pre-existing immunity is the most important concern for any vaccine development, especially for VV-based vaccine candidate. Previous studies revealed that the pre-existing immunity reduced antibody response of vaccinated animals to some degree. Gudmundsdotter et al. revealed that pre-existing immunity to VV did not reduce the proportion of individuals who responded to HIV-1, but did lower the magnitude of responses (16). Another research also confirmed this conclusion. Altenburg et al. also showed that orthopoxvirus-specific pre-existing immunity reduced the induction of antigen-specific antibodies under specific conditions and completely prevented induction of antigen-specific T cell responses by rMVA-based vaccination. However, importantly, protective efficacy of an MVA-based influenza vaccine against a homologous challenge was not impaired in the presence of orthopoxvirus-specific pre-existing immunity (1). Therefore, the pre-existing immunity likely impaired the consequence of immunizations for target antigen after repeated vaccination with same VV-based immunogen. Alternative prime boost immunization strategy (recombinant virus plus protein) was adopted to avoid the negative effect of pre-existing immunity (33). In summary, the immunological outcomes might be influenced upon repeated immunization with single recombinant VV expressing CPV-VP2; therefore, a new immunization strategy should be considered to promote the antibody responses by optimizing immunization regimens.
Despite that multiple CPV vp2-based vaccine candidates including VP2-based VLP (28,40), VP2 subunit (31), and DNA replicon-based VP2 candidates (6, 7), even recombinant virus-based CPV vaccine (24), have been developed already, whether such CPV vaccine candidates provide sterile protection for challenged dogs remain still elusive. Our findings provided solid evidence that recombinant virus expressing CPV2 vp2 conferred not only robust immune response but also complete immune protection for dogs upon virulent CPV2 challenge.
Conclusions
Our findings in mice and dogs indicated that VV- CPV2-VP2 has an attractive potent to promote systematic responses against CPV2 infection. The current results further strengthen the evidence that the VV that encodes exogenous antigen could be used as a promising vaccine candidate.
Footnotes
Acknowledgment
We sincerely thank Dr. Shengbo Cao at the Huazhong Agricultural University for editorial assistance.
Compliance with Ethics Guidelines
The animal study was approved by the ethics committee of the Wuhan University of Bioengineering, China (permit no. WUB20181031). All animal studies and methods were permitted by ARRIVE guidelines.
Authors' Contributions
Y.L. and Y.L. conceived and designed the experiments. W.Z. and X.W. carried out the experiments. Y.L. and W.Z. wrote the article. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from National Natural Science Foundation of China (Grant No. 31700873), Hubei Provincial Natural Science Foundation of China, Grant Nos. 2018CFB449 and 2017CFB240, and High-level Scientific Research Foundation for the introduction of talent of Wuhan University of Bioengineering (Grant No. 2017KQ01).