
Abstract
Cystic fibrosis (CF) is the most common, life-limiting, inherited disease in Caucasian populations. Currently in the UK, there are approximately 8000 people with CF and 3 million healthy carriers. It is an autosomal recessively inherited condition that causes multisystem disease, particularly affecting the lungs and gastrointestinal tract. CF occurs in 1 in 2500 live births so with the advent of national newborn screening for this condition, GPs are likely to face more consultations relating to CF. The aim of this article is to give an overview of CF including its genetics, screening programme and diagnosis, clinical features, current management and future therapies.
The GP curriculum and cystic fibrosis
Cystic fibrosis (CF) is one of the key diseases listed in the knowledge base for
The cause of CF
CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome seven. This codes for the CFTR protein, which is a cyclic adenosine monophosphate-regulated chloride channel present in epithelial cell membranes. Absent or reduced CFTR function causes reduced secretion of chloride and consequently water from inside to outside the cell. Increased sodium resorption due to lack of negative feedback from the CFTR to the epithelial sodium channel compounds this problem. The result is dehydrated epithelial surface liquid and consequent damage to tubular structures: the airways, gut, exocrine pancreas, biliary tree and vas deferens.
Over 1600 CFTR gene mutations have been reported. The most common mutation worldwide is the deletion of phenylalanine at codon 508 (phe508del), which occurs in 75% of CF patients in the UK. The mutant CFTR protein produced cannot fold into its proper shape and is destroyed before it reaches the cell surface. Other mutations result in abnormal CFTR production or defective channel function. There is a spectrum of phenotypic severity due not only to this genetic heterogeneity but also to environmental factors and to variability in other ‘modifer’ genes whose protein products impact on organ systems affected by CF. The most severe mutations cause complete absence of functional CFTR and CF symptoms occur in infancy. Milder mutations reduce CFTR quantity or function and disease may only become apparent in later life with patients diagnosed with CF as adults.
Genetics
People with CF have two copies of the mutant CFTR gene. One in 25 people in the UK are carriers of CF. They are asymptomatic and have one normal and one mutant CFTR gene. It has been postulated that carriers are so prevalent possibly because in the past being a carrier led to a survival advantage following infections of typhoid and cholera as less chloride was lost in the diarrhoea.
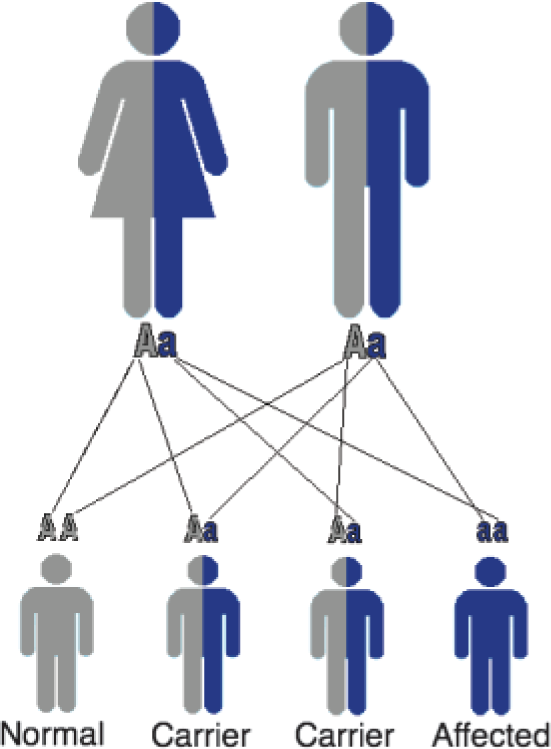
Carriers have a 50% chance of passing on the mutant CFTR gene to a child. If both parents are carriers, in each pregnancy, there is:
a 25% chance their child will have CF a 50% chance that their child will be a carrier a 25% chance that their child will be unaffected with two normal CFTR genes (Fig. 1)
Autosomal recessive inheritance.
Screening and diagnosis
Newborn blood spot screening
Screening for CF has been offered since 2007 for all newborns in the UK. Screening reduces delays in diagnosis and studies have demonstrated benefits in lung function, nutrition, cognitive function and survival for those affected by CF.
At day 5–8 after birth, the midwife carries out a heel prick on the newborn to spot blood onto a standard card. This is analysed for immunoreactive trypsinogen (IRT) produced by the pancreas, which is raised in CF. Raised IRT is not diagnostic of CF due to false positives so if the IRT level is greater than the 99.5th centile, DNA analysis on the blood spot is used to screen for four common CFTR gene mutations. Babies with two CFTR mutations are presumed to have a diagnosis of CF. If none or only one of the common DNA alterations is found initially, further testing is required to determine the likelihood of CF (Fig. 2).

CF screening algorithm.
It is important to realize that even if the screening test for CF is negative, there is a small chance that the child could still have CF. This is because there are false negatives for IRT and also if gene testing is done when IRT is high, the child may have two rare genes and therefore be screened negative while still having CF. Therefore, if a child presents later with features of CF and has been screened negative as a newborn, it is important not to exclude the diagnosis and to arrange appropriate investigations.
Screening follow-up
Following screening, when CF is suspected, the local CF clinic informs the primary care team and arranges to inform the parents at a home visit, sometimes in conjunction with a member of the primary care team. The parents are then offered an appointment the next day at the CF centre and advised that their child needs further tests to confirm the diagnosis.
Parents will also be told if their child is a carrier of CF and warned that occasionally there are rare alterations of the CF gene that are not recognized by the screening test so there is a small chance that their child will have CF. Therefore, if they have any concerns about their child's health, they should see their GP.
If the child has CF or is a carrier, this has implications for other family members. The family may wish to be referred to the regional genetics centre for genetic counselling to discuss further testing for family members (cascade screening), which can be performed on blood or saliva samples. Not all family members may wish to know whether they are a carrier and indeed, unwelcome issues of paternity have sometimes surfaced.
Antenatal screening
Antenatal screening is available to test whether a foetus has CF. This is considered if:
the parents already have a child with CF, both parents are known to be carriers of CF causing mutations or if the foetus has echogenic (very bright) bowel on antenatal ultrasound.
Prior to testing and preferably prior to pregnancy, parents should be seen by a genetic counsellor from the regional genetics service. Chorionic villus sampling (CVS) can be performed from week 10 of pregnancy onwards, allowing the parents to consider the option of an early termination if the foetus is affected. There is a risk of miscarriage of 1–2% with CVS. Amniocentesis can be performed later in pregnancy between 15 and 18 weeks and has a 1% risk of miscarriage. However, the results take several weeks so many couples prefer to opt for the earlier CVS.
Preimplantation genetic diagnosis
If both parents are carriers, embryos created using in vitro fertilization (IVF) techniques can be tested for CF at a very early stage of development and only embryos free from CF are then implanted into the mother's uterus. There are currently 12 centres in the UK licensed by the Human Fertilisation and Embryology Authority to carry out preimplantation genetic diagnosis.
Diagnostic tests
The measurement of sweat electrolyte levels, usually chloride, may help confirm the diagnosis of CF. Pads soaked in pilocarpine to stimulate sweat production are secured, usually to the lower arm. Then a small electrical current is passed through the pad to increase sweating further. After 5 minutes, the pads are removed and a piece of filter paper or plastic coil is secured over the stimulated area for 20–30 minutes to collect the sweat (Fig. 3). Parents and patients can be reassured that no needles are involved and the test is not painful, although some people experience a tingling sensation.

Macroduct system for sweat testing.
Patients with classic CF have a raised sweat chloride concentration of greater than 60 mmol/l. However, patients with borderline (30–60 mmol/l) or normal (less than 30 mmol/l) sweat chloride levels may still be diagnosed with non-classic CF if they also have:
a CF phenotype in at least one organ system one disease causing mutation on each CFTR gene CFTR dysfunction measured by nasal potential difference measurement
Clinical features
CF is a multisystem disease. Its clinical features and their approximate time of onset are shown in Fig. 4. Screening was only introduced nationally in 2007 and indeed can miss the diagnosis of CF, therefore it is important for GPs to be aware of the different clinical features of CF so that the diagnosis is made early as this can improve outcomes.
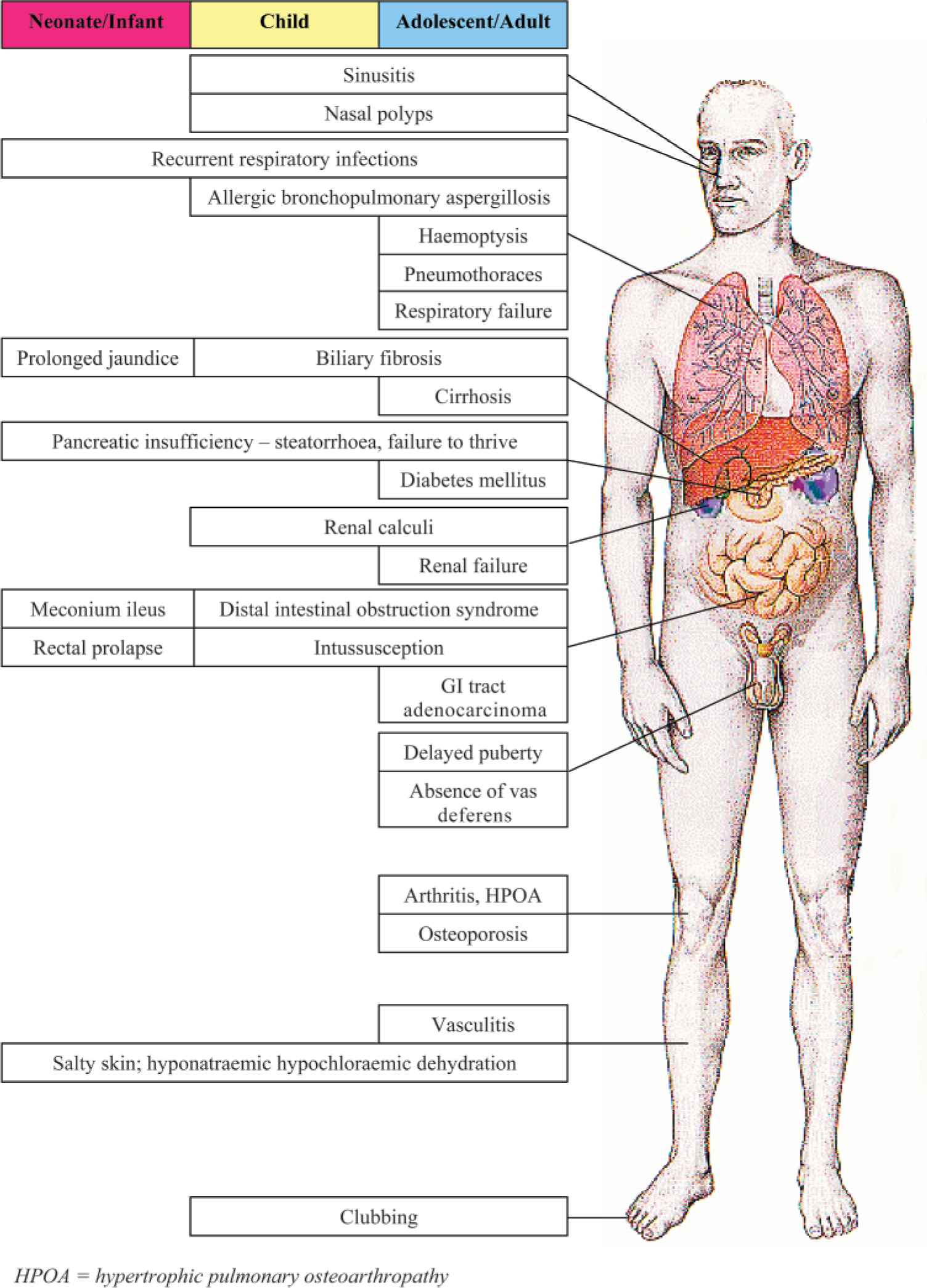
Clinical features of CF and approximate age of onset.
Pulmonary manifestations and management
In CF, the airway surface liquid bathing the cilia is dehydrated with a reduction in airway surface liquid height from 7 to 3 mm. The cilia are compressed and their function compromised so that mucociliary clearance of inhaled pathogens fails. In addition, the inflammatory response to pathogens is greatly increased. Chronic lung infection and inflammation are complicated by acute infective exacerbations and lead to progressive lung damage with bronchiectasis (irreversible dilatation of the airways) and eventually respiratory failure. Ultimately, this is the cause of death in most people with CF.
Airway clearance techniques
In early disease, the aims of management are to prevent infection and maintain good lung function. Regular aerobic exercise should be encouraged from diagnosis as increased shear stress across the cilia, due to increased airflow on exercise, increases airway surface liquid height through a CFTR-independent mechanism. Exercise may therefore be a fundamental disease-modifying therapy. Adults should aim for 20–30 minutes of aerobic exercise three to five times per week.
Chest physiotherapy is important to clear the sticky excess lung secretions. It should start from diagnosis as it helps to maintain respiratory function, even in those with mild chest involvement. Most patients do two sessions per day and up to four during exacerbations. Sessions may last 10–15 minutes if there are few secretions and up to an hour if there are many secretions. A physiotherapist will advise and teach the most appropriate methods for each patient. Methods include
postural drainage (positioning to aid drainage of secretions) percussion (of the chest with cupped hands to loosen secretions) active cycle of breathing techniques—breathing control (relaxed breathing), thoracic expansion exercises (deep breathing) and forced expiration techniques (huffing to move secretions) positive expiratory pressure (PEP) mask, which gives a small amount of back pressure during expiration to open the airways and clear the mucus oscillating PEP via a flutter valve or acapella high-frequency chest wall oscillation (vest therapy)—machine sends pulses of air into a vest causing vibrations that help move mucus
After education by the physiotherapist, the carer for the child performs the physiotherapy. From age 2, breathing exercises can be introduced as games. Most children start to perform part of their own physiotherapy by the age of 9 and all of it by the teenage years, although they may need help in an exacerbation.
Bacterial infections
Staphylococcus aureus is the main bacteria that infects the airway early in life. The Cystic Fibrosis Trust recommends that all children diagnosed with CF should be on continuous antistaphylococcal antibiotic prophylaxis with flucloxacillin. Pseudomonas aeruginosa is usually the main infecting bacteria by the late teenage years. Chronic infection with P. aeruginosa is associated with a poorer outcome. Eradication of new Pseudomonas should always be attempted using regimens such as 6 weeks of high-dose ciprofloxacin (750 mg twice daily for adults) plus 3 months of nebulized antipseudomonal antibiotics or intravenous antibiotics if oral and nebulized treatment fails. When Pseudomonas infection becomes chronic, regular nebulized antibiotics (e.g. tobramycin or colomycin) reduce bacterial load. This can be time-consuming but faster nebulizers, e.g. eflow and iNeb, are now available.
The lungs of CF patients are susceptible to infection with many environmental gram-negative pathogens. Of note, infection with organisms of the Burkholderia cepacia complex, in particular Burkholderia cenocepacia, is associated with a poorer prognosis. This was the first organism noted to be transmitted from one CF patient to another. Subsequently, cross-infection with other organisms has occurred. Therefore, all CF patients should be segregated by microbiological cohort, and individual segregation within those cohorts should be encouraged. As treatment of traditional CF pathogens has improved, new challenges have emerged such as the increase in highly antibiotic resistant non-tuberculous mycobacterial infection. Treatment of acute and chronic pulmonary infection in CF should be directed by a CF specialist centre with appropriate microbiological support.
Infective exacerbations
A patient with CF should be treated with antibiotics if they present with an infective exacerbation, indicated by any of:
Increased cough or change in character of cough Increased sputum production Increased shortness of breath Fever, decrease in appetite or weight loss Acute change in chest examination, e.g. new crackles or wheeze Decrease in forced expiratory volume in 1 second (FEV1) Acute change on chest X-ray
Antibiotics should be given even if a viral upper respiratory tract infection is suspected as they are associated with the onset of secondary bacterial infections.
Patients should liaise directly with the CF specialist centre when they are unwell. Otherwise, the GP should take a sputum sample or cough swab and liaise with the CF centre about the most appropriate antibiotic protocol. Two weeks of a broad-spectrum oral antibiotic may be appropriate in a mild exacerbation but if there is no improvement with oral antibiotics or the exacerbation is more severe, the patient should be admitted for intravenous antibiotics, preferably to the CF centre. Some patients then prefer to finish their course of intravenous (IV) medication at home, given appropriate teaching and community support.
Viral infections
Respiratory viral infections may precipitate up to 40% of chest exacerbations. Annual influenza immunization is recommended for CF patients aged over 6 months and for their immediate family. Respiratory syncytial virus (RSV) causes bronchiolitis in infants and trials of prophylaxis with palivizumab, a monoclonal antibody to RSV, are underway in CF. Montelukast, a leukotriene receptor antagonist, may be used for a month post-RSV bronchiolitis as there is some evidence that it reduces cough.
Airway hyperreactivity
Airway hyperreactivity occurs in about 25% of patients with CF and responds to beta 2 agonists and inhaled steroids.
Allergic bronchopulmonary aspergillosis
The fungus Aspergillus is commonly found in the sputum in CF and in 5–10% of patients, a hypersensitivity reaction to it leads to allergic bronchopulmonary aspergillosis (ABPA) with raised IgE, Aspergillus-specific IgE and eosinophils and positive Aspergillus precipitins. This presents with cough, wheeze, shortness of breath and sputum plugs with brown/black flecks. Sometimes there is fever, myalgia and malaise. Following diagnosis, treatment is with oral corticosteroids and an antifungal such as itraconazole to reduce the allergic response and prevent progression of bronchiectasis.
Chronic pulmonary treatment
Multiple therapies (Table 1) are available for the treatment of lung disease, which, in addition to airway clearance techniques, can be time-consuming for the patient, but help to slow progression of the disease.
Pulmonary therapies

End-stage lung disease
In end-stage lung disease, complications include haemoptysis, pneumothorax and respiratory failure. Haemoptysis may be fatal if severe and may be prevented with bronchial artery embolization or more rarely lobectomy. Pneumothoraces require a chest drain or pleurodesis if recurrent or persistent, although this may affect suitability for future transplantation.
Lung transplant
Lung transplant has the potential to extend and improve the quality of life of suitable candidates. It is currently considered when life expectancy is less than 2 years despite maximum medical therapy and, in adults, FEV1 is less than 30% predicted. Five year survival after transplant is less than 50% in children and slightly better in adults (50% at 6 years).
Non-pulmonary manifestations and management
Upper airway disease
Chronic rhinosinusitis is common in CF as mucus is thicker and may fill the sinuses and become infected with S. aureus or P. aeruginosa. Nasal polyps result from chronic inflammation in the nasal airways. In childhood, nasal polyps are nearly always due to CF and are an indication for a sweat test. By adulthood, 50% of CF patients have nasal polyps.
Chronic rhinosinusitis can be helped by regular use of a steroid nasal spray, regular saline nasal douches and with intermittent antibiotics. Nasal polyps can be reduced in size with intranasal steroid drops or sprays. Drops should be limited to 4 weeks use due to their higher systemic absorption. If medical treatment fails, referral to Ear nose and throat (ENT) for polypectomy or functional endoscopic sinus surgery is the next step. Unfortunately, 50% of polyps recur within 2 years.
Pancreatic insufficiency
Pancreatic insufficiency occurs in up to 90% of newborn CF patients, due to the viscous secretions plugging the pancreatic ducts and causing pancreatic injury. Pancreatic enzyme secretion is therefore inadequate and causes fat malabsorption, characterized by steatorrhoea (frequent, pale, oily, offensive stools), poor growth, abdominal pain, deficiency of fat-soluble vitamins (A, D, E and K) and essential fatty acids and occasionally rectal prolapse. Faecal pancreatic elastase is measured to confirm pancreatic insufficiency, then treatment should be commenced with fat-soluble vitamins and oral pancreatic enzyme replacement therapy, e.g. Creon, under the supervision of a CF specialist dietitian. Enzymes are required with each fat- or protein-containing meal or drink.
Those with pancreatic insufficiency require a high-calorie and high-fat diet. The dietitian will advise on optimizing nutrition, which may involve in infants, supplementing breast milk with fortified formula, adding fat to their solids and supplementing salt. In older children onwards, if the intake of calories is inadequate, high-calorie shakes or supplements or nocturnal feeds via a gastrostomy tube may be required. Up to 10% of people with CF do not have pancreatic insufficiency; however, they are at increased risk of symptomatic pancreatitis.
Gastrointestinal symptoms and management
About 10% of neonates with CF present with meconium ileus shortly after birth due to sticky meconium causing intestinal obstruction. This is treated with Gastrografin enemas or surgery. Distal intestinal obstruction syndrome (DIOS) may occur in adolescence or adulthood when the distal small intestine or caecum becomes obstructed by putty-like faecal material. Keeping well hydrated can reduce the risk of DIOS. Medical management with stool softeners, oral N-acetylcysteine or bowel cleansing agents such as Gastrografin or KleenPrep is usually successful and laparotomy is only occasionally required. Simple constipation is also common in CF if the energy dense diet is low in fibre and responds to dietary advice and laxatives.
Crohn's disease and coeliac disease are both more common in CF. The latter should be suspected if there is still malabsorption despite adequate pancreatic enzyme replacement. Appendicitis is also more common in CF but often presents in a more indolent fashion due to development of large appendix abscesses. Intussusception may be a cause of intermittent colicky pain and fever. Clostridium difficile infection is rare but tends to present with no diarrhoea and with mild abdominal pain but with high fevers and inflammatory markers and features of a pan-colitis. CF bowel issues should be evaluated in the first instance by the specialist CF centre as presentations are peculiar to CF and careful perioperative chest care is vital.
Gastro-oesophageal reflux is common in CF and may adversely affect lung function, possibly through aspiration. Treatment is with proton pump inhibitors, prokinetics and as a last resort surgical fundoplication.
Liver disease
In CF, the abnormal chloride transport makes bile more concentrated, which can lead to plugging of the intrahepatic bile ducts. This first leads to focal biliary cirrhosis, seen in about 25% of patients with CF, which is clinically insignificant. However, with time the plugging of the bile ducts becomes more diffuse and around 7% develop multilobular biliary cirrhosis, which may progress to portal hypertension, splenomegaly, oesophageal varices and liver failure in 3%. The first sign of CF in a neonate is sometimes obstructive jaundice but this usually resolves spontaneously during the first few months of life.
Early liver disease, picked up from an ultrasound scan or persistent abnormal liver function tests, benefits from ursodeoxycholic acid (UDCA). This is a hydrophilic bile acid that improves bile flow and protects against toxic hydrophobic bile acid. Used early at high doses of 20 mg/kg/day, there is evidence that it halts the progression of focal cirrhosis.
Liver transplantation is the only potentially curative treatment for liver cirrhosis and should be offered to those with progressive liver failure whose lung function is adequate. Five year survival rates are around 75%.
Diabetes
With loss of pancreatic function, there is a gradual onset of insulin deficiency, leading first to impaired glucose tolerance then progressing to frank diabetes mellitus. The UK Cystic Fibrosis Trust suggests an oral glucose tolerance test annually after the age of 12 years. The average age of onset of diabetes is 18–21 years and by the age of 25, 30% of CF patients have diabetes. Initially for some, oral agents that stimulate insulin secretion, such as sulphonylureas, may be sufficient but in general, insulin is the treatment of choice. Patients need to maintain a high-calorie diet so diabetic care is undertaken by endocrinologists and dietitians with a special interest in CF. Attending courses such as DAFNE (dose adjustment for normal eating) is helpful. An annual diabetic review is advised to screen for any micro- or macrovascular complications.
Fertility
Only about 1% of males with CF are fertile. Most are born with absent vas deferens or less commonly atresia of the vas deferens. However, spermatogenesis is normal so IVF with aspirated sperm is an option.
Women are generally of normal fertility but should be given the option of genetic counselling before becoming pregnant. Pregnancy is not advised for women with an FEV1 less than 50% predicted nor for those with portal hypertension or poorly controlled diabetes. Adult CF centres should have specialist high- risk antenatal care pathways arranged with their local obstetric service. Female infertility occurs in up to 20% of women with CF due to thick cervical mucus or secondary amenorrhoea due to malabsorption or chronic disease. IVF may therefore be necessary due to infertility or for preimplantation genetic diagnosis.
Sweat gland
Excess sweat is lost from sweat glands (as well as from the gastrointestinal tract) and can lead to a metabolic alkalosis with hypochloraemia, hyponatraemia and hypokalaemia (pseudo-Bartter syndrome). The patient may need volume replacement with normal saline and potassium supplements. Furthermore, in hot weather or with excess sweating, the patient may need salty snacks or salt tablets and adequate fluids.
Musculoskeletal
Low bone mineral density occurs in about 25% of young adults with CF due to a range of factors including poor absorption of calcium and vitamin D, use of corticosteroids and raised proinflammatory cytokines caused by recurrent chest infections. Dual energy X-ray absorptiometry (DEXA) scanning at age 10 years and then every 1–3 years is recommended to check for low bone mineral density. Prevention involves adequate dietary calcium and vitamin D intake with the dietitian's input, weight-bearing exercise and avoiding smoking and excess alcohol. Bisphosphonates are useful in osteoporosis to inhibit bone resorption.
Episodic arthritis affects up to 8% of adults with CF, causing an acute onset mono- or polyarthritis, usually affecting the large joints in an asymmetric pattern. Episodes usually last up to 10 days and settle spontaneously. There is no link with the degree of pulmonary disease. Treatment is usually symptomatic with non-steroidal anti-inflammatory drugs (NSAIDs) and occasionally oral steroids. A small subgroup develops more chronic erosive polyarthritis and disease-modifying antirheumatic drugs (DMARDs) may be considered.
Hypertrophic pulmonary osteoarthropathy (HPOA), characterized by clubbing (Fig. 5) and chronic periostitis of the long bones, occurs in up to 7% of those with CF, usually after 10 years of age. It causes polyarthritis of the knees, ankles and wrists with tenderness of the ends of the long bones. It worsens with pulmonary exacerbations so treatment involves treating the underlying lung disease as well as NSAIDs, occasionally corticosteroids and, in resistant cases, intravenous pamidronate.

Clubbing. The picture is accompanied by a poem by the patient, Scott Petersen, illustrating the impact of CF on his life.
Organization of care and the role of the GP
If a child presents with any of the above combination of symptoms, referral to a paediatrician is appropriate who will organize the diagnostic tests, after discussion with the family. Following diagnosis, all CF patients are seen at a CF centre with follow-up appointments at least every 3 months. At the annual review, the individual's condition and treatment are assessed and optimized by the multidisciplinary team: specialist CF consultant, nurse, physiotherapist, dietitian, psychologist and social worker. Transition from paediatric to adult services can be a difficult time and specific transition clinics have been introduced to try to smooth the handover of care.
Patients with CF and their families consult the GP in two different ways: as ‘lay’ patients about non-CF problems and as ‘expert patients’ for help with CF-related problems. They soon build up knowledge of the management of common CF problems and may often contact the CF centre directly for advice, bypassing the GP. If the patient does see the GP about CF-related problems, the local CF centre will have guidelines, e.g. for preferred antibiotics and dosage, and would encourage GPs to contact them for further advice on management decisions.
The most common complaint from patients about primary care is about prescriptions, including inadequate supplies of, for example, essential enzyme replacement therapy. An efficient repeat prescription service, which ensures that adequate supplies of medication are supplied, is valued by patients. Funding for expensive drugs should be available from the Primary Care Trust via shared care agreements with the CF specialist centre. GPs can also help encourage adherence to treatment while understanding that day-to-day treatment is time-consuming. For example, one study found the average length of time spent on treatment activities was 108 minutes/day.
Often the tertiary specialist centre is a long way from the patient's home so the GP can provide the patient and family with details of local sources of support, e.g. carers' groups. Families can also be directed towards appropriate sources of help and information as in Box 1. The GP can provide valuable support during difficult times such as at diagnosis and during the transition from paediatric to adult care. Supporting home intravenous therapy also helps those patients who wish to minimize their time spent in hospital. In addition, the GP can highlight entitlements such as The Blue Badge Scheme that enables those with mobility problems to park close to where they need to go. The GP should also provide support for the whole family and practical help, such as ensuring routine appointments for family members do not clash with the CF sufferer's daily routine. All family members should be offered annual influenza vaccinations. Finally, towards the end of life, GP input and liaison with community palliative care teams are invaluable to support those who wish to die at home.
Useful information for GPs, patients and families.
Cystic Fibrosis Trust: www.cftrust.org.uk—UK charity with information for patients and doctors Cystic Fibrosis Medicine: www.cysticfibrosismedicine.com—clinical information for patients and doctors from the Leeds Regional Adult and Paediatric Cystic Fibrosis Units UK Newborn Screening Programme Centre: http://newbornbloodspot.screening.nhs.uk-patient information leaflets on screening, suspected CF and carriers of CF Genetic Interest Group: www.gig.org.uk—support for patients affected with genetic disorders Human Fertilization and Embryology Authority: www.hfea.gov.uk—information on UK fertility clinics UK Cystic Fibrosis Gene Therapy Consortium: www.cfigenetherapy.org.uk—information on gene therapy research Cystic Fibrosis Foundation: www.cff.org—US foundation with patient information Breathing Room: www.thebreathingroom.org—community of adults with CF (US based)
www.cysticfibrosis.com—patient forum
Future therapies
Mutation-specific therapies
Class I mutations of the CFTR gene, affecting about 10% of CF patients, have a premature stop codon in the messenger ribonucleic acid (mRNA) so an abnormally short CFTR protein is produced or no protein at all. A molecule, PTC124, which can allow normal transcription is currently in clinical trials.
With Class II mutations of the CFTR gene, such as the commonest mutation Phe508del, the protein is misfolded, causing it to be degraded within the cell. Small molecule correctors of misfolded CFTR that allow it to reach the cell membrane are being sought by high throughput screening and one, VX-809, is in Phase II trials.
With Class III and IV mutations, the CFTR protein reaches the membrane but does not work properly. ‘Potentiators’ improve the function of the mutant CFTR. For example, VX-770, which acts on the CFTR protein to open chloride channels, is currently in Phase III clinical trials. Phosphodiesterase inhibitors such as aminophylline are also effective in increasing CFTR chloride conductance.
Ion transport modulation
Drugs are being developed to modulate the abnormal ion transport that occurs in CF. Parion 55-02, currently in clinical trials, inhibits the excessive sodium reabsorption of the uninhibited epithelial sodium channel. Other drugs in development act to improve chloride secretion via CFTR-independent mechanisms, e.g. Moli 1902, INS 37217, SPI-8811 and denufosol. Bronchitol acts similarly to hypertonic saline to increase airway surface liquid height and mucociliary clearance.
Anti-inflammatory therapies
Phase II trials of oral N-acetylcysteine, inhaled glutathione and oral sildenafil are underway in search of better tolerated anti-inflammatory therapies.
Inhaled antibiotics
Several Phase II and III studies are underway to produce a wider range of nebulized antibiotics and also antibiotics delivered by inhaler that may be less time-consuming and better tolerated by patients.
Gene therapy
Gene therapy in CF aims to insert a healthy copy of the CFTR gene into the affected cells. The gene can be transferred into cells using a virus vector or by coating the gene with liposomes that adhere to the surface of the cell and encourage the gene to enter. While it is already technically possible to transfer the CFTR gene into airway cells in humans, two major challenges need to be addressed for gene therapy to provide clinical benefit to patients. The first is to improve the efficiency of the gene transfer into cells. The second is to make gene expression last longer than the 1–2 weeks currently achieved.
Prognosis
In the 1930s, when CF was first recognized, most babies with CF were only expected to live a few months. With improving treatment, life expectancy has continually improved. Currently, over half of those affected should live beyond the age of 35 years and it is predicted that children born with CF today should have a median life expectancy of 50 years even if there were no further advances in treatment.
Key points
CF is the commonest autosomal recessive disease in the UK with 1 in 25 people being carriers CF is caused by a mutation in the CFTR gene. As the CFTR protein is expressed throughout the body, CF causes a multisystem disease, particularly affecting the respiratory and digestive tracts. National neonatal screening for CF was introduced in 2007 Aggressive treatment with antibiotics is key in preventing lung damage. Sputum cultures are important in guiding treatment and antibiotics should not be delayed by lack of systemic features, e.g. fever, during an exacerbation. Airway clearance techniques are important for all CF patients, even if there are no clinical symptoms CF care is best delivered by specialist CF centres