
Abstract
Abstract
Culturing cells in vitro can produce a uniform population for the study of cellular differentiation, which is especially useful for the quantification of gene expression or the observation of subcellular structures. In zebrafish, a handful of immortalized cell lines have been used for these purposes, despite being heavily selected by passaging. Methods for primary cell culture of zebrafish embryonic blastomeres have been previously reported, but require combining a large number of genetically heterogeneous embryos, meaning that subsequent cell cultures are not clonal. Without genetically uniform cultures, this model system cannot exploit the wealth of available embryonic lethal mutants in zebrafish. We therefore describe methods for the generation of zebrafish embryonic blastomere cell cultures from single genetically characterized embryos. We examined myogenic differentiation and gene expression in single-embryo cultures from early wild-type embryos, as well as embryos containing an embryonic lethal mutation of unc45b, a myosin chaperone known to be required for sarcomere organization during myogenesis. We also demonstrated the practical usefulness of this technique by experimentally manipulating expression of specific genes in individual embryos before cell culture using standard tools of zebrafish biology such as morpholino-oligonucleotide gene knockdown and transgene-mediated gene expression.
Introduction
In contrast, primary culture uses cells or tissues taken directly from explants, without the immortalization that occurs after multiple passages. Primary cell cultures from embryonic blastomeres have been used to examine mechanisms of differentiation in species as diverse as sea urchins 9 and humans. 10 In the zebrafish, primary cell culture methods using blastula-stage embryos have been previously described,11–15 but are not often used in developmental research, despite the usefulness of examining differentiating cells in cultured monolayers. This is largely because zebrafish embryos are both small and transparent enough for the resolution of individual cells,16–18 and so the differential benefits of cell culture systems are reduced when compared to other organisms. However, cells in monolayer are preferable for imaging subcellular structures and processes, which is not feasible in live zebrafish embryos beyond the first few cell divisions, when blastomeres are syncytial or nondeterminate. Techniques for live cell imaging at the subcellular level in whole zebrafish embryos exist,19,20 but are technologically demanding and therefore beyond the reach of most laboratories.
One of the advantages of zebrafish as a developmental model is the availability of many hundreds of strains carrying mutations in genes of molecular or developmental interest.16,21 Given this wealth, it would be useful to develop primary cell culture methods for genetically characterized zebrafish to examine, for example, myogenic differentiation in specific mutant or transgenic cells. This would require the use of primary embryonic cell cultures derived from genetically uniform cell populations, containing the mutations or transgenes of interest. However, existing methods for primary embryonic cell culture in zebrafish require the dissociation of large numbers of early blastula-stage embryos, well before phenotypic identification of mutants or transgene carriers would be possible.11,14,15 This effectively prohibits the use of heterozygous carrier strains for embryonic lethal mutations, which comprise the majority of useful mutants, since genetically uniform sets of embryos cannot be obtained without phenotypic screening. We have therefore explored the feasibility of generating differentiated cell cultures from single zebrafish embryos, which provide genetically identical populations of cells that can be individually genotyped from unattached cells in the culture medium.
As proof of principle, we have examined spontaneous myogenic differentiation of our single-embryo cultures by both phenotypic and genotypic analysis. Spontaneous myogenesis of dissociated blastula cultures has been previously reported by expression of muscle-specific markers in culture,14,22 though not at the subcellular level. We examined single-embryo cultures for similar marker expression, particularly the subcellular patterns of actin and myosin expression characteristic of differentiated myocytes. 23 To demonstrate the usefulness of the single-embryo zebrafish embryonic blastomere (seZEB) culture system, we also examined cultures derived from embryos possessing an embryonic lethal mutation in the unc45b gene.24,25 Recent studies have implicated unc45b as an essential molecular chaperone, responsible for the proper folding of nascent muscle myosin heavy chain (MHC).26,27 This gene is an excellent marker of proper sarcomere formation, as muscles that develop in unc45b−/− mutants lack the characteristic banding pattern that arises from sarcomere organization in striated muscle cells.24,25 Finally, we performed microinjection of antisense oligonucleotides or DNA gene expression constructs into zebrafish embryos before dissociation for cell culture, demonstrating that the seZEB culture system can be used to examine the effects of specific manipulations of gene expression on subsequent myogenic differentiation in culture.
Materials and Methods
Zebrafish maintenance
The inbred strain AB, developed by George Streisinger, 28 was obtained from the Zebrafish International Resource Center. For convenience, this strain is considered wild type (WT) for all subsequent experiments. The unc45b mutant strain steif, developed by Christine Etard, 25 was a generous gift from the laboratory of Uwe Strähle (Karlsruhe, Germany). Stable transgenic fli1-GFP (full name Tg(fli1a:EGFP)y1, zfin ID = ZDB-GENO-011017-4) was obtained from the Zebrafish Stock Center. A stable transgenic unc45b-EGFP strain was generated in our lab from AB embryos by microinjection as previously described. 29 All adult zebrafish were bred and maintained according to standard procedures, 12 and kept at 28°C on a day/night cycle of 14 h light/10 h dark. Adults were housed in a cycled-water aquatic facility and fed twice daily with brine shrimp. Embryos were raised at 28°C in the standard embryo medium 12 for up to 5 days.
Embryonic cell culture (seZEB)
Zebrafish embryos at 3 h postfertilization (hpf) were sterilized, dechorionated as previously described,14,22 and dispensed individually into microfuge tubes before being dissociated in 25% trypsin/EDTA with triturgation. The cells were pelleted by centrifugation and resuspended in 20 μL of minimal culture medium (50% Dulbecco's modified Eagle's medium and 50% Leibowitz's L-15 medium, with 2 mM L-glutamine, 0.8 mM CaCl, and antibiotics as described below) containing 10% fetal bovine serum to halt trypsin digestion. Each single-embryo suspension was then seeded as a 20 μL spot onto laminin-coated eight-well glass chamber slides (Nunc). Cells were permitted to attach for 2 h at 28°C in a 5% CO2 incubator and then fed with 200 μL of the appropriate culture medium (Table 1). Attachment proceeded for 24 h, after which the medium was changed to remove floating unattached or dead cells for genotyping. For larger-scale RNA collection of differentiating WT cells, 10–20 embryos per well were dissociated simultaneously and plated as 50 μL aliquots.
DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; L-Gln, L-glutamine.
Cell culture additives
Several different media were used to establish optimal culture conditions (Table 1). The basal culture medium was made up of 25%/25%/50% Dulbecco's modified Eagle's medium/L-15/Hank's saline, with 10% final volume fetal bovine serum, 2 mM L-glutamine, and 0.8 mM CaCl. All culture media were supplemented with antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin, 100 μg/mL kanamycin, 50 μg/mL gentamycin, and 25 μg/mL amphotericin B). The enriched medium contained 10% whole-embryo extract by volume. Embryo extract was made as previously described,14,22 then centrifuged to remove tissue debris, and sterile-filtered. Supplemental HEPES/saline buffer consisted of 3.5 mg/mL HEPES, 344 μg/mL KH2PO4, 285 μg/mL K2HPO4, 375 μg/mL NaOH, 170 μg/mL NaHCO3, and 12.5 mM sodium pyruvate, final medium pH of 7.4. Purified human or bovine insulin was added to a final concentration of 5 μg/mL.
Zebrafish embryonic tail muscle cultures
Three-day-old zebrafish embryos were washed twice in sterile Hank's saline and sterilized by incubation for 2–3 min in cold 0.5% bleach. Excess bleach was removed by two further washes in sterile Hank's saline. The embryos were dechorionated manually in the sterile embryo medium containing 0.1 mg/mL tricaine, and pinned down with ultrafine needles for dissection. The ectoderm/skin was removed using fine forceps and the developing striated tail muscle was removed in short strips. Aggregated muscle tissue from 20 embryos was dissociated in 25% trypsin/EDTA with triturgation and plated on laminin-coated eight-well glass chamber slides (Nunc). Cultures were fed with Fish-Specific Enriched Medium (detailed above) and cultured for several days at 28°C in a 5% CO2 incubator, before fixation and imaging.
Derived cleaved amplified polymorphic sequence genotyping (dCAPS)
Single-embryo culture DNA samples were obtained by centrifuging unattached and/or dead cells from the removed culture medium at high speed and dissolving the pellet in 20 μL of lysis buffer (50 mM KCl, 10 mM Tris, 5 mM EDTA, 0.01% gelatin, 0.5% IGEPAL, and 0.1% Tween) for subsequent genotyping. Polymerase chain reaction (PCR) was subsequently performed using primers specific for the steif mutation in unc-45b (containing a single-nucleotide polymorphism [Table 2]) using the following conditions: 94°C, 3 min (94°C, 30 s; 51.5°C, 40 s; 72°C, 1 min) for 40 cycles, with final extension at 72°C for 5 min. Single-nucleotide polymorphism identification of mutant PCR products was achieved by restriction digestion at 37°C with EcoRI and subsequent 2% agarose gel electrophoresis. Amplification and digestion of DNA from phenotypically identified 3-day-old mutant and WT embryos were used as controls.
Cell culture reverse-transcription PCR
Samples were collected in 100 μL of Trizol reagent (Invitrogen) at timed intervals from the point of re-feeding (after 24 h of cell attachment), which was designated time = 0. Cells were lysed by triturgation and stored in Trizol at −20°C. RNA was extracted in 2/5ths volume of chloroform and precipitated by the addition of 1/2 volume of isopropanol. The RNA was centrifuged for 10 min at 12,000 g at 4°C, washed in 70% ethanol, and air-dried before resuspension in nuclease-free diethylpyrocarbonate-treated water. One-step reverse-transcription PCR (RT-PCR) was then performed, using a Superscript III RT-PCR kit (Invitrogen) according to the manufacturer's directions, using primers for various developmental markers (Table 2), and the following PCR conditions: cDNA synthesis at 50°C for 30 min; 94°C for 2 min (94°C for 15 s; 55°C for 30 s; 68°C for 1 min) for 40 cycles; final extension of 68°C for 5 min. Negative controls used non-RT-mix lacking reverse transcriptase, whereas positive controls used whole-embryo cDNA from multiple stages.
Immunocytochemistry and histology
Cell cultures on glass chamber slides were fixed at 4°C in 1:1 methanol:acetone for 10 min, allowed to air-dry, and stored at −20°C. For green fluorescent protein (GFP) transgenic strains, cultures were fixed at room temperature in 2% paraformaldehyde for 15 min, washed twice in phosphate-buffered saline (with 0.1% Triton X-100), and stored at 4°C. Dehydrated cultures were rehydrated in phosphate-buffered saline (PBS) blocking solution with 5% bovine serum albumin for a minimum of 2 h before staining. Histochemical stains included 1:50 Alexa 568 or 546 phalloidin (Invitrogen) and 1 μg/mL DAPI (Sigma). Muscle-myosin immunofluorescent staining was accomplished by primary antibody incubation with 1:20 F59 or S46 anti-MHC (Developmental Studies Hybridoma Bank) for 1 h, followed by four washes in PBS. Secondary antibody staining occurred for 1 h with 1:1000 Alexa 488 anti-mouse (Invitrogen). Cultures were washed three more times in PBS and subsequently viewed under phase-contrast or fluorescence microscopy.
Microinjection of DNA
Antisense, morpholino-modified oligonucleotides (morpholinos or MOs) and DNA expression constructs were injected into the yolk of 2-cell to 8-cell stage embryos. Each embryo was injected with 5–10 ng of DNA or morpholino dissolved in Danieau buffer [58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, and 5 mM HEPES, pH 7.6] in a total volume of 10 nL. Embryos were subsequently kept at 27°C until the 500–1000 cell (high) stage and cultured by the seZEB method described above. Morpholinos injected included p53–GCGCCATTGCTTTGCAAGAATTG 24 and unc45b–ATCTCCAATTCTCCCATCGTCATT. 30 The GFP expression construct consisted of the eGFP gene and fused to an 1140 bp fragment upstream of the unc45b gene, cloned into the Tol2 plasmid vector. Tol2 vector alone was used for control injections.
Results
Optimization of cell culture conditions for seZEB cultures
ZEB cultures have previously been used for the study of myogenesis,22,31 but it is unknown whether the culture conditions used in those experiments were optimal for myogenic differentiation, or whether these conditions would be suitable for differentiation of cultures derived from single embryos. Factors that commonly influence myogenesis in cell culture include the attachment substrate32,33 and media additives such as growth factors and nutritional supplements.34,35 We adapted the protocols of Collodi et al. 11 and Norris et al. 22 for the culturing of single zebrafish embryos on eight-well glass chamber slides, as depicted in Figure 1A. Initial experiments focused on the optimization of culture media and cell substrates to ensure myogenic differentiation in ZEB cultures. We noted three criteria required for optimal myogenic cell differentiation: First, strong cell–substrate attachment is required to promote a flattened, elongated cell morphology. Mesenchymal cells that are normally capable of undergoing myogenic differentiation when cultured on a laminin substrate often fail to do so when attached to fibronectin/poly-lysine substrates.33,36 We therefore attempted to establish single-embryo cultures on various common cell culture substrates, including bulk rat-tail collagen, poly-L-lysine, and laminin (Fig. 1B). Of these, laminin substrates were most efficient for cell attachment of early blastomeres, as measured by counting total attached cells after 4 days of culture.
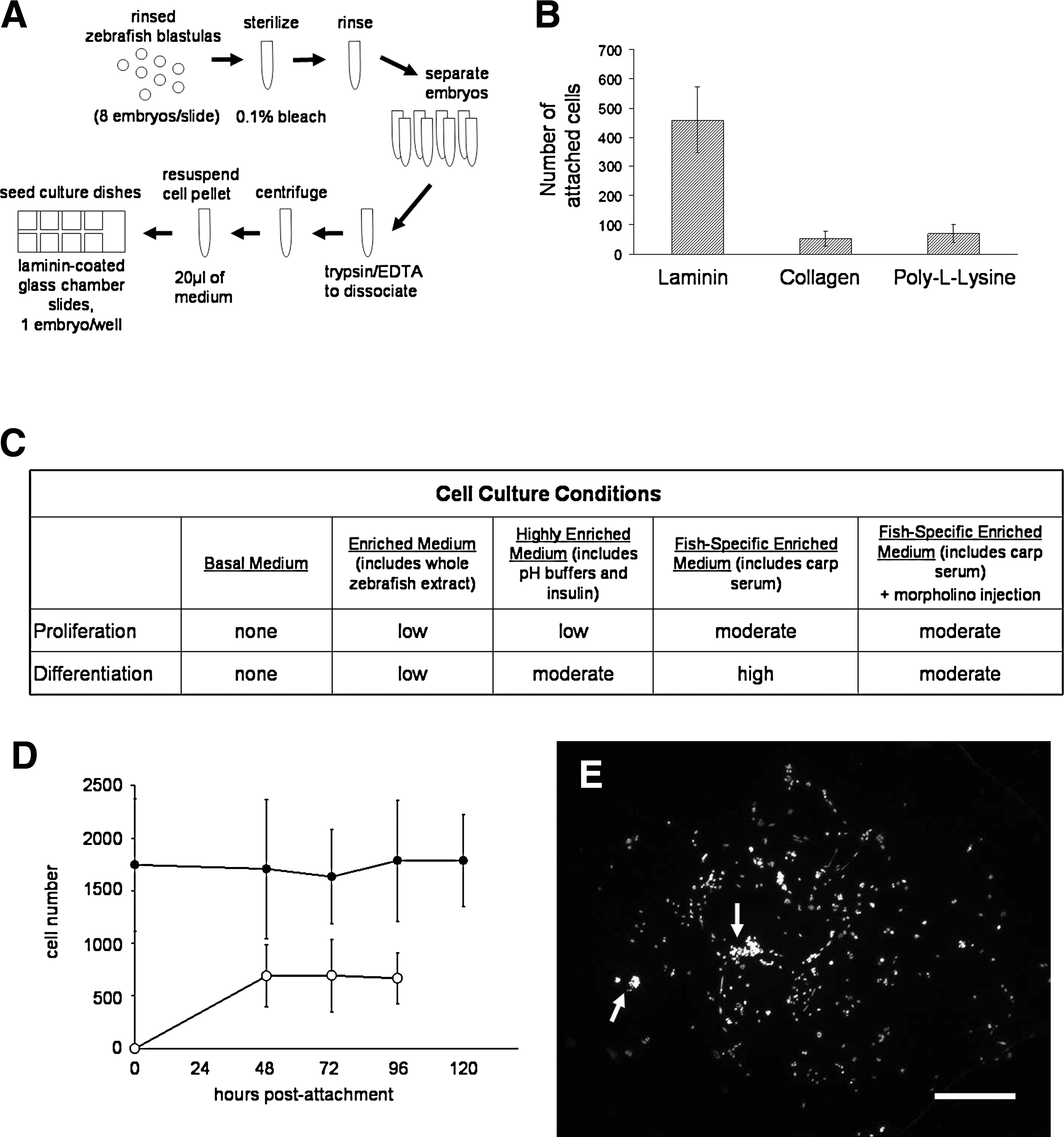
Optimal substrates and media for single-embryo zebrafish embryonic blastomere (seZEB) culture.
Second, nutritional and trophic factors in mesenchymal cell cultures must favor sufficient proliferation to ensure cell–cell communication between attached cells, and finally, growth factors that favor myogenic differentiation must be present in the culture medium. In the basal medium (Table 1), cells failed to proliferate, and phenotypic differentiation was not observed (Fig. 1C). Some proliferation was seen under nutritionally enriched conditions (enriched medium and highly enriched medium; Table 1 and Fig. 1C), attended by an increased rate of differentiation where cell density was highest. The addition of supplemental insulin gave slightly better results (Fig. 1C). Insulin-like growth factors are trophic factors known to increase both cell proliferation and differentiation in cultured myoblasts. 35 The further addition of carp serum gave the best results of all media tested (Fig. 1C), presumably because it contains numerous fish-specific trophic factors. The rate of differentiation in cultures containing both carp serum and insulin was very high, with morphologically distinct myocytes detectable in almost every culture. When larger numbers of embryos were used to obtain confluent cell cultures (data not shown), the rate of differentiation was lower than for single-embryo cultures under the same culture conditions, possibly due to inhibitory signaling effects from overcrowding or decreased substrate surface area. Cell proliferation in highly enriched media was sufficient to overcome initial cell death after bleaching of embryos (Fig. 1D), as determined by counts of adherent cells in each culture after DAPI staining (Fig. 1E). Relatively constant cell numbers were maintained up to the point of cellular differentiation, which took place in our cultures between 2 and 4 days after attachment. Differentiated cells in highly enriched media constituted between 20% and 50% of the total cell count (Fig. 1D), lower than but comparable to the rates of differentiation reported in mammalian C2C12 cell cultures (50–90%).37,38
Characterization of seZEB cell cultures
Initial cell attachment in single-embryo cultures resulted in the formation of isolated clusters of blastomeres of varying sizes (Fig. 2A), which remained rounded and poorly attached for several hours thereafter. Cell death was observed during the sterilization and dissociation process, but proliferation was sufficient to keep cell numbers relatively constant (Fig. 2B), and floating or dead cells were removed. Surviving cells proliferated slowly under these conditions, even in the most enriched media (Fig. 1C). Proliferation was restricted to regions of higher cell density, resulting in piled-up clusters of cells by 2 days of culture (arrows in Fig. 1E), which had a flattened, fibroblast-like phenotype. Cells began fusing and elongating over the next 24 h to form isolated bundles of elongated cells (Fig. 2B, arrows), which displayed an apparent myogenic phenotype. Multinucleated cell bundles were similar in appearance to those formed in differentiating mammalian C2C12 cells39,40; however, fusion of myocytes was often incomplete, resulting in elongated or partially fused nuclei rather than larger numbers of small, discrete nuclei (Fig. 2G). Unlike C2C12 cells, proliferating ZEB cells were not contact-inhibited, and continued to pile up during myoblast fusion and subsequent differentiation. Comparison of the progression of myoblast fusion in ZEB cells to C2C12 cells indicated that proliferation and fusion in ZEB cultures were likely concurrent, rather than sequential.
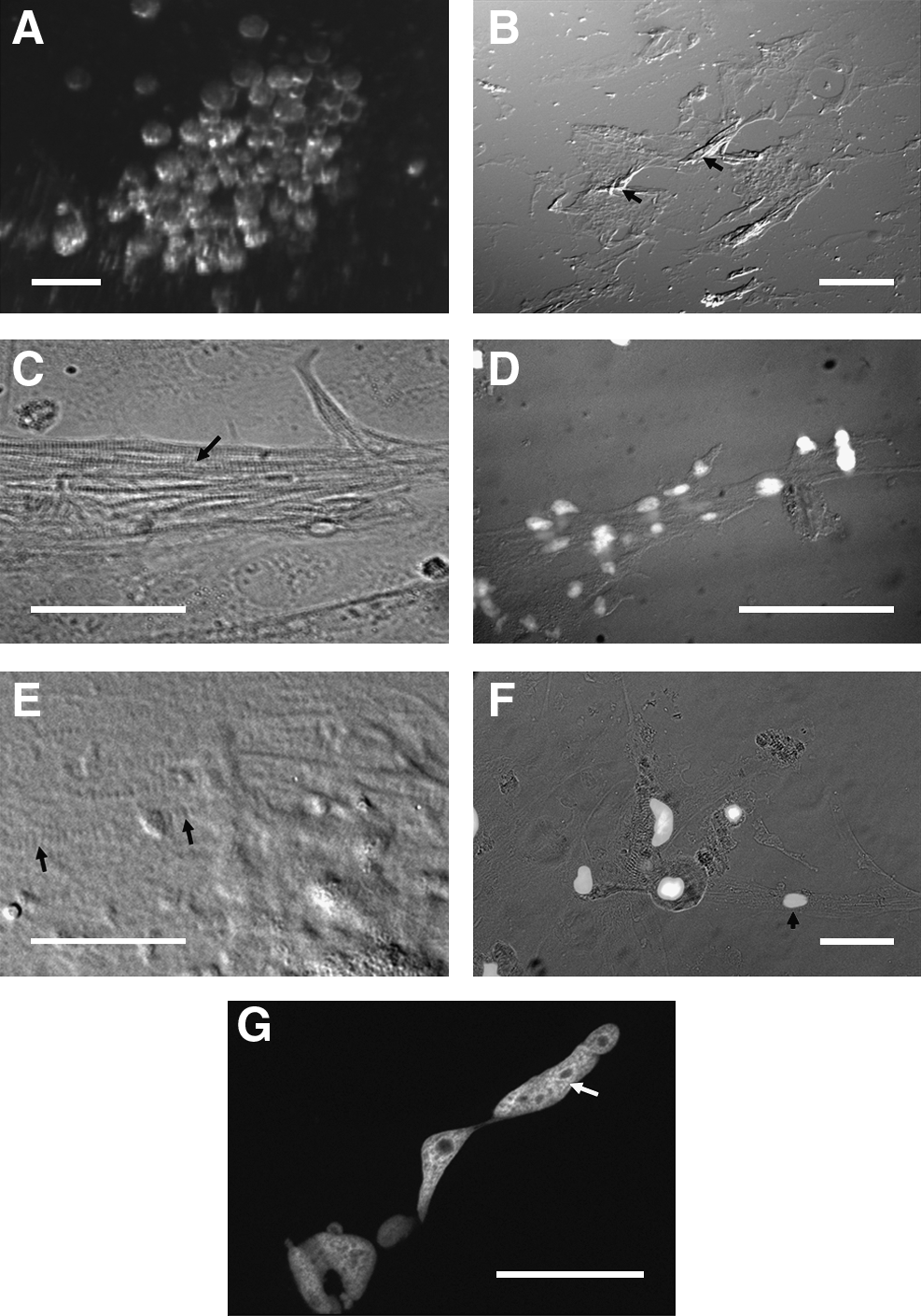
Phenotypic characterization of seZEB cultures. Single-embryo zebrafish cell cultures were established on eight-well chamber slides.
The cultures at this point contained two morphologically distinct types of myocyte-like cells (Fig. 2C–F). Elongated myotube-like cell bundles were detected toward the periphery of the cultures (Fig. 2C) or in cell-dense clusters throughout the culture (Fig. 2B), whereas cells between these bundles remained flattened, often in contact with their neighbors, but with numerous filamentous cell extensions (Fig. 2E, arrows). Both the myotubes and the filamentous extensions of flattened cells contained the highly identifiable banding patterns characteristic of sarcomere arrangement in developing skeletal muscle (arrow in 2C). Elongated cells were determined to be multinucleated (Fig. 2D, G), whereas flattened myoblast-like cells toward the center of the cultures often remained single-nucleated (Fig. 2F, arrowhead), despite the presence of sarcomere bands.
The banded cell extensions in both types of myocytes stained positive for muscle markers such as actin (Fig. 3A, C) and MHC (Fig. 3E). Actin staining in myocyte-like cells was present in a distinctive banded pattern that is characteristic of developing sarcomeres. This pattern was detected in both the single-nucleated, flattened myocytes (Fig. 3A inset, 3B) and the elongated, multinucleated myocyte bundles (Fig. 3C, D). Further, staining with the fast-twitch-specific MHC antibody F59 (Fig. 3E) revealed that expression of MHC was limited to cells with myocyte-like banding patterns (Fig. 3E), whereas the undifferentiated cells in between did not express detectable myosin (arrows). Most importantly, costaining for myosin and actin together (Fig. 3F) revealed clearly separate, alternating red and green bands, which correspond to the A band and I band, respectively, of functional sarcomeres (3F inset). When a different myosin antibody (S46) was used against the slow-twitch-specific isoform of MHC, no difference was noted in staining patterns (data not shown). This is not unexpected, since early embryonic muscle in zebrafish has been shown to coexpress both isoforms. 41
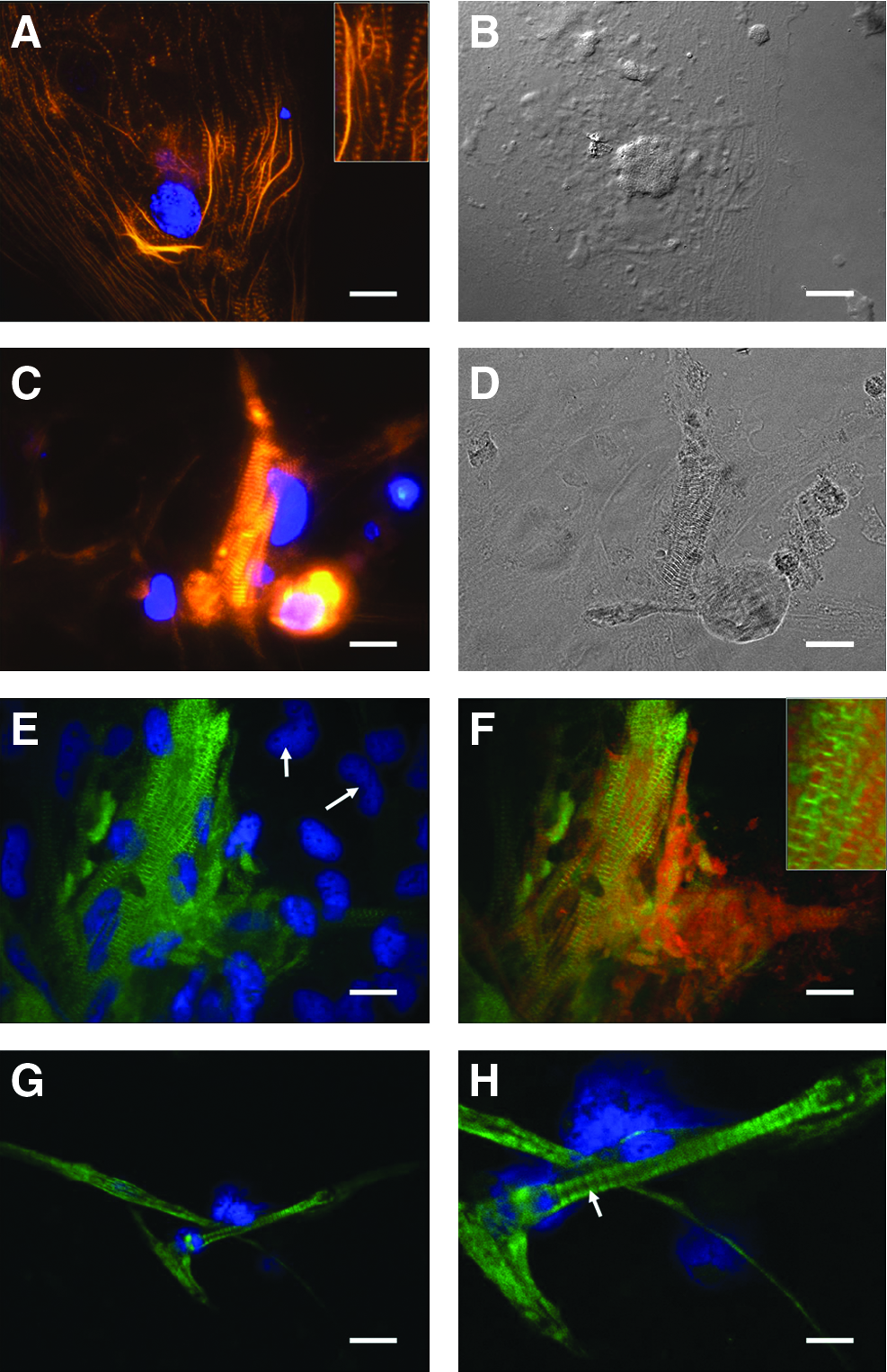
Molecular characterization of cells derived from seZEB cultures. Single-embryo cultures were stained for several markers of myocyte differentiation. Actin expression in seZEB cultures is shown by Alexa 546-phalloidin staining
To compare these ZEB cells morphologically with fully differentiated myocytes, we cultured developing tail muscle tissue from 3-day-old zebrafish embryos in our fish-specific enriched medium. The phenotypes of these cells (Fig. 3G) appeared very similar to those of our seZEB cultures, with elongated cells containing multiple nuclei, and the banding patterns after myosin antibody staining (Fig. 3H) were identical (arrow).
Gene expression during myogenesis in seZEB cell cultures
Although the patterns of myosin and actin expression in single-embryo cultures were characteristic of myocyte differentiation, we examined expression of other muscle cell markers, to provide further evidence that the seZEB culture system is optimized for myogenic differentiation. Other cell types may differentiate in single-embryo cultures, especially endothelial cells, which are also known to form from cells of fibroblastic lineage. 42
Unc45b is a myosin chaperone required for proper sarcomere assembly and cell function in striated muscles.24–27 It is normally localized to sarcomeres, where it may be involved in dynamic folding and re-folding of the myosin motor domain during muscle contraction. 43 As a rapid method of detecting muscle-specific cellular differentiation, we examined unc45b-GFP transgene expression in seZEB cultures. Cultures from zebrafish embryos containing this transgene displayed GFP expression only in phenotypically differentiated myocyte bundles (Fig. 4A, B). DAPI nuclear staining revealed numerous isolated fibroblast-like cells (arrows) that did not express GFP, both at the periphery of cultures (Fig. 4A) and between the myoblast bundles toward the center of cultures (Fig. 4B), as was expected. GFP expression was restricted to large, flattened cells that displayed characteristic actin staining patterns (Fig. 4C) typical of differentiating myocytes. To test for endothelial cell differentiation, we also cultured single embryos containing a fli1-GFP transgene. Fli1 is a marker of early differentiating endothelial cells, 44 which are also known to develop from squamous fibroblast-like mesenchymal cells in culture. 45 In fli1-GFP cultures, no signal was detected in any cell (Fig. 4D), although developing embryos expressed significant GFP in differentiating endothelial tissues (not shown). This is consistent with the hypothesis that our cell culture method specifically favors myogenic differentiation over other pluripotent fibroblast fates.
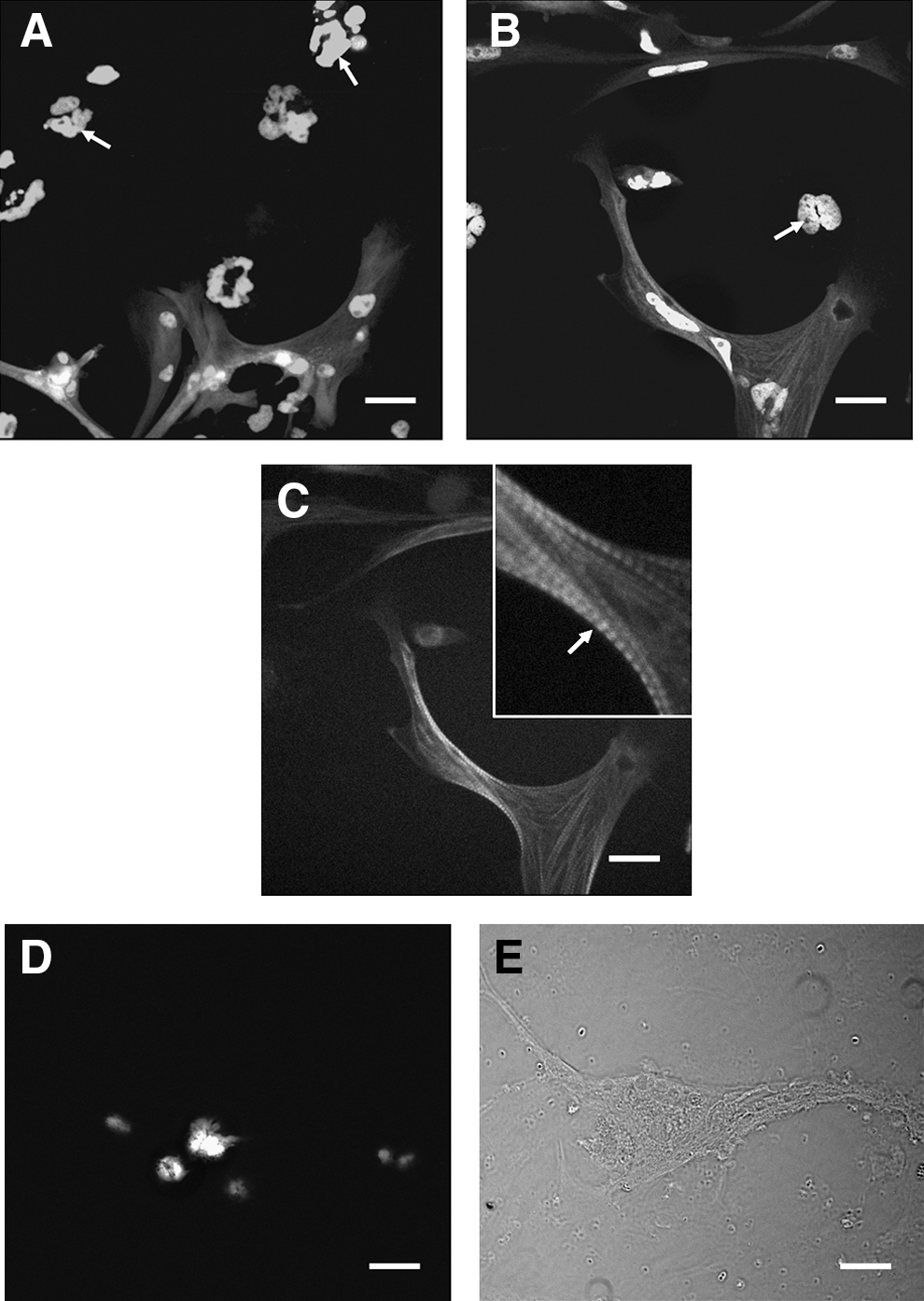
Expression of transgenic markers in seZEB cultures. Single-embryo cell cultures were established using embryos from unc45b-GFP or fli1-GFP transgenic zebrafish, which fluoresce in muscle tissue and endothelial tissue, respectively.
As an independent assay to confirm that ZEB cultures grown under our optimized culture conditions specifically favored myogenic differentiation, we performed RT-PCR analysis using primers for a set of developmental marker genes (Table 2) over a 48-h time-course of cell culture. We first examined expression of the endothelial transcription factor fli1. The absence of PCR product generated from these primers (Fig. 5A, top row) further supports the conclusion that these culture conditions were optimized for nonendothelial differentiation. Expression of endothelial marker transcripts was not detected even after 96 h of culture (not shown). By contrast, expression levels of the muscle-specific transcription factor myoD and the myosin chaperone unc45b were both high, especially at early time-points (Fig. 5A, second and third rows). The neural-specific transcription factor HUC and the housekeeping gene gapdh were used as negative and positive controls, respectively (Fig. 5A, fourth and fifth rows). Neural marker was not detected, indicating that the cells in ZEB cultures were exclusively of mesenchymal fate, as spontaneous neuronal differentiation occurs only in ectodermal cells. Positive control RT-PCR using whole-embryo extract from combined embryonic stages (4 hpf to 3 days) demonstrated expression of all markers (Fig. 5B).
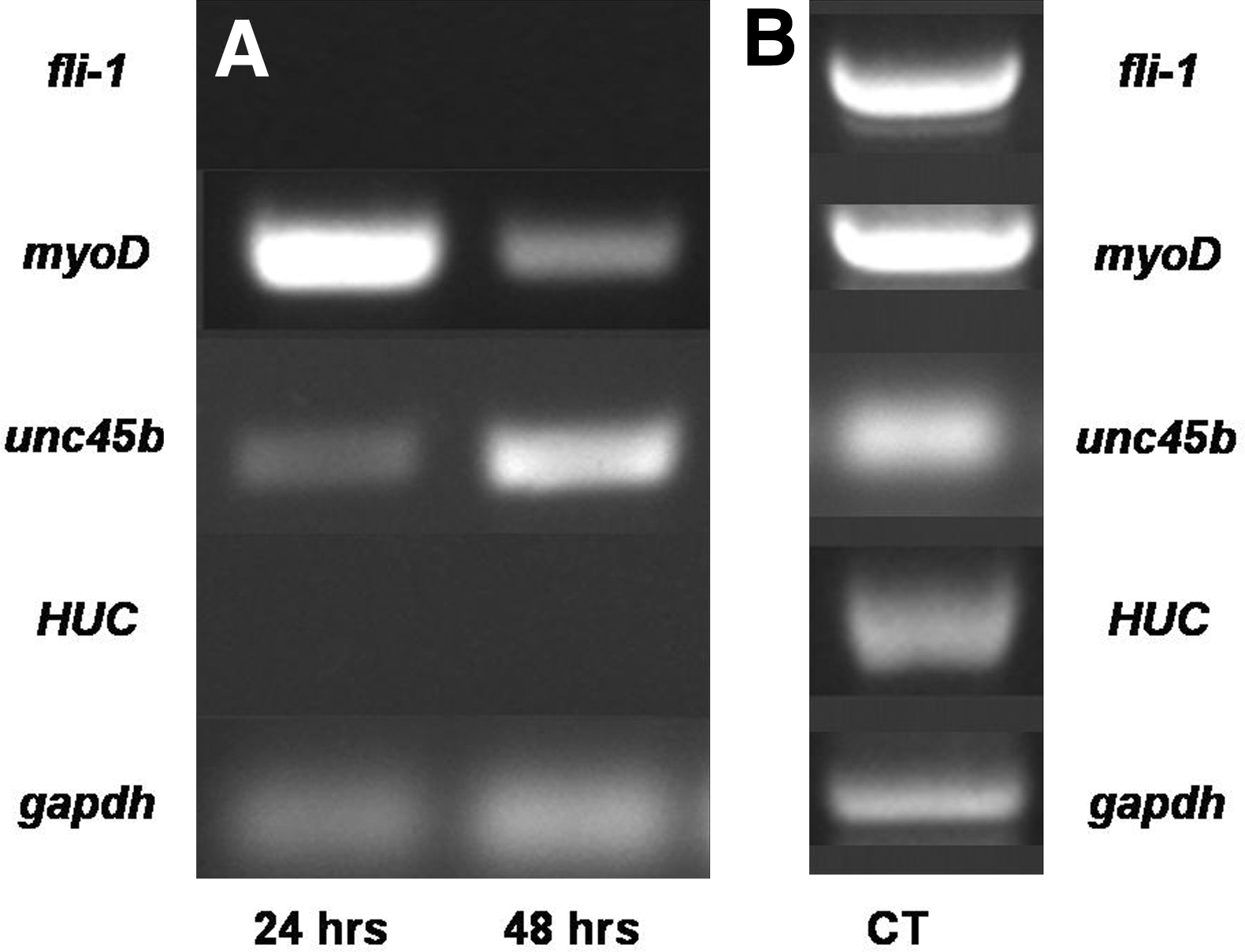
Expression of markers for endothelial, neuronal, and myogenic differentiation in ZEB cultures. ZEB cultures derived from multiple WT embryos were established and collected at 24-h intervals to obtain RNA for analysis by RT-polymerase chain reaction (PCR).
Myogenesis in seZEB cell cultures from mutant embryos
To test the applicability of our seZEB culture system as a model for genetic studies, we examined the progress of myogenic differentiation in cultures derived from zebrafish mutants that are reported to have deficiencies in muscle development. Specifically, we used heterozygous unc45b mutant zebrafish (steif) to generate seZEB cultures lacking Unc45b myosin chaperone function. Homozygous steif embryos at later stages are characterized by a lack of motility, poorly defined skeletal muscle birefringence, and extreme heart edema, with poorly organized sarcomeres, 25 as shown in Figure 6A. We derived single-embryo cultures from the progeny of heterozygous steif parents, which should consist of 25% homozygous steif embryos and 75% phenotypically WT embryos, including steif heterozygotes. Obviously, at the stage at which embryos are dissociated for blastomere culture, we were unable to phenotypically distinguish these two classes of embryos. Therefore, cell culture supernatant from steif seZEB cultures was drawn off and replaced after 24 h of cell attachment, and cells in this supernatant were used for genotyping. Dead or unattached cells in the media were collected by centrifugation for DNA extraction and PCR amplification using the dCAPS method. This allowed us to identify the genotypes (mutant, WT, or heterozygous) of the cells in the cultures before examination of cellular morphology and marker expression (Fig. 6B). Genotyping revealed that the expected 1:2:1 ratio of WT, heterozygous, and homozygous mutant embryos was obtained from heterozygote crosses (Fig. 6C).
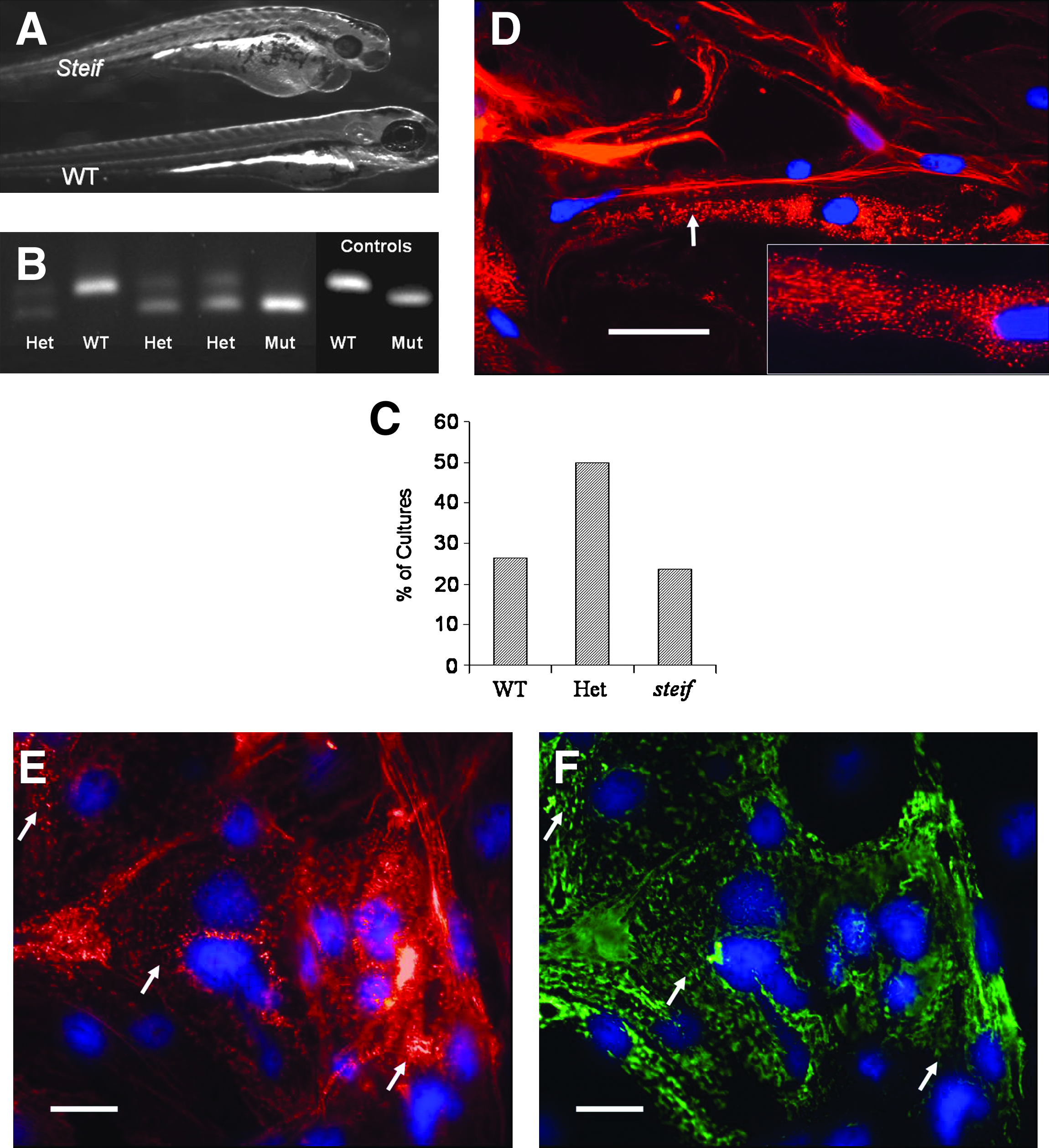
Marker expression and genotyping of seZEB cultures from unc45b mutant embryos. Single-embryo cultures of ZEB cells were established from embryos obtained by crossing steif mutant heterozygote parents.
Given the muscle-specific defects of steif mutant embryos, it was unsurprising that myofibrillogenesis was severely reduced in cells derived from cultured steif embryonic blastomeres (Fig. 6D–F). Mutant cells underwent apparent myogenic differentiation in these cultures, with peripheral cells forming long fibril-like bundles (Fig. 6D, E) that were often multinucleated and morphologically similar to myofibrils, but did not contain the characteristic pattern of sarcomere banding. Actin staining demonstrated that many of these fibril-like processes contained poorly organized punctate regions of actin expression (arrows in Fig. 6D, E), possibly corresponding to regions of failed sarcomere assembly. Further, when cells were costained with myosin antibodies, a similar punctate pattern of expression was detected (Fig. 6F). The disorganized patterns of actin and myosin expression colocalized in these cultures throughout the cell body (compare arrows in 6E and F).
After 4 days of culture, the percentage of genotyped WT cultures in all experiments that were viable and contained phenotypically identifiable myocytes with characteristic banding patterns was over 66% (Fig. 7A, first column, n = 256). Of genotyped steif cultures, however, fewer were viable (Fig. 7A, second column, n = 64) and almost none contained fully differentiated myocytes with identifiable striations. Further, after blind-scoring a number of apparently homozygous steif cultures (determined by lack of visible striations and punctate patterns of actin staining), identification was confirmed by dCAPS genotyping at over 90% accuracy (Fig. 7B).
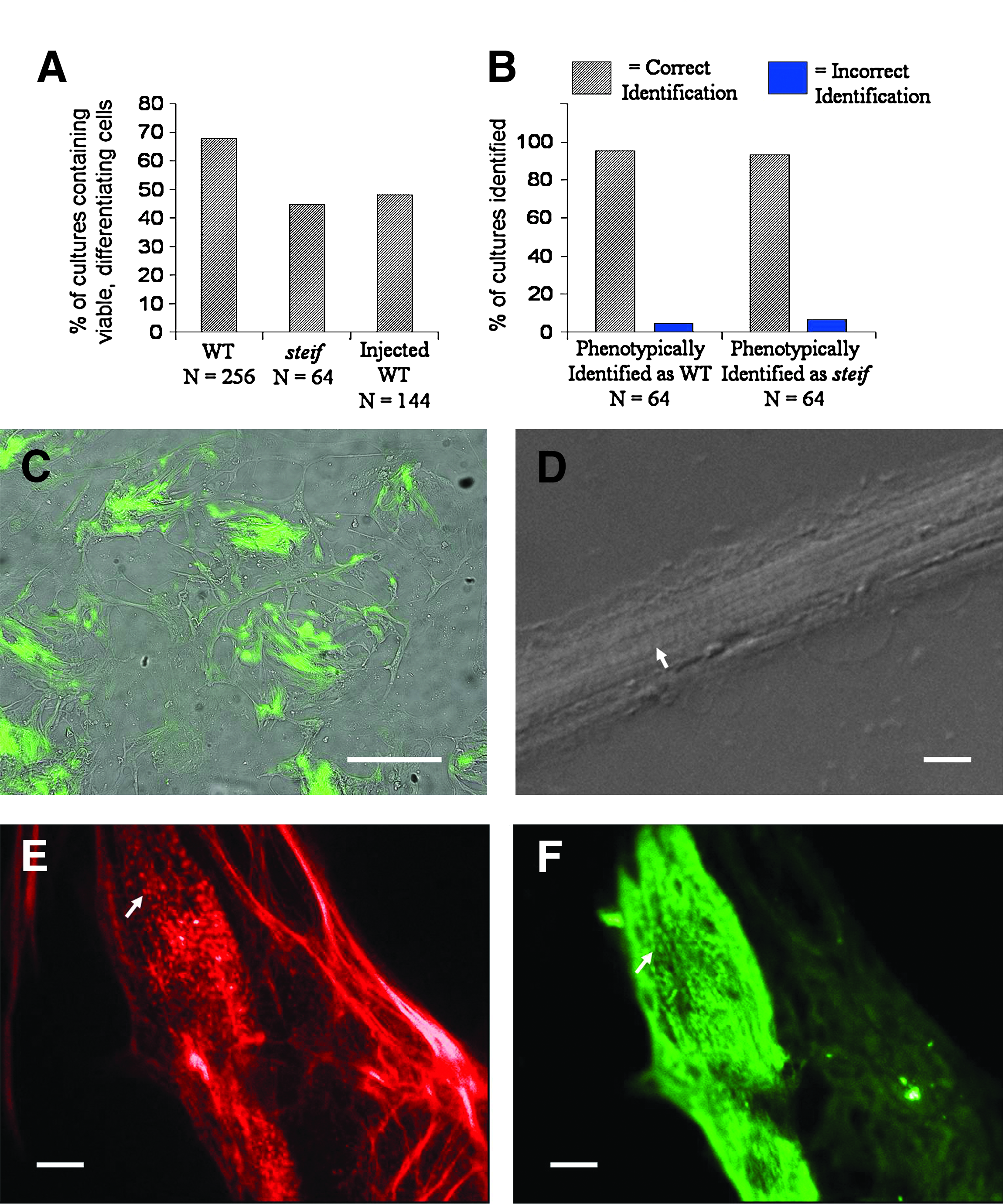
Microinjection of embryos with antisense morpholino-oligonucleotides or gene expression constructs followed by seZEB culture. The percentage of cultures with viable, differentiating cells was measured for genotyped WT, steif, and injected embryos
A useful aspect of zebrafish as a developmental model is the ease with which gene expression can be manipulated in embryos by microinjection of antisense oligonucleotides (to downregulate gene expression) or in vitro synthesized mRNA or DNA expression constructs (to upregulate gene expression). If seZEB cultures could be created from microinjected embryos, it would be possible to examine the effects of changes to specific gene expression on subsequent myogenesis. Therefore, we injected individual embryos at the two- to four-cell stage with antisense morpholino-oligonucleotides (MOs) or DNA expression constructs before seZEB cell culture. Microinjection of embryos before dissociation resulted in only a slight reduction in proliferation and differentiation (Fig. 1C). Further, viability of cultures from injected embryos was lower than uninjected WT embryos by only ∼15% (Fig. 7A, third column, n = 144). Injection with a plasmid DNA containing GFP under the control of the unc45b upstream region was used to determine whether gene expression can be upregulated in the seZEB culture system. After 2 days of culture, GFP expression was detected in differentiating myocytes (Fig. 7C), but not in cells injected with an empty vector (not shown). Additionally, injection with MO was used to determine whether gene expression can be effectively downregulated in this culture system. Injection with a p53 MO (routinely coinjected in zebrafish gene-knockdown experiments to reduce apoptosis) resulted in reduced viability (Fig. 7A) but did not prevent the differentiation of myocytes (Fig. 7D) identifiable by the formation of elongated cell bundles with visible striations (arrow). Further, injection of single embryos with an unc45b MO resulted in a phenocopy of the steif mutant cellular phenotype, with punctate patterns of actin (Fig. 7E) and myosin (Fig. 7F) staining (arrows). Nearly all viable cultures derived from unc45b MO-injected embryos displayed this phenotype (n = 56).
Discussion
Previous studies have made use of the ZEB culture protocols established by Collodi et al. 11 for the study of zebrafish muscle cells in culture, although little emphasis has been put on the molecular mechanisms of myogenic differentiation in these cells. In other vertebrate developmental models, cellular differentiation studies have generally focused on whole-embryo studies, or easy-to-maintain cell lines such as mouse C2C12 or tumor-derived cells. While fibroblast-like cell lines of this type are highly competent for myogenic differentiation in culture, these cells have the disadvantage of being selected for immortality (or at least continued proliferation after repeated passaging), which likely result in altered genetic programs and/or aneuploidy. The variation from WT gene expression in these lines is generally not examined. 8 By contrast, primary cell culture using ZEB protocols seems a preferable method for the study of gene expression in cultures undergoing myogenic differentiation. Primary cell culture methods are well established for many vertebrates, and some of these protocols have been extensively used for the study of cellular differentiation. However, zebrafish have the advantage of being a relatively inexpensive and therefore widely used model for genetic manipulation. The use of microinjection to introduce RNA or DNA for overexpression studies, transgene vectors for genomic insertion, or antisense oligonucleotides for knockdown experiments makes these organisms one of the most versatile genetic models currently in use.46,47 Further, the wide availability and wealth of mutant and transgenic zebrafish lines means that a large number of genes thought to be involved in myogenic differentiation could be studied using our single-embryo cell culture system.
Undetermined mesenchymal cells from vertebrate embryos have been shown to assume a fibroblast-like phenotype in cell culture and undergo spontaneous myogenic differentiation, under a variety of culture conditions.34,42,48 Even mature, differentiated cells of mesenchymal origin can be induced to undergo dedifferentiation and subsequent myogenesis in culture. 48 Some culture conditions, however, can minimize or eliminate myogenic differentiation in mesenchymal cell cultures. For example, too high or too low cell density,49,50 trophic factors such as transforming growth factor-β family members,35,51 and improper cell culture substrates 36 can all affect myogenic differentiation and even prevent myogenesis from occurring in culture. Our results establish a set of favorable conditions for single-embryo cultures to undergo myogenic differentiation in terms of culture method, substrate, and media. Laminin has long been recognized as an ideal cell culture substrate for myocytes,32,33 and seems to better promote myogenic differentiation in the ZEB culture system than alternative substrates such as poly-L-lysine or collagen. Additionally, although previous studies have made use of few medium supplements for ZEB primary culture, most commonly bovine serum and zebrafish embryo extract alone,13–15 we found that both insulin and carp serum had a profound impact on the proliferation and differentiation of ZEB cultures, to the extent that a single blastula-stage embryo was capable of dividing to the point of cell-cell contact, allowing the differentiation of myocytes. We have also established that seZEB cultures did not express ectodermal markers, such as genes involved in early neural development, consistent with a uniformity of mesenchymal cell type. Although ectoderm-inducing factors are present in the zebrafish yolk even before fertilization, 52 the dilution of these factors during the trypsinization of the embryo may prevent ectodermal or chordamesodermal specification. It is also known that embryonic fibroblast cultures of mesenchymal origin can be induced to undergo other types of cellular differentiation, including endothelial42,45 and hematopoietic differentiation. 53 Under the conditions and methods outlined here, endothelial markers are not detected, further demonstrating the uniformity of cell type in single-embryo cultures under our specific culture conditions. Some myogenic cells were flattened and single-nucleated, whereas others were multinucleated cell bundles. Slow muscle precursors in zebrafish are larger, flattened, and single-nucleated, and found at the periphery of the myotome, 54 and the two myogenic cell populations in our culture system could thus represent regionalized slow-muscle and fast-muscle differentiation. However, immunofluorescence using fast-muscle- or slow-muscle-specific anti-MHC antibodies resulted in identical staining patterns in both types of myocytes. This may indicate that our ZEB myocytes express both isoforms, as has been shown in early embryonic muscle in zebrafish. 41
The myosin chaperone Unc45b is a useful marker of early muscle differentiation, as its expression begins at ∼9 hpf in whole embryos, corresponding to the onset of muscle differentiation and the organization of sarcomeres in striated muscle.24,25 Indeed, in our culture system, expression of transcripts for Unc45b and the early muscle transcription factor MyoD was detectable at high levels within 24 h of plating, indicating only a short (12–18 h) delay in developmental timing of gene expression due to cell stress and the attachment period. The absence of functional Unc45b is known to lead to decreased myofibrillogenesis and sarcomere organization in steif mutant zebrafish 25 and unc45b morphants, 24 which is consistent with our results showing actin disorganization in cultured cells. Despite the early onset of unc45b expression during myogenesis in culture, cells in mutant cultures became elongated and multinucleated, and displayed increased actin expression, confirming the observation that zygotic Unc45b is not required for the earliest stages of myogenic differentiation. Further, the predicted genetic segregation of mutant alleles matched the observation of phenotypic characteristics in the seZEB culture system, as confirmed by genotyping. This demonstrates that the mutations, although zygotic lethal when homozygous in whole embryos, did not impair the ability of cells in seZEB cultures to undergo myogenic differentiation to the point of phenotypic identification. Therefore, zebrafish strains possessing early embryonic lethal mutations in genes of interest could potentially still be used for the study of cellular differentiation in vitro by our methods. This represents a major advantage of the seZEB culture system, which permits the examination of mutant cells in culture that could not be obtained from early embryonic lethal larval fish, or from bulk cultures of mixed mutant and nonmutant cells.
The microinjection of antisense oligonucleotides for the knockdown of genes involved with myogenesis before cell culture is a useful tool for developmental genetics, and the injection of unc45b morpholino resulted in a phenocopy of the steif mutant phenotype in cells derived from nearly 100% of embryos. Any stress from microinjection resulted in only a 15%–20% reduction in viability and differentiation, and this indicates the feasibility of routine knockdown experiments in the seZEB culture system. Moreover, microinjection with a GFP reporter construct before culture resulted in detectable GFP expression under the control of a myocyte-specific promoter. These experiments show that both upregulation and downregulation of gene expression can be manipulated in zebrafish embryos before cell culture with a high success rate. Further, it is possible to drive ZEB cultures toward alternate cell fates, such as smooth muscle, 22 by the addition of specific trophic factors to the cell culture media (although we did not test this in the experiments described here). This indicates that it would be possible to examine the effects of specific gene expression on differentiation in different types of muscle cells. Sun et al. 55 have also observed the differentiation of ZEB cells into morphologically identifiable neurons and astrocytes after 2 weeks of culture in the presence of sodium selenite on poly-D-lysine substrates, and Collodi et al. 56 have detected hepatocyte P450 in ZEB cultures treated with 10 nM 2,3,7,8-tetrachlorodibenzo-P-dioxin. Although these methods are not well characterized outside of mammalian cells, and attachment on nonlaminin substrates was limited in our seZEB cultures (Fig. 1B), these studies suggest that it may be possible to expand the usefulness of the seZEB culture system for studies of genetic mechanisms of differentiation and development in a wide variety of cell and tissue types. Future studies may therefore focus on the development of zebrafish-specific markers and methods for characterizing the differentiation of seZEB cell cultures into alternate cell fates.
The ZEB culture system will be a useful tool for the study of myogenic differentiation at the cellular level, requiring little in the way of technologically advanced methods. All images of sarcomeres in this study were acquired using fluorescence or differential interference contrast microscopy, rather than more expensive and complicated methods such as scanning confocal microscopy or fluorescence correlation spectroscopy, which are generally necessary to observe subcellular structures in situ.17,19,20 Further, the ability to examine cellular differentiation in isolated, pluripotent cells in monolayer, which can be easily and instantly treated with pharmacological reagents and viewed in real time, without immobilizing live embryos, is of singular value. However, the limitations of this technology are clear: cells in culture are by definition removed from their normal embryonic context, and are therefore only useful for studying processes that are cell-autonomous and for which culture conditions have been at least partially defined.
Footnotes
Acknowledgments
The authors would like to thank Andrew Waskiewicz for the use of his zebrafish aquatic facility, which is supported by an MRS grant from the Natural Sciences and Engineering Research Council of Canada (NSERC), as well as the gift of fli1-GFP transgenic lines. The authors would also like to thank Heather McDermid for the use of her cell culture facilities, Uwe Strähle for generously providing steif mutant strains, and Mike Belosevic for the gift of carp serum and fish cell culture additives. Finally, the authors would like to thank Daniel Brewster for dissection of 3-day embryonic tail muscle and Eva Guvnowski for creation and injection of the unc45b-GFP transgene construct. This work was also supported by an NSERC Discovery grant.
Disclosure Statement
No competing financial interests exist.