
Abstract
Abstract
The small-molecular compound miglustat (N-butyldeoxynojirimycin, Zavesca®) has been approved for clinical use in type 1 Gaucher disease and Niemann–Pick type C disease, which are disorders caused by dysfunction of the endosomal-autophagic-lysosomal system. Miglustat inhibits a number of enzymes involved in glycoconjugate and glycan metabolism, including β-glucosidase 2 (GBA2), which is exceptionally sensitive to inhibition by miglustat. GBA2 is a glucosylceramide-degrading enzyme that is located on the plasma membrane/endoplasmic reticulum, and is distinct from the lysosomal enzyme glucocerebrosidase (GBA). Various strands of evidence suggest that inhibition of GBA2 contributes to the therapeutic benefits of miglustat. To further explore the pharmacology and biology of GBA2, we investigated whether the zebrafish homolog of GBA2 has similar enzymatic properties and pharmacological sensitivities to its human counterpart. We established that zebrafish has endogenous β-glucosidase activity toward lipid- and water-soluble GBA2 substrates, which can be inhibited by miglustat, N-butyldeoxygalactonojirimycin, and conduritol B epoxide. β-Glucosidase activities with highly similar characteristics were expressed in cells transfected with the zebrafish gba2 cDNA and in cells transfected with the human GBA2 cDNA. These results provide a foundation for the use of zebrafish in screening GBA2-targeting molecules, and for wider studies investigating GBA2 biology.
Introduction
A
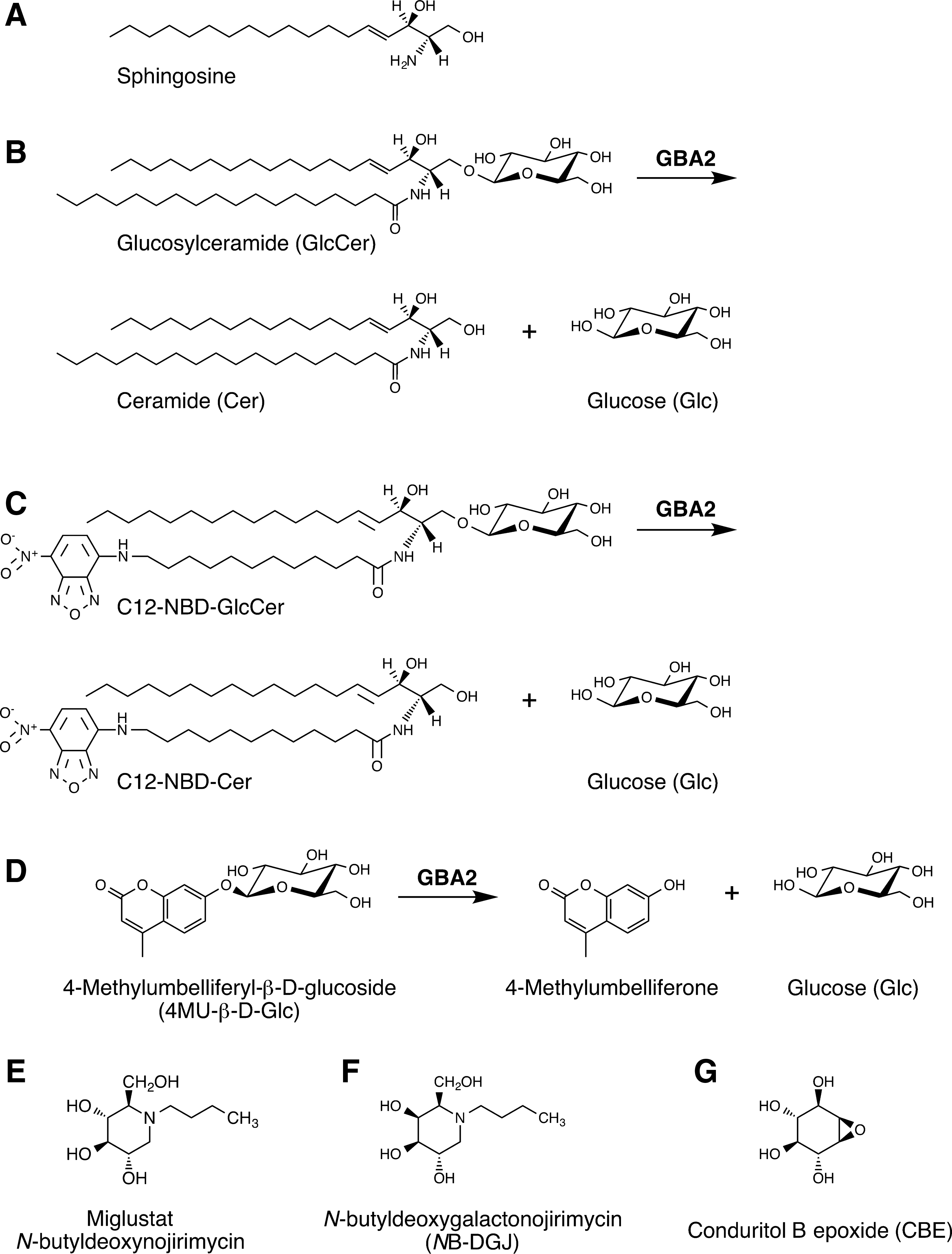
Structures of sphingolipids, β-glucosidase 2 (GBA2) substrates, and inhibitors.
Sphingosine-1-phosphate is involved in multiple intracellular and extracellular signal transduction processes, including the regulation of T lymphocyte trafficking and the maintenance of vascular integrity.5–7 Glycosphingolipids also have various biological roles and are associated with a number of diseases,8,9 including mammary epithelial cell adhesion, 10 embryonal gastrulation, 11 development of the central nervous system, 12 insulin signaling, 13 maturation of invariant (type I) natural killer T-cells, 14 spermatogenesis, 15 lysosomal storage disorders, 16 infantile-onset epilepsy, 17 and polycystic kidney disease. 18
Sphingoid base synthesis and subsequent metabolic steps take place on the endoplasmic reticulum (ER), including the production of ceramide. Remarkably, various enzymatic conversions between nonglycosylated sphingolipids, anabolic and catabolic reactions, can be catalyzed by a number of isoenzymes. In particular, numerous ceramidases and sphingomyelinases have been identified, and these enzymes are present on the plasma membrane and multiple organelles. Together, the enzymes metabolizing nonglycosylated sphingolipids form a highly interconnected, dynamic network. 2
Glucosylceramide (GlcCer) (Fig. 1B), is synthesized from ceramide at the cytoplasmic side of the Golgi apparatus,19,20 from where it is transported to the cytoplasmic face of the ER and plasma membrane.21,22 The enzyme β-glucosidase 2 (GBA2), by virtue of its extra-lysosomal localization, is thought to act on this pool of GlcCer, producing ceramide and glucose.23–25 In this way, GBA2 connects the pool of GlcCer located on the ER and plasma membrane with the metabolic network of nonglycosylated sphingolipids. Because of the metabolic connectivity of these sphingolipids, it is difficult to assign specific biological roles to individual sphingolipid species, including GlcCer.
Newly made GlcCer is also translocated from the cytoplasmic side to the luminal face of the Golgi, where its carbohydrate is extended with additional sugars. The resulting complex glycosphingolipids are transiently exposed at the cell surface, before they are endocytosed and degraded in endolysosomes, via stepwise removal of sugars. In the course of lysosomal glycosphingolipid degradation, GlcCer re-emerges as a metabolic intermediate, and is, in turn, cleaved into glucose and ceramide. The latter step is catalyzed by the enzyme glucocerebrosidase (acid β-glucosidase, GBA).26,27 GBA2 and GBA are thus both GlcCer-degrading enzymes, but they are present at different cellular locations, and act on different cellular pools of GlcCer. Structurally, GBA2 and GBA are unrelated enzymes and belong to different glycohydrolase families, GH116 and GH30, respectively. 28
Genetic deficiencies in GBA result in Gaucher disease, characterized by accumulation of GlcCer and glucosylsphingosine. The most common form, type 1 Gaucher disease, primarily affects cells of the mononuclear phagocyte system in the liver, spleen, and bone marrow. 29 GBA2 is mutated in a genetic disorder referred to as SPG46 (named after SPastic Gait locus #46), which is characterized by the combination of spastic paraplegia and cerebellar ataxia.30–33 Other symptoms of SGP46 include axonal neuropathy, thin corpus callosum, cerebellar and cerebral atrophy, dysarthria, and cognitive impairment. SPG46 patients were first diagnosed early in life (median age 7, range 1–20).30–33 At present, it is not understood how mutations in GBA2 cause ataxia/paraplegia, nor are adequate therapies available for SPG46, resulting in life-long disability and poor quality of life.
Recently, we showed that five nonsense and five missense GBA2 mutants associated with SPG46 are enzymatically inactive, with some of the mutants being expressed at very low levels, suggesting that a lack of GBA2 enzyme activity underlies the pathology seen in SPG46. 34 Remarkably, none of the Gba2 gene-disrupted murine studies have described neurological or neurodegenerative symptoms.25,35–37 In contrast, transcriptional silencing of the gba2 homolog in zebrafish caused motor neuron axons to develop abnormal branches compared to the axonal outgrowth in controls. 31 Also, in touch-response tests, gba2-targeted zebrafish embryos displayed slower movements and shorter escape distances. 31 Further, skin fibroblasts derived from Gba2-deficient mice exhibit enhanced actin polymerization along with microtubule persistence, and display more filopodia and lamellipodia. 36 These findings may be indicative of the cell biological basis of the neuronal pathology seen in SPG46, considering that microtubules are affected in other forms of spastic paraplegia. 38 It remains to be established whether the cytoskeleton is affected in cerebellar and motor neurons of SPG46 patients expressing mutant forms of GBA2.
Miglustat (Zavesca®, N-butyldeoxynojirimycin) is an alkylated imino sugar resembling glucose (Fig. 1E), and it can inhibit a number of enzymes participating in glycoconjugate and glycan metabolism,39–42 including GBA2,24,43–46 depending significantly on dose/concentration. Miglustat is approved in over 40 countries for clinical use in type 1 Gaucher disease and Niemann–Pick type C (NPC; not approved for NPC in the United States).47–49 In mouse models of NPC disease and Sandhoff disease, which are both lysosomal glycosphingolipid storage disorders with a neurodegenerative course, miglustat suppresses central nervous system inflammation, delays the loss of motor control and muscle strength, and extends lifespan.50,51 At the same time, miglustat increases the level of glucosylceramide in the brain.50,51
On the basis of these findings it was suggested that the beneficial effects of miglustat in these mouse models are attributed in part to inhibition of Gba2.50,51 Likewise, disruption of the Gba2 gene reduces disease severity in mouse models of type 1 Gaucher disease 35 and of NPC disease, 52 suggesting that specifically targeting GBA2 is an attractive strategy in both of these diseases. Gba2-deficiency lowers serum levels of IL-2, IL-6, granulocyte colony-stimulating factor, and CXCL1/KC, and prevents the type 1 Gaucher-associated elevation of IL-1α and IL-12. 35 Further, Gba2-deficiency extends the average lifespan of NPC mice from 73 to 83 days, and delays the loss of coordination and onset of ataxia by 15 days. 52 At present it is not understood why Gba2 deficiency ameliorates the two distinct pathologies of type 1 Gaucher and NPC disease. Nevertheless, targeting GBA2 appears a promising strategy for diseases caused by dysfunction of the endosomal-autophagic-lysosomal system, prompting a call for the development of specific GBA2-inhibiting compounds, 35 and suggestions of lead compounds for this purpose. 52
Ever since it was established that, in contrast to all other glycosylated sphingolipids, GlcCer is the only glycosphingolipid that is synthesized at the cytoplasmic side of the Golgi apparatus,19,20 the biological significance of this topological exception was not clear. Recently, the characterization of GBA2 as an extra-lysosomal GlcCer-degrading enzyme, the recognition of GBA2 as an effective pharmacological target in mouse models of lysosomal storage diseases, and the identification of mutations in the GBA2 gene in patients presenting with ataxia and paraplegia, provide the first tangible clues for biological roles of GlcCer present outside of the lysosome. Further studies on GBA2 are therefore warranted. To this end, the zebrafish is well suited, as this species has been recognized as a robust model organism for modeling neurodegenerative conditions, including motor neuron diseases, and for high-throughput drug screens.53,54 Human and zebrafish GBA2 are highly conserved at the amino acid level (66% identical, 79% similar; Fig. 2). All GBA2 residues mutated in SPG46 are conserved in zebrafish gba2, with the exception of Arg870 (p.Arg870*), which corresponds to Gly818.

Alignment of amino acid sequences of human GBA2 (gi|24308251|ref|NP_065995.1), mouse gba2 (gi|240120073|ref|NP_766280.2), and the zebrafish gba2 cDNA we obtained. The latter is identical to gi|125821356|ref|XP_687652.2, except for residues #229, 308, 366, and 437, which are indicated with an asterisk (*). Amino acids that are fully conserved between these three species are shaded in black, and similar amino acids in gray. Two domains conserved among members of the glycosyl hydrolase (GH) family 116 are indicated with dots (amino acids 151–455 in human GBA2) and dashes (amino acids 521–886 in human GBA2) above the sequences, Glyco_hydr_116N (Pfam PF12215) and DUF608 (Pfam PF04685), respectively. Active site residues are outlined with a box, the catalytic nucleophile (E527 in human GBA2) and acid/base catalyst (D677 in human GBA2).
In previous studies we established that human GBA2 is not only sensitive to inhibition by miglustat, but also by a distinct alkylated imino sugar, N-butyldeoxygalactonojirimycin (NB-DGJ; Fig. 1F),24,46 and by an inositol-derived epoxide, conduritol B epoxide (CBE; Fig. 1G). 46 Here, we demonstrate for the first time that the zebrafish homolog of GBA2 has similar enzymological properties and pharmacological sensitivities as its human counterpart, paving the way for further functional studies and pharmacological screens.
Materials and Methods
Animals and animal care
Zebrafish (AB strain) were maintained, bred, and developmentally staged as described. 55 All zebrafish studies were approved by the Dalhousie University Committee on Laboratory Animals (Protocol No. I15-18).
cDNA cloning
Whole adult zebrafish (≥1 year) were euthanized by immersion in 2 mg/mL 3-aminobenzoate, methanesulfonic acid (MS-222, Tricaine; Sigma). RNA was isolated from brain tissue using RNeasy™ Mini Kit (Qiagen), according to the manufacturer's instructions. Full-length first-strand cDNA was synthesized using Superscript™ II Reverse Transcriptase (Invitrogen) with Oligo(dT)12–18 primer mix, according to the manufacturer's instructions. gba2 cDNA was amplified by polymerase chain reaction (PCR) using Phusion Hot Start High-Fidelity DNA Polymerase (Thermo Scientific), forward primer gba2 attB1-F (5′
Cell culture
SH-SY5Y cells were obtained from the American Type Culture Collection (ATCC), and cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. SH-SY5Y cells were transfected with the cDNA encoding zebrafish gba2 (C-terminally FLAG-tagged) in the pCMV6-NeoR using TransIT-2020 transfection reagent (Mirus), according to manufacturer's instructions. Stably transfected cells were selected by subculturing the cells at a 1:10 ratio 24 h post-transfection in 300 μg/mL G418 sulfate (Geneticin; Life Technologies) for 2–3 weeks. Surviving colonies were picked by trypsinization, and expanded in the presence of G418. Mock-stably transfected cells were resistant to G418, but did not overexpress any form of GBA2.
Enzyme assays
Frozen cell pellets were thawed, resuspended in distilled water, diluted in 100 mM citric acid/200 mM disodium hydrogen phosphate buffer, pH 5.8, and homogenized by passing through QIAshredder centrifugation devices (17,000 g, 2 min; Qiagen). Whole adult zebrafish were euthanized by immersion in 3-aminobenzoate, rinsed, and stored at −80°C. Upon use, zebrafish were thawed, gutted, mechanically homogenized in three volumes of citric acid/phosphate buffer, pH 5.8 using a Tissue-Tearor (BioSpec Products), and centrifuged at 500 g for 10 min to remove debris. β-Glucosidase activity toward N-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-
β-Glucosidase activity toward 4-methylumbelliferyl-β-
Western blotting
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting onto polyvinylidene fluoride (PVDF) membrane was performed as described. 46 Primary antibodies were mouse anti-FLAG monoclonal antibody (clone M2; 1:1000; Sigma), and rabbit mAb anti-GAPDH (clone 14C10, 1:40,000; Cell Signaling). Blots were developed with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Immunofluorescence
For immunofluorescence staining, SH-SY5Y-derived cell lines were grown on glass coverslips, fixed with 4% paraformaldehyde for 30 min, washed with 5 mM NH4Cl, permeabilized in 0.3% Triton X-100 for 25 min, and blocked in 1% BSA for 30 min (all solutions in phosphate-buffered saline). Cells were incubated with rabbit polyclonal anti-DYK antibodies (1:2000; Cell Signaling), and a mouse anti-annexin II monoclonal antibody (1:1000, clone 5; BD Biosciences), and Alexa Fluor® 594- and Alexa Fluor® 488-labeled secondary antibodies (Invitrogen). Slides were mounted in Vectashield (Vector), and imaged with a Zeiss LSM510 confocal scanning fluorescence microscope.
Statistics
All quantitative date are presented as average + or +/− standard deviation. Enzyme activity data were tested for significance via one-way ANOVA and Dunnett's or Tukey's post hoc tests for multiple comparisons. IC50 values were calculated via nonlinear regression and fitting enzyme activity data to a sigmoidal dose–response (variable slope) function or a two-sites model. Statistical tests and curve fitting were performed with Prism 6.0f software (GraphPad); p < 0.05 was considered statistically significant.
Results
GBA2-like activity in zebrafish
To establish whether the zebrafish expresses a GlcCer-degrading enzyme activity with similar characteristics as human GBA2, we incubated zebrafish homogenates with the fluorescent GlcCer analog C12-NBD-GlcCer (Fig. 1C), which resulted in its conversion to C12-NBD-Cer (Fig. 3A, B). The zebrafish-mediated deglycosylation of C12-NBD-GlcCer was inhibited by 97% and 90% in the presence of 10 μM miglustat (Fig. 1E) and 300 μM NB-DGJ (Fig. 1F), respectively (Fig. 3A, B).
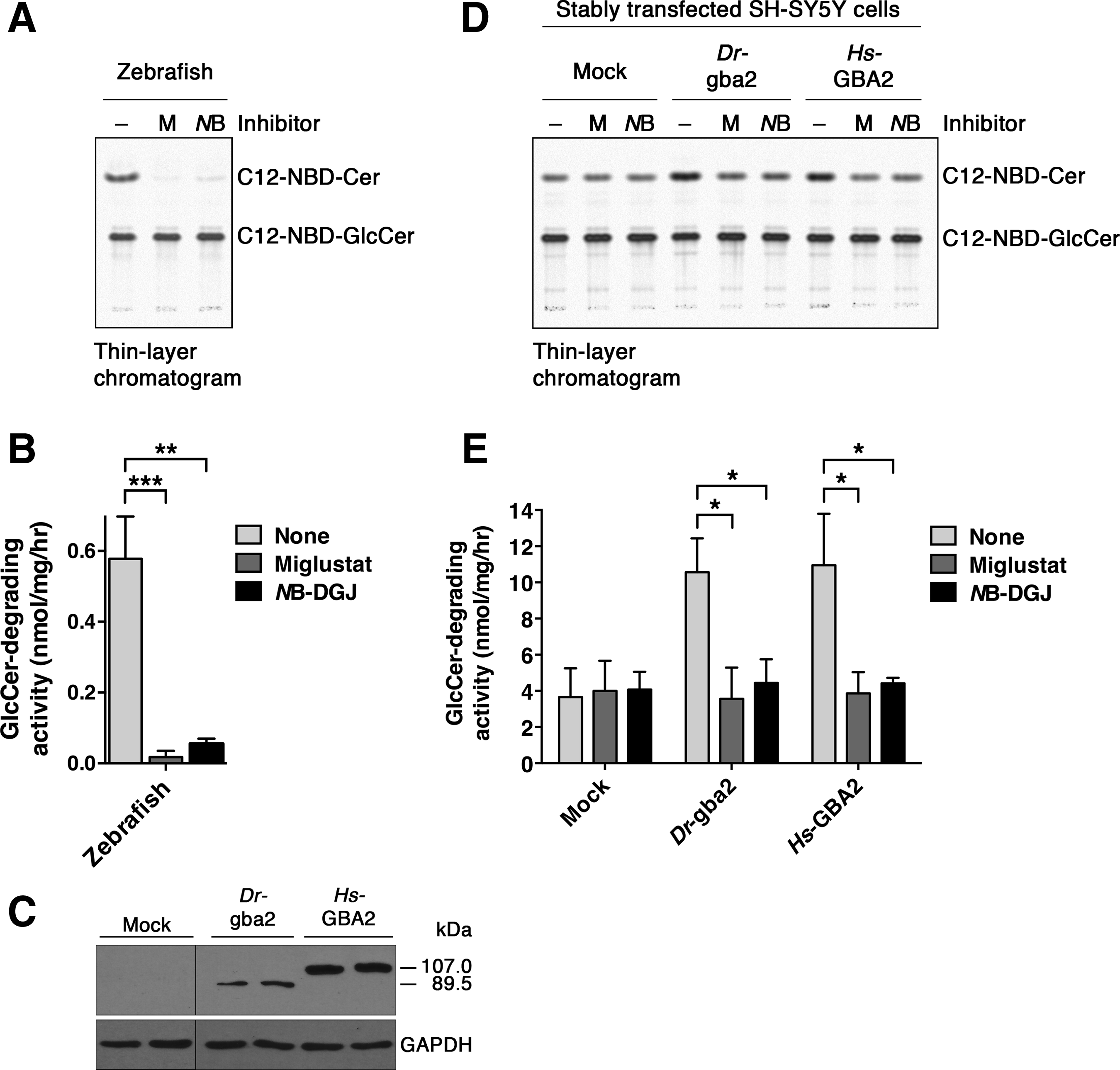
β-Glucosidase activity of zebrafish gba2 toward the fluorescent GlcCer analog C12-NBD-GlcCer.
Cloning and expression of zebrafish gba2
To determine whether zebrafish gba2 is responsible for the C12-NBD-GlcCer-degrading activity detected in zebrafish homogenate, we cloned the gba2 cDNA via a PCR-based strategy. We extracted a 3.6 kb cDNA that encodes the same amino acid sequence as NCBI Reference Sequences XP_005172389.1, XM_682560.6, and XP_687652.2, except for residues #229, 308, 366, and 437, where we found Val, Ser, Arg, and Val, respectively. Val229 and Val437 match corresponding residues in the human and mouse GBA2 sequences, while Ser308 and Arg366 align to Glu and Lys, respectively (Fig. 2). Zebrafish gba2 belongs to glycohydrolase family 116, 28 which is characterized by two large conserved domains, Glyco_hydr_116N (Pfam PF12215) 56 in the N-terminal half, and DUF608 (Pfam PF04685) in the C-terminal half of the protein (Fig. 2).
Because mutations in the GBA2 gene are associated with complex neurological/neurodegenerative pathologies,30–33 and because pharmacological inhibition of Gba2 in the brain is therapeutically effective in mouse models of neurodegenerative lysosomal storage diseases,50–52 we aimed to study GBA2 and its zebrafish homolog in a neuron-like cell line. For this purpose we chose SH-SY5Y cells, which were originally derived from a human neuroblastoma, 57 an early childhood tumor originating in the neural crest. SH-SY5Y cells resemble undifferentiated catecholaminergic neurons/neuroblasts, expressing proliferative cell nuclear antigen, and immature neuronal markers, including nestin.58,59 SY-SY5Y cells are widely used to model dopaminergic neurons in Parkinson's disease studies. Previously, we stably expressed human GBA2 in SH-SY5Y cells (hereafter Homo sapiens-GBA2, or Hs-GBA2 cells). 46 Now, having obtained the cDNA encoding zebrafish gba2, we stably transfected SH-SY5Y cells with this cDNA. The cells (hereafter Danio rerio-gba2, or Dr-gba2 cells) expressed zebrafish gba2 with an apparent molecular weight of 89.5 kDa, close to its nominal mass of 97.8 kDa (Fig. 3C).
Characterization of zebrafish gba2 enzyme activity
Having observed endogenous C12-NBD-GlcCer-degrading activity in zebrafish homogenate, we next assessed whether the zebrafish gba2 cDNA encodes an enzyme with a similar enzymatic activity. Thus, we compared the level of C12-NBD-GlcCer-degrading activity of Dr-gba2 cells with that of mock-transfected SH-SY5Y cells, and with that of SH-SY5Y cells stably transfected with the human GBA2 cDNA. Relative to mock-transfected cells, the C12-NBD-GlcCer-degrading activity was two times higher in Dr-gba2 cells and in Hs-GBA2 cells (Fig. 3A, B). The cDNA-mediated enzyme activities of Dr-gba2 and Hs-GBA2 cells were 12-fold higher than the endogenous activity of zebrafish homogenate. In the presence of miglustat or NB-DGJ, the enzyme activities of Dr-gba2 and Hs-GBA2 cells were reduced to the endogenous levels of SH-SY5Y cells (Fig. 3A, B).
We further evaluated the β-glucosidase activities of Dr-gba2 and Hs-GBA2 cells using the fluorogenic substrate 4-methylumbelliferyl-β-
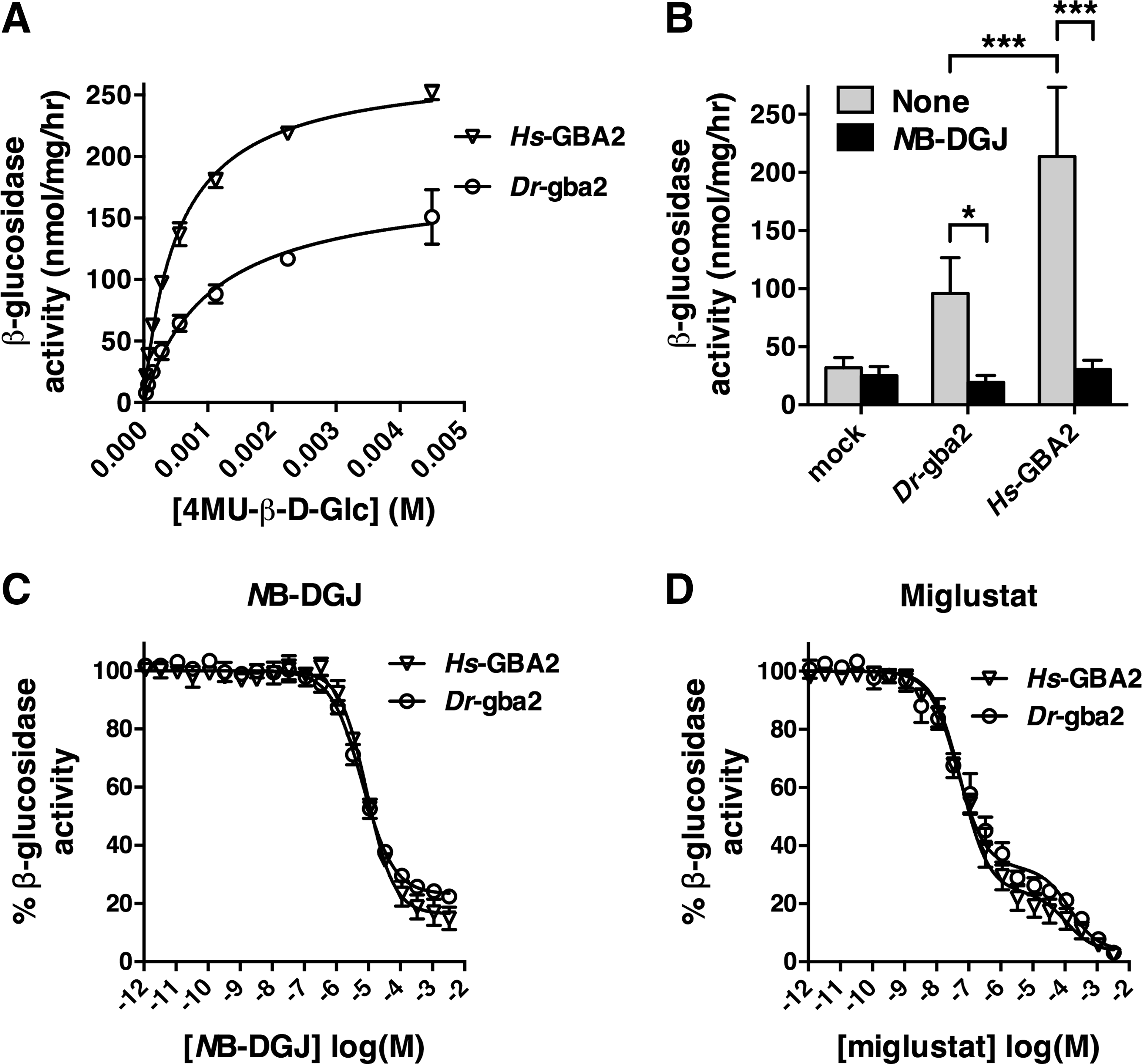
Activity of zebrafish gba2 toward the fluorogenic substrate 4MU-β-
Further, NB-DGJ reduced the enzyme activities of Dr-gba2 and Hs-GBA2 cells to control values (Fig. 4B). The IC50 values of NB-DGJ toward the enzyme activities of Dr-gba2 and Hs-GBA2 cells were comparable, 6.4 and 9.1 μM, respectively (Fig. 4C), as the corresponding IC50 values of miglustat, 44 nM for Dr-gba2 cells, and 58 nM for Hs-GBA2 cells (Fig. 4D). The residual β-glucosidase activity remaining at the highest NB-DGJ concentration (Fig. 4C) needs to be attributed to GBA, which is endogenously present in SY-SY5Y cells and is not affected by NB-DGJ. 46 Conversely, because miglustat inhibits both GBA2 and endogenous GBA, 46 the residual β-glucosidase activity remaining at the highest miglustat concentration is close to zero (Fig. 4D). Further, the biphasic shape of the miglustat titration curve (Fig. 4D) reflects the considerable difference between the miglustat sensitivities of GBA2 and GBA. 46
It is well established that the lysosomal enzyme GBA is irreversibly inhibited by CBE (Fig. 1G), which functions in this respect as a suicide substrate, forming a covalent bond with the enzyme during the course of the catalytic reaction mechanism.27,60 Characteristic of this type of mechanism-based enzyme inhibition is its time-dependent nature, as the apparent IC50 decreases over time. 61 Previously, we established that human GBA2 is also inactivated by CBE in a reaction mechanism-based manner, with an increasing proportion of the enzyme reacting with CBE over time, although with slower kinetics compared with GBA. 46 Therefore, to determine whether zebrafish gba2 is sensitive to inhibition by CBE in a comparable way, we preincubated zebrafish homogenate, along with Dr-gba2 and Hs-GBA2 cells with 2.5 mM CBE for 5 and 60 min, before the addition of the substrate. CBE reduced the C12-NBD-GlcCer-degrading activity in all samples after 5 min of preincubation, and reduced the enzyme activity further after 60 min of preincubation (Fig. 5). The level of inactivation was stronger in zebrafish homogenate and in mock-transfected SH-SY5Y cells, compared with the cells overexpressing zebrafish and human GBA2 (Fig. 5). Clearly, CBE had a similar impact on zebrafish gba2 and human GBA2, reducing the enzyme activity in a time-dependent manner.
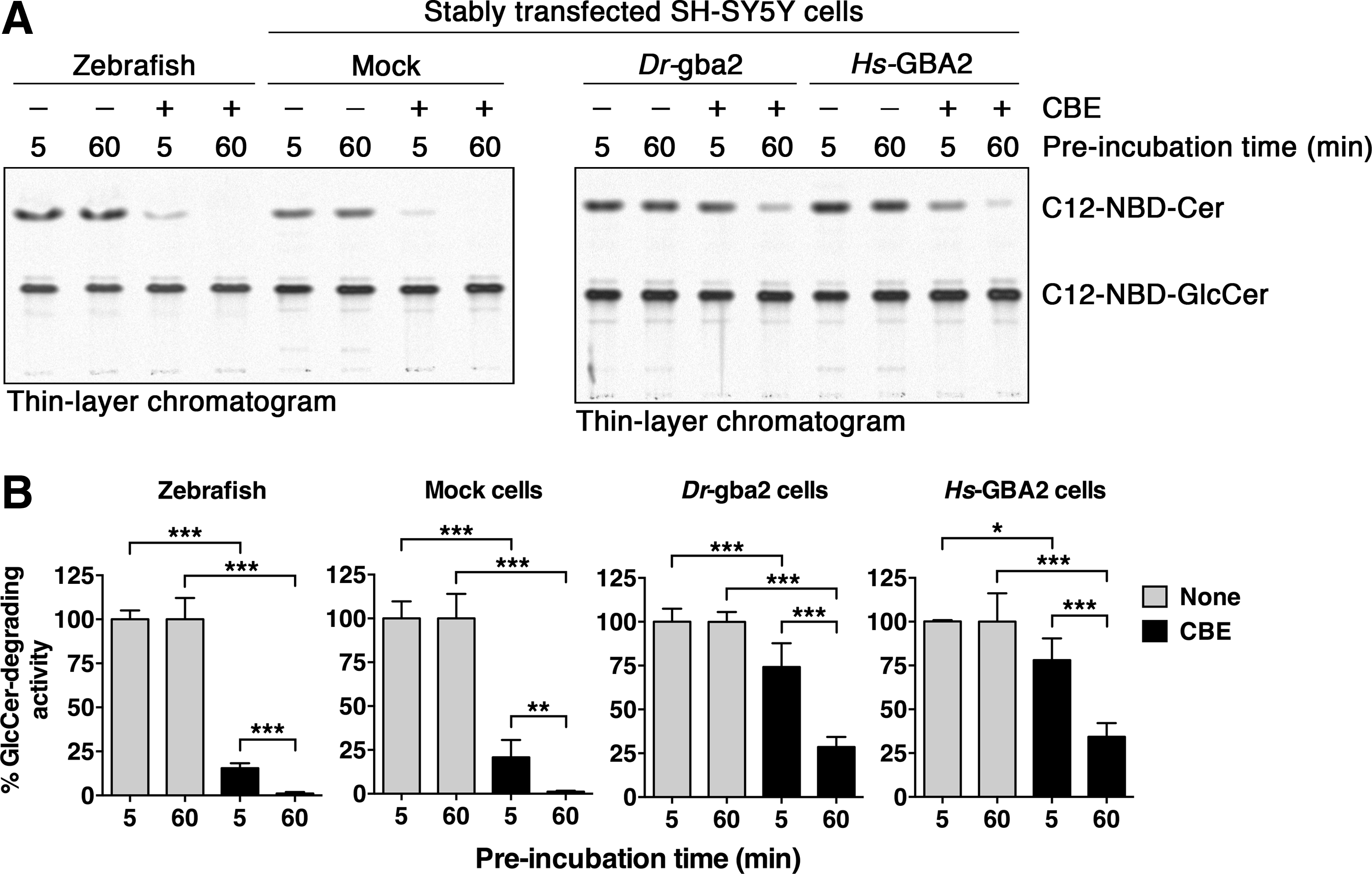
Inactivation of zebrafish gba2 by CBE.
Cellular localization of zebrafish gba2
Finally, we compared the cellular localization of zebrafish gba2 and human GBA2 by immunostaining and confocal fluorescence microscopy. Both homologs were distributed throughout the cell, and they had a similar distribution as annexin II, a phospholipid-binding protein associated with the plasma membrane62,63 (Fig. 6). In combination with biochemical evidence that human GBA2 is a membrane-associated protein,23,43,44 the observed immunofluorescence staining patterns suggest that human and zebrafish gba2 were both associated with the plasma membrane.
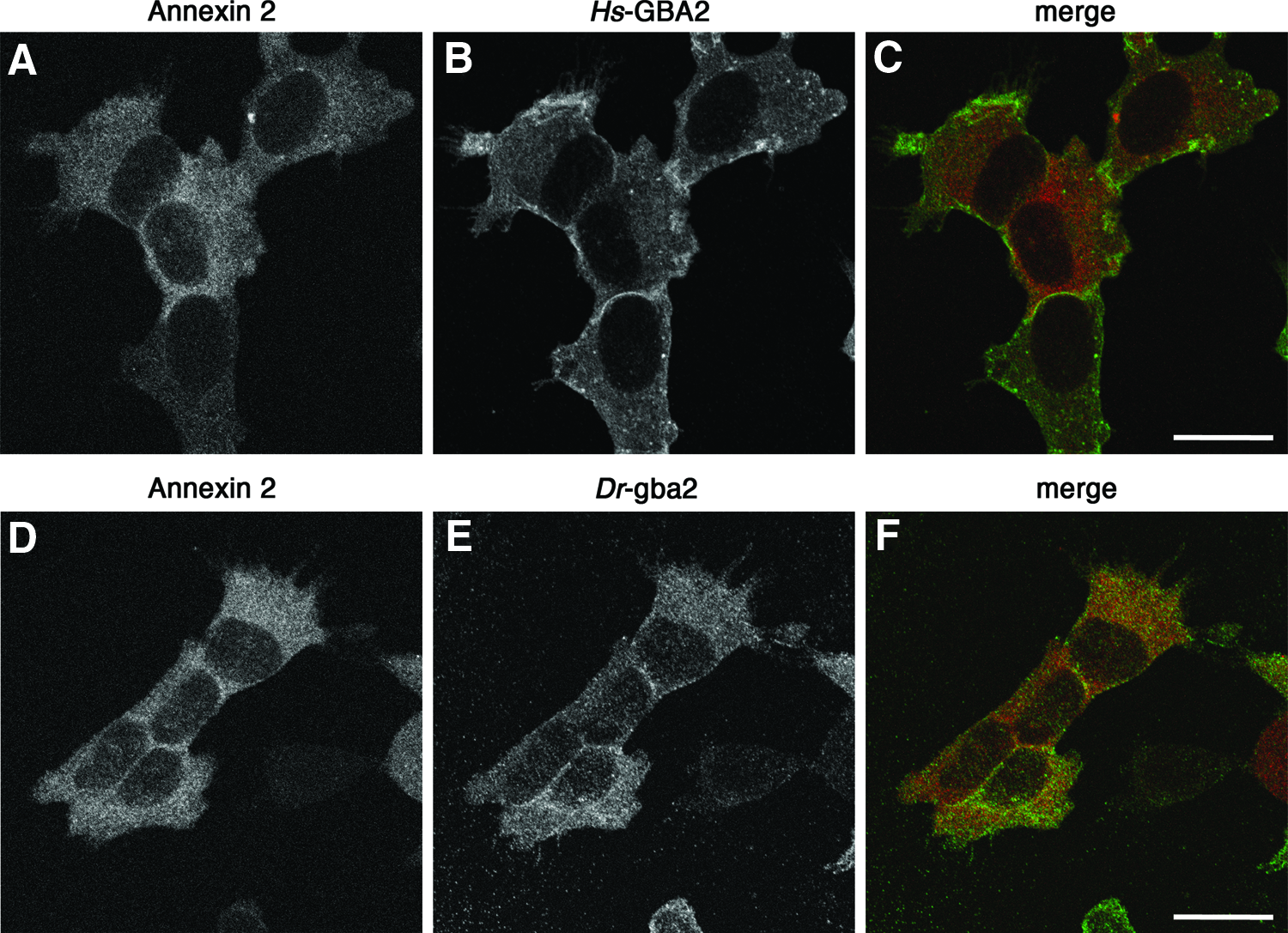
Immunolocalization of human and zebrafish GBA2. Stably transfected SH-SY5Y cells expressing FLAG-tagged human GBA2 (Hs-GBA2 cells)
Discussion
We have compared the endogenous GlcCer β-glucosidase activity in adult zebrafish with that of cells that are stably transfected with zebrafish gba2 cDNA. The endogenous zebrafish enzyme activity and that of the transfected cells were comparable in that they were almost completely inhibited by 10 μM miglustat and 300 μM NB-DGJ. These two enzyme activities were also susceptible to inactivation by CBE in a time-dependent manner, which is typical for the formation of covalent bonds with irreversible inhibitors. 61 The combined sensitivity to inhibition by low-concentration miglustat, NB-DGJ, and CBE characterizes the GlcCer-degrading activity in zebrafish as gba2, and differentiates it from its lysosomal counterpart GBA, which is refractory to NB-DGJ, 46 and relatively insensitive to miglustat.41,44,45
Zebrafish gba2 was similarly sensitive to inhibition by NB-DGJ and miglustat as human GBA2. The IC50 values of these inhibitors toward the fish enzyme were close to the IC50 values measured against the human enzyme. In comparison to previous studies,24,46 the IC50 value of NB-DGJ measured here toward human GBA2 was consistent. In contrast, the IC50 value of miglustat measured here toward human GBA2 (58 nM) was higher compared to previous measurements (5–20 nM).43,46 It is currently unclear what underlies this difference. Further, the majority of the β-glucosidase activity measured in mock-transfected SH-SY5Y cells using either C12-NBD-GlcCer or 4-methylumbelliferyl-β-
The relative levels of β-glucosidase activities measured in SH-SY5Y cells overexpressing zebrafish gba2 and human GBA2 differed, depending on the substrate used. Employing C12-NBD-GlcCer, Dr-gba2, and Hs-GBA2 cells had similar levels of enzyme activity, while using 4-methylumbelliferyl-β-
Taken together, our results demonstrate that the catalytic activity of zebrafish gba2 is comparable to its human homolog, with the possibility of a difference in substrate preference. This study provides therefore an underpinning for the use of zebrafish in screening gba2-targeting molecules. The latter hold promise as therapeutics in diseases that are caused by dysfunction of the endosomal-autophagic-lysosomal system. 64 These diseases include not only the rare heritable lysosomal storage disorders mentioned above, such as Sandhoff, NPC, and type 1 Gaucher disease, but also more common neurodegenerative disorders, including Alzheimer's and Parkinson's.65,66 In addition, this study provides a biochemical foundation for the use of zebrafish as a model for SPG46, an approach that may offer new insights in ataxias and paraplegias, conditions for which currently few therapies are available.
Further insights into the pathology of SPG46 may be obtained using genome-edited zebrafish that express mutant forms of gba2 corresponding to those found in SPG46 patients. To this end the CRISPR/Cas9 methodology appears attractive, as it can be efficiently applied in zebrafish. 67 This approach may also be used to generate zebrafish models of lysosomal glycosphingolipid storage disorders, to complement the recently described zebrafish model for type 2 Gaucher disease, developed with the TALEN methodology. 68 Zebrafish models of lysosomal glycosphingolipid storage disorders would be amenable for behavior- or survival-based drug screening studies using large numbers of GBA2-inhibiting compounds. Here, we have demonstrated that zebrafish gba2 is functionally and pharmacologically similar to human GBA2. This finding satisfies one of the conditions for the translation of gba2-targeting studies in zebrafish to human patients.
Footnotes
Acknowledgments
This study was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grant no. 386633-2011) (S.S. and A.C.v.d.S.), Scotia ScholarsOM Award from the Nova Scotia Health Research Foundation (S.S.), a Research Investigatorship from the IWK Foundation (Halifax) (A.C.v.d.S.), and the Peggy Davison Clinician Scientist Award from Cancer Care Nova Scotia (J.N.B.). We are also grateful to the Genome Canada IGNITE Project (Orphan Diseases: Identifying Genes and Novel Therapeutics to Enhance Treatment) for access to the Tecan Infinite M200 PRO plate reader.
Disclosure Statement
No competing financial interests exist.