
Abstract
Recent in vitro cell-free studies have shown that cytochrome c release from mitochondria is a critical step in the apoptotic process. The present study examined the expression of cytochrome c protein after transient focal cerebral ischemia in rats, in which apoptosis was assumed to contribute to the expansion of the ischemic lesion. In situ labeling of DNA breaks in frozen sections after 90 minutes of middle cerebral artery (MCA) occlusion showed a significant number of striatal and cortical neurons, which were maximized at 24 hours after ischemia, exhibiting chromatin condensation, nuclear segmentation, and apoptotic bodies. Cytosolic localization of cytochrome c was detected immunohistochemically in the ischemic area as early as 4 hours after 90 minutes of MCA occlusion. Western blot analysis of the cytosolic fraction revealed a strong single 15-kDa band, characteristic of cytochrome c, only in the samples from the ischemic hemisphere. Western blot analysis of the mitochondrial fraction showed a significant amount of mitochondrial cytochrome c in nonischemic brain, which was decreased in ischemic brain 24 hours after ischemia. These results provide the first evidence that cytochrome c is being released from mitochondria to the cytosol after transient focal ischemia. Although further evaluation is necessary to elucidate its correlation with DNA fragmentation, our results suggest the possibility that cytochrome c release may play a role in DNA-damaged neuronal cell death after transient focal cerebral ischemia in rats.
Cytochrome c, a water-soluble peripheral membrane protein of the mitochondria, is known to be an essential component of the mitochondrial respiratory chain (Boyer et al., 1977). Its function is to transport electrons from the coenzyme QH2–cytochrome c reductase complex to the cytochrome c oxidase complex in the electron transport chain. Recently, it has been suggested that cytochrome c participates in apoptosis in a cell-free system (Liu et al., 1996). Mitochondria are assumed to be involved in apoptosis by releasing cytochrome c to the cytoplasm where it activates caspase 3 (CPP32), a molecule of the interleukin-1β-converting enzyme (ICE) family, which has been shown to then trigger apoptosis (Liu et al., 1996). Consistent with this finding, microinjection of cytochrome c has been shown to result in apoptosis (Li et al., 1997). More recent evidence shows that the expression of Bcl-2 acts to inhibit cytochrome c translocation, thereby blocking CPP32 activation and the apoptotic process, not only in a cell-free system but also in intact cells (Kluck et al., 1997; Yang et al., 1997). However, the involvement of cytochrome c in apoptosis is totally unknown in vivo. In the present study, we sought to clarify this point by examining the localization of cytochrome c protein after focal cerebral ischemia in rats, in which apoptosis is known to be involved (Linnik et al., 1993; Tominaga et al., 1993; Li et al., 1995a,b,d; Charriaut-Marlangue et al., 1996; Choi, 1996; Chopp and Li, 1996; Gillardon et al., 1996; Asahi et al., 1997) and which contributes to the expansion of the lesion (Du et al., 1996). Also in the present study, in situ labeling of DNA breaks after 90 minutes of middle cerebral artery (MCA) occlusion showed a significant number of striatal and cortical neurons, which were maximized at 24 hours after ischemia. Using immunohistochemistry and Western blotting on the same ischemia/reperfusion model, we examined the subcellular distribution of cytochrome c protein expression after transient focal ischemia. The results show that cytoplasmic expression of cytochrome c was detected only in the ischemic brain as early as 4 hours. Mitochondrial cytochrome c of the ischemic brain was decreased at 24 hours after ischemia, compared with that of the nonischemic brain. However, there was no alteration of the subcellular distribution of cytochrome oxidase, a mitochondrial respiratory chain protein. These data provide the first evidence that cytochrome c is being released from the mitochondria to the cytoplasm after focal cerebral ischemia. Although further evaluation is necessary to elucidate its correlation with ischemic apoptosis, our results suggest the possibility that increased cytosolic cytochrome c may play some role in DNA-damaged neuronal cell death after transient focal cerebral ischemia.
MATERIALS AND METHODS
Focal cerebral ischemia
Adult male Sprague-Dawley rats (250 to 280 g) were subjected to transient focal ischemia by intraluminal MCA blockade with a nylon suture as described (Yang et al., 1994). The rats were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases (Table 1), samples for analysis being taken immediately after cannulation, 10 minutes after occlusion, and 10 minutes after reperfusion. After a midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. A 22.0-mm 3-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 90 minutes of MCA occlusion, blood flow was restored by the withdrawal of the nylon suture.
Physiologic variables during and after ischemia
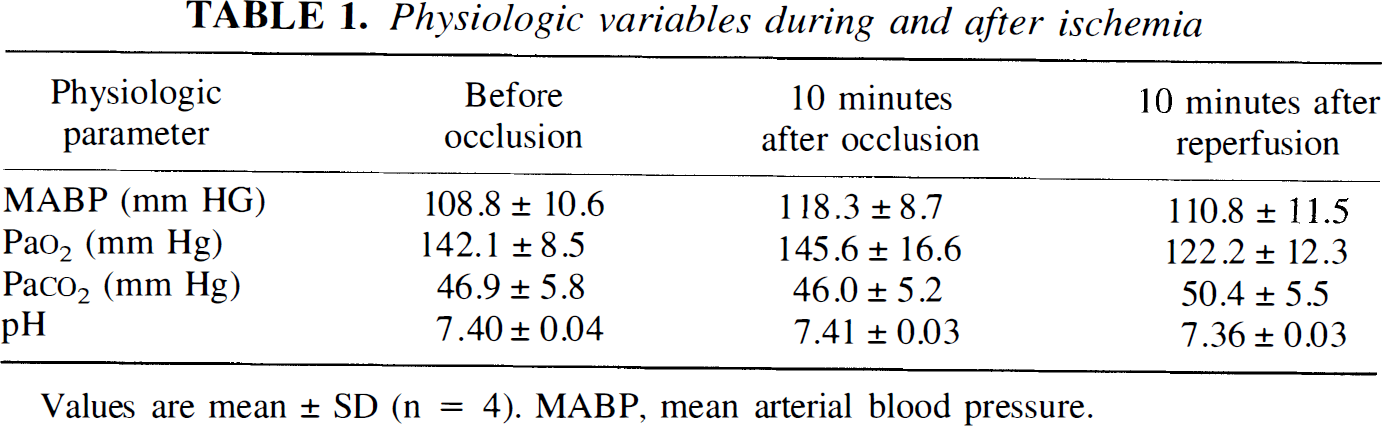
Values are mean ± SD (n = 4). MABP, mean arterial blood pressure.
In situ labeling of DNA fragmentation
At 4, 24, or 72 hours after 90 minutes of MCA occlusion (n = 4, each), the brains were removed and rapidly frozen. They were sectioned with a cryostat into a thickness of 20 µm from the anterior side to the posterior side. Brain sections at the level of the caudate putamen that showed typical infarction were stained using an in situ technique to detect the DNA free 3'-OH ends by terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end labeling (TUNEL) reaction as described (Kondo et al., 1997). Briefly, frozen brain sections were fixed for 30 minutes in 3.7% formaldehyde in 0.1 mol/L phosphate-buffered saline (PBS), pH 7.4. The slides were placed in 1 × terminal deoxynucleotidyl transferase (TdT) buffer (Life Technologies, Gaithersburg, MD, U.S.A.) for 15 minutes, followed by reaction with TdT enzyme (Life Technologies) and biotinylated 16-dUTP (Boehringer Mannheim, Indianapolis, IN, U.S.A.) at 37°C for 60 minutes. The slides were then washed in 2×SSC (150 mmol/L sodium chloride, 15 mmol/L sodium citrate, pH 7.4) for 15 minutes, followed by a washing in PBS two times for 15 minutes. Avidin-biotin horseradish peroxidase solution (ABC kit, Vector Laboratories, Burlingame, CA, U.S.A.) was applied to the sections for 30 minutes, then the slides were washed for 15 minutes with 0.175 mol/L sodium acetate. Staining was visualized using 0.025% diaminobenzidine and 0.075% H2O2 in PBS with 0.4 mg/mL nickel sulfate. The slides were rinsed with water, stained with methyl green for 10 minutes, dehydrated, and mounted.
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin and subsequently with 4% formaldehyde in 0.1 mol/L PBS (pH 7.4) after 4, 24, and 72 hours reperfusion after ischemia. Brains were removed, postfixed for 12 hours, sectioned at 50 µm on a vibratome, and processed for immunohistochemistry. The sections were reacted with mouse monoclonal antibody against rat cytochrome c (7H8.2C12, Pharmingen, San Diego, CA, U.S.A.) at a dilution of 1:500. Immunohistochemistry was performed using the avidin-biotin technique, and then the nuclei were counterstained with hematoxylin solution for 10 minutes. As a negative control, sections were incubated in the absence of primary antibodies. For histologic assessment, alternate slices from each brain section were stained with cresyl violet.
Western blot analysis
Protein extraction of both the mitochondrial and cytosolic fraction was performed as described (Liu et al., 1996), with modifications. Approximately 900 mg of tissue of both ischemic and nonischemic hemispheres, or alternatively, 150 mg of ischemic brain and nonischemic brain from the corresponding contralateral brain, were cut into pieces after 4, 24, and 72 hours reperfusion, homogenized gently by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ, U.S.A.) in 7 volumes of cold suspension buffer (20 mmol/L HEPES-KOH [pH 7.5], 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride, 2 µg/mL aprotinin, 10 µg/mL leupeptin, 5 µg/mL pepstatin, and 12.5 µg/mL N-acetyl-leu-leu-norleucinal). The homogenates were first centrifuged at 750g at 4°C, and then at 8,000g for 20 minutes at 4°C; 8,000g pellets were used as the mitochondrial fraction. The supernatant was further centrifuged at 100,000g for 60 minutes at 4°C. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, U.S.A.). The primary antibodies were either a 1:1,000 dilution of 7H8.2C12 rat cytochrome c monoclonal (Pharmingen) or 1 µg/mL of 20E8C12 cytochrome oxidase subunit IV monoclonal (Molecular Probes, Eugene, OR, U.S.A.). Western blots were performed with horseradish peroxidase-conjugated antimouse immunoglobulin G using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, England). Rat heart cytochrome c (Sigma, St. Louis, MO, U.S.A.) was used as a positive control. Densitometric analysis was made on the results of the mitochondrial fraction from both ischemic and nonischemic brain (n = 2 each). The film was scanned by GS-700 imaging densitometer (Bio-Rad), and the results were quantified using the multi-analyst software program (Bio-Rad).
RESULTS
Physiologic data
All rats survived the transient MCA occlusion. There were no significant differences in blood pressure, blood gas, and pH among groups (Table 1).
DNA fragmentation was detected after transient MCA occlusion
To elucidate whether DNA fragmentation is induced in 90 minutes of MCA occlusion and subsequent reperfusion, we examined in situ labeling of DNA breaks in the infarcted brain sections (Fig. 1). TUNEL staining does not label normal neuronal cells in the noninfarcted area (data not shown). In contrast, two different patterns of staining are observed in the neuronal cells in the infarcted area (Fig. 2B—D). Some neuronal cells in the infarcted area are densely labeled in their nuclei, using TUNEL staining, accompanied by small particles around the nuclei that resemble apoptotic bodies (arrows in Fig. 2B and C). These cells are compatible with those in the apoptotic cell death process as described previously (Charriaut-Marlangue and Ben-Ari, 1995; Li et al., 1995a,b; Kondo et al., 1997). Besides these typical TUNEL-positive cells, slightly TUNEL-stained cells are also observed (Fig. 2D, arrowheads). These cells show diffuse nuclear and cytoplasmic TUNEL staining, which is consistent with necrotic cells, as reported (Charriaut-Marlangue and Ben-Ari, 1995; Li et al., 1995a,b; Kondo et al., 1997). Only the densely labeled cells (Fig. 2B and C) are considered to be TUNEL-positive in this study.
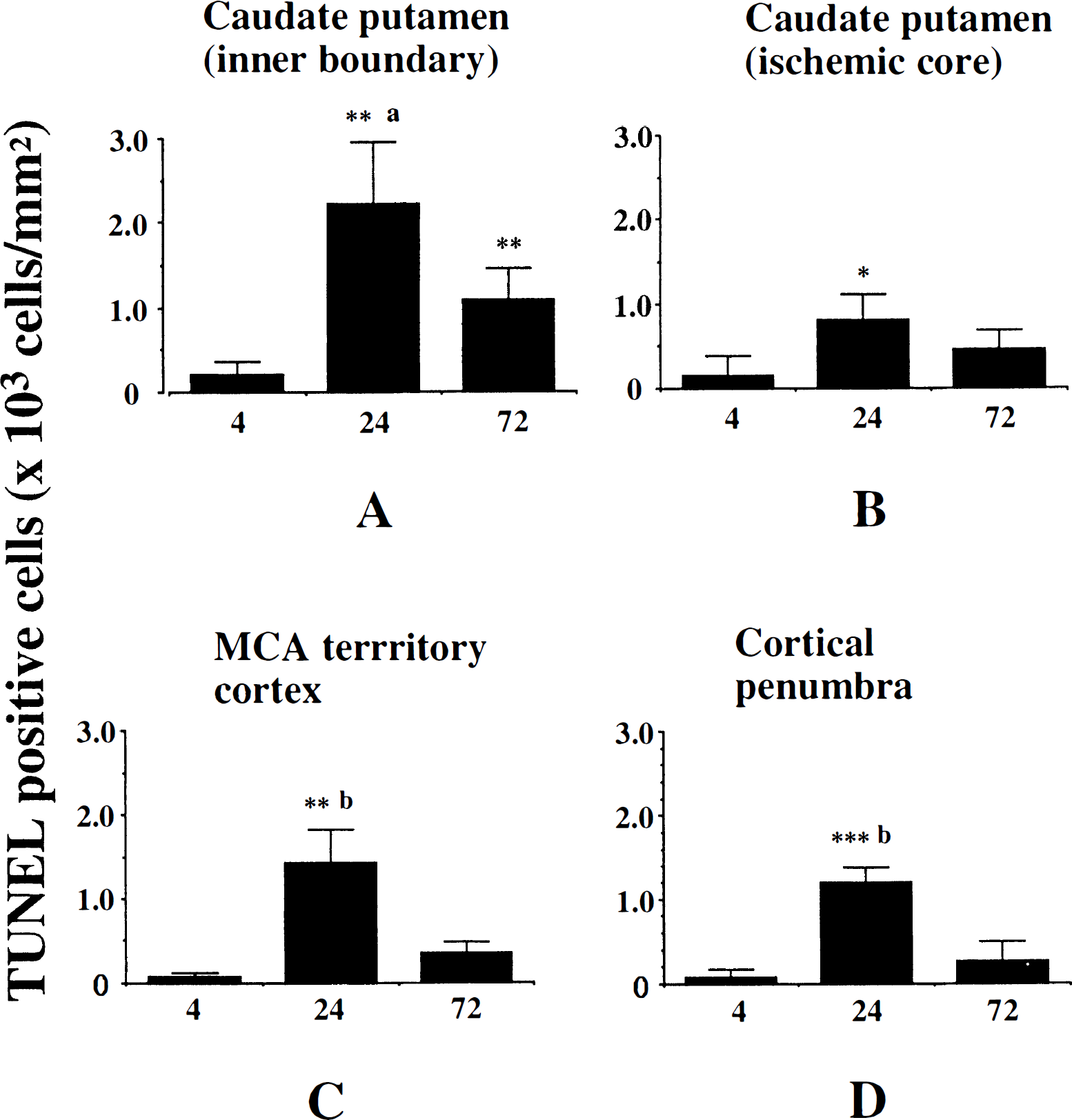
Temporal change of the number of TUNEL-positive cells in various brain regions.
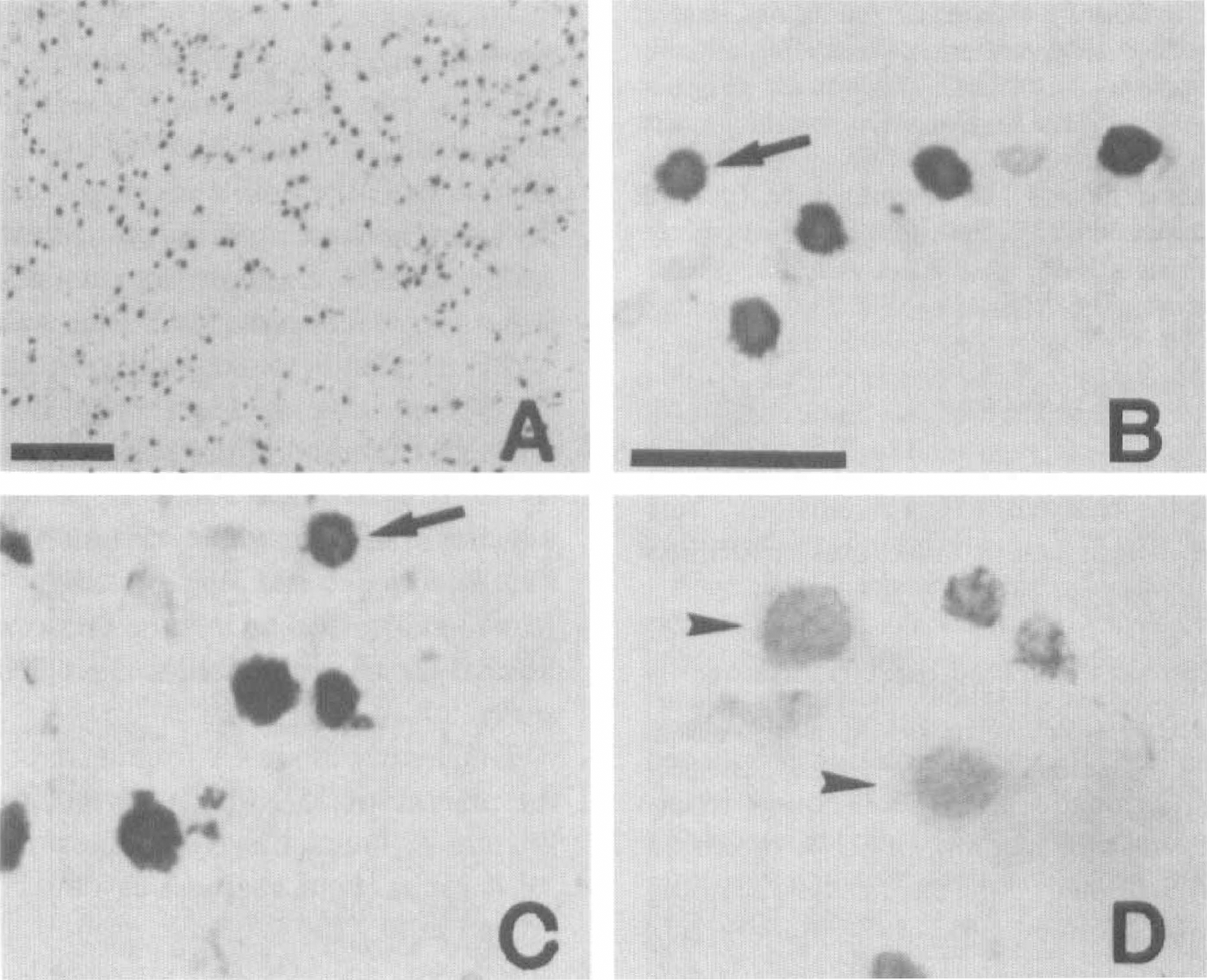
Photomicrographs showing TUNEL-positive cells in the inner boundary of the caudate putamen (
To quantify the DNA-fragmented cells after ischemia, the number of TUNEL-positive cells was counted at 4, 24, and 72 hours after 90 minutes of MCA occlusion (n = 4 each). Four regions, as defined previously (Murakami et al., 1997a), were used to assess the anatomic distribution of TUNEL-positive cells (inner boundary of caudate putamen, ischemic core of caudate putamen-lateral caudate putamen, MCA territory cortex, and cortical penumbra). The number of TUNEL-positive cells in each region was counted in a high-powered field (×400) by a investigator who was blinded to the studies, and expressed as number/mm2. Results were expressed as mean ± standard deviation. The statistical significance of differences between the time points in each group was evaluated by analysis of variance. A P value less than 0.05 was considered to be significant. As illustrated in Fig. 1, only a small number of TUNEL-positive cells were detected at 4 hours after ischemia. In all regions, they were significantly increased at 24 hours compared with those at 4 hours. In all regions except the ischemic core of the caudate putamen, the number of TUNEL-positive cells was significantly decreased at 72 hours after ischemia compared with that at 24 hours (Fig. 1). These results are compatible with those of previous reports of the temporal profile of apoptotic neurons after focal ischemia in rats (Li et al., 1995a) and mice (Kondo et al., 1997; Murakami et al., 1997a). TUNEL-positive cells were observed dispersed throughout in the entire MCA area. In particular, the inner boundary of the caudate putamen had a larger number of TUNEL-positive cells. This regional predominance is consistent with the results of previous reports (Li et al., 1995b; Kondo et al., 1997; Murakami et al., 1997a).
Cytoplasmic expression of cytochrome c was detected in ischemic brain after transient MCA occlusion
The cresyl violet stain was used to determine the area of the infarcted tissue in the frozen coronal sections. The infarcted area apparently appeared at the caudate putamen and piriform cortex as early as 4 hours after reperfusion, and extended to the MCA territory cortex 24 to 72 hours after reperfusion (data not shown).
Cytochrome c protein expression after 90 minutes of transient focal ischemia and subsequent reperfusion was analyzed by immunohistochemistry. The data of the cytoplasmic cytochrome c expression are summarized in Table 2. Homogeneous cytoplasmic immunoreactivity of cytochrome c was visible as early as 4 hours after reperfusion in the whole ischemic area (Fig. 3B). After 24 hours of reperfusion, the population of immunoreactive cells was increased, and the intensity of cytoplasmic immunostaining was significantly enhanced with a slight background, compared with that observed after 4 hours of reperfusion (Fig. 3D). More immunoreactive cells were observed in the core of the caudate putamen and MCA territory cortex than in the inner caudate putamen and cortical penumbra. At 24 hours, we observed several cells presenting nuclear fragmentation, one of the characteristic features of apoptosis, as shown by hematoxylin counterstaining (Fig. 3E). However, no remarkable correlation between these cells and immunoreactive cells was shown in the present study. After 72 hours of reperfusion, most of the cells had been destroyed and the background immunoreactivity was increased (Fig. 3F). There was no immunoreactivity in the contralateral hemisphere (Fig. 3C) nor in the control specimens, which were treated without primary antibody (Fig. 3A). The absence of immunoreactivity in the contralateral hemisphere is considered to be caused by the thorough fixation of the brain by formaldehyde, which prevents the antibody from reaching the mitochondrial intermembrane space, but not the cytosol. In fact, immunohistochemistry with frozen sections results in dotted cytosolic immunoreactivity of cytochrome c in control brain (data not shown).
Summary of cytoplasmic cytochrome c protein expression after 90 minutes MCA occlusion
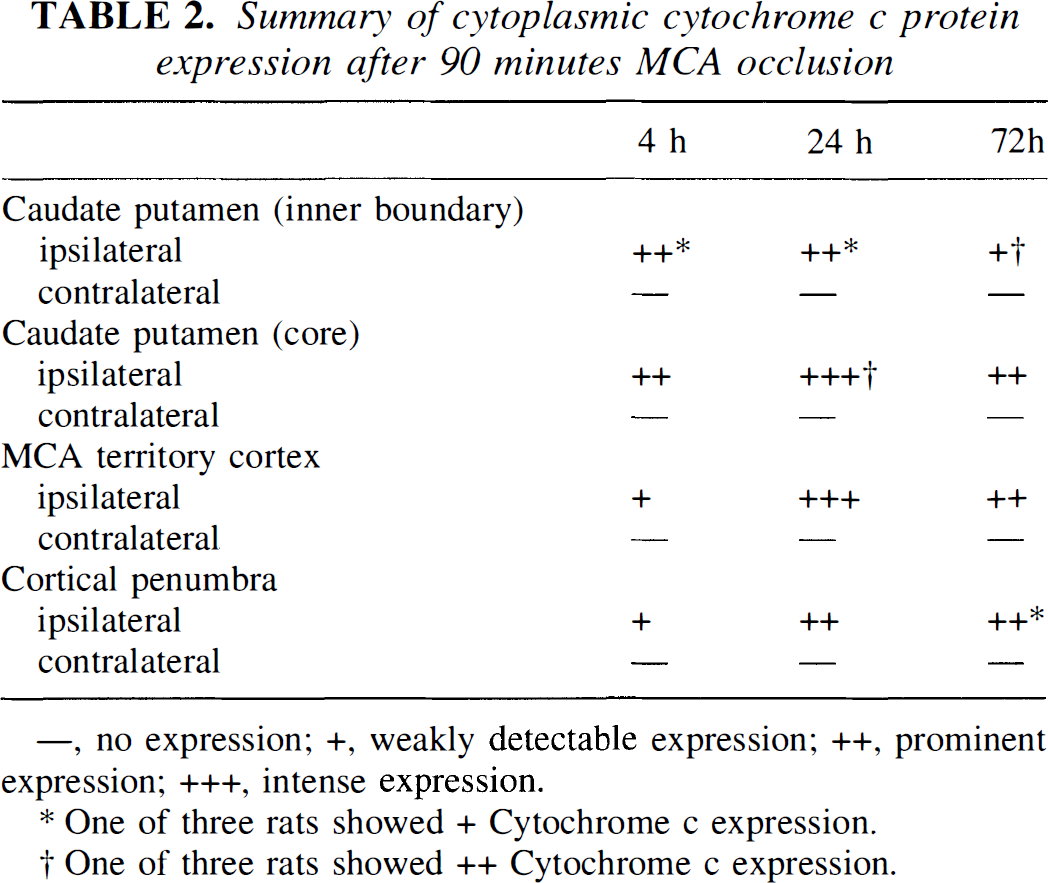
—, no expression; +, weakly detectable expression; ++, prominent expression; +++, intense expression.
One of three rats showed + Cytochrome c expression.
One of three rats showed ++ Cytochrome c expression.
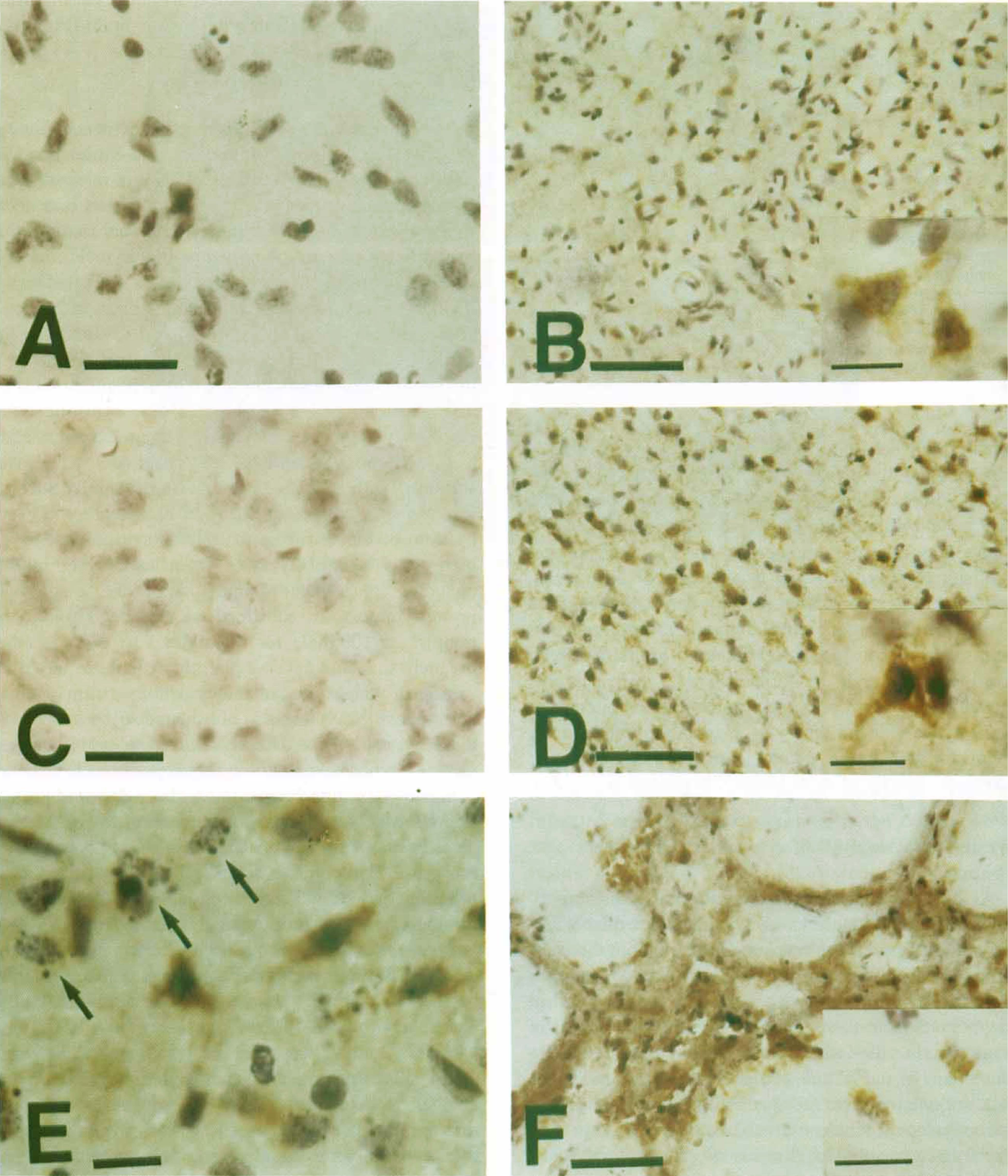
Cytochrome c immunostaining with hematoxylin counterstaining in coronal brain sections from rats after 4 and 24 hours of reperfusion after 90 minutes of ischemia.
Western blot analysis of cytosolic cytochrome c expression
As shown in Fig. 4A, cytochrome c immunoreactivity was evident as a single 15-kDa band, detected by Western blot analysis of the cytosolic fraction from the ischemic hemisphere, as early as 4 hours after 90 minutes MCA occlusion (lane 3), whereas there was no band in the nonischemic sample (lane 2). The characteristic single band in the ischemic sample was sustained at 24 hours (lane 5) and 72 hours (lane 7) after ischemia. These data not only confirm the specificity of the monoclonal antibody for cytochrome c used in this study, but also show that cytosolic localization of cytochrome c was significantly increased after transient ischemia. Furthermore, a significant amount of mitochondrial cytochrome c was detected by Western blot in nonischemic brain (mean optical density, 8.71; n = 2), which was decreased in ischemic brain after 24 hours ischemia (mean optical density, 5.83; n = 2). Correspondingly, the cytosolic fraction from the same sample showed a marked increase of cytochrome c in ischemic brain (Fig. 4B). However, cytochrome oxidase was strongly expressed in the mitochondrial fraction, but not in the cytosolic fraction of both ischemic and nonischemic brain (Fig. 4B).
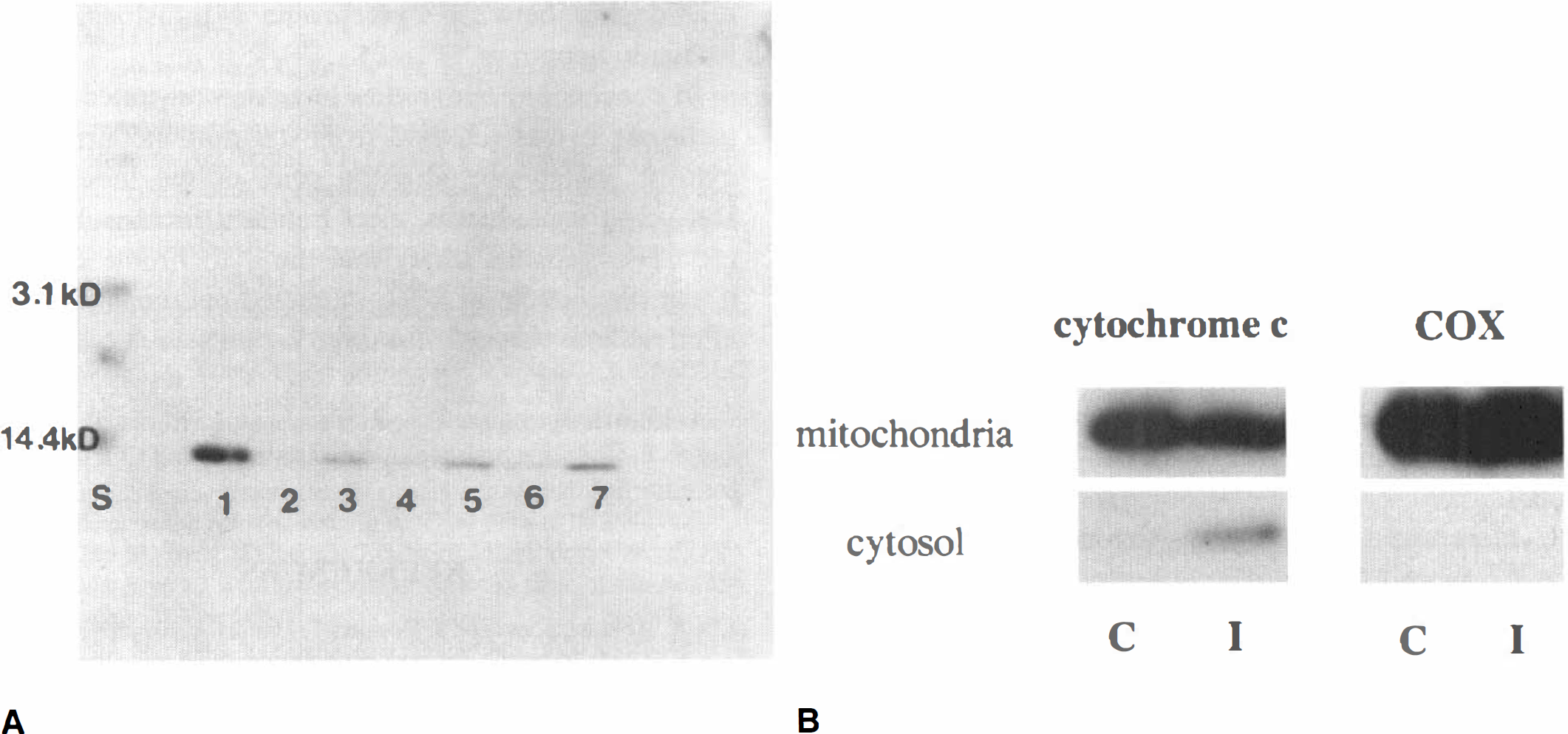
Western blot analysis of cytochrome c and cytochrome oxidase.
DISCUSSION
The current study provides the first evidence that cytochrome c, an essential component of the mitochondrial respiratory chain (Boyer et al., 1977), is released from mitochondria into the cytoplasm after transient focal ischemia. This observation derives from our findings that a significant amount of mitochondrial cytochrome c is detected in nonischemic brain (mean optical density, 8.71), which is decreased in ischemic brain at 24 hours after ischemia (mean optical density, 5.83), and that the cytosolic fraction from the same samples shows a marked increase of cytochrome c in ischemic brain (Fig. 4B). Also, by immunohistochemistry, cytosolic cytochrome c was detected only in the ischemic brain as early as 4 hours after ischemia (Figs. 3 and 4A), which was sustained until 24 and 72 hours after ischemia. Such an increase in cytosolic localization of cytochrome c appears to be derived from the mitochondrial inner membrane, and is unlikely to be from some other source, including injury-induced upregulation and a defect in protein import. In addition, we ultracentrifuged the samples at 100,000g for 60 minutes to exclude other organelles, including endoplasmic reticulum and ribosomes, in which most of the cytochrome c involved in the process of synthesis is considered to be located. In this way, we believe that we could exclude this biosynthetic cytochrome c as thoroughly as possible. We cannot completely exclude the possibility that a defect of protein import also contributes to the increase of cytosolic cytochrome c, and further evaluation is necessary to clarify this point. However, we believe that a decreased amount of mitochondrial cytochrome c, which is consistent with the increase of cytosolic cytochrome c (Fig. 4B), substantiates the validity of our observation that cytochrome c release from mitochondria contributes mainly, if not exclusively, to the increased cytosolic cytochrome c after ischemia.
Several authors have reported that cerebral ischemia could cause impairment of mitochondrial function (Abe et al., 1995; Almeida et al., 1995; Lee, 1995). Mitochondria are also known as the site where oxygen free radicals, especially superoxide anions, are produced during cerebral ischemia (Piantadosi and Zhang, 1996), and free radicals play a major role in the expansion of neuronal injury after both focal (Kinouchi et al., 1991; Yang et al., 1994; Chan, 1996; Kondo et al., 1997; Murakami et al., 1998) and global ischemia (Murakami et al., 1997b; Kawase et al., 1997). Thus, nonspecific mitochondrial degradation such as osmotic lysis and swelling may occur after ischemia/reperfusion injury and may therefore contribute to the increase of cytosolic cytochrome c. We do not exclude this possibility completely; however, we believe that not only mitochondrial degradation but also the translocation of cytochrome c contributes to the increase of cytosolic cytochrome c for the following reasons. First, as shown by Western blot analysis in Fig. 4B, cytochrome c was decreased in the mitochondria and correspondingly increased in the cytosol after ischemia, whereas cytochrome oxidase showed no alteration of the subcellular distribution after ischemia. This result suggests the intactness of the mitochondrial fraction isolated from the ischemic brain. From this observation, we believe the decrease of mitochondrial cytochrome c may indicate that cytochrome c release from the intact mitochondria occurred after ischemia. Second, immunohistochemistry showed cytoplasmic expression of cytochrome c only in the ischemic lesion, and cytoplasmic immunoreactivity was predominantly found in the cells presenting intact morphology at 4 hours after ischemia (Fig. 3B). Third, we have recently developed the in situ technique for evaluating mitochondrial membrane potential by rhodamine 123 probe (Murakami et al., 1998). Our previous study showed that the number of rhodamine-positive cells, which is considered to indicate the intact mitochondrial membrane potential, was significantly decreased after 24 hours ischemia, but not after 4 hours ischemia in wild-type mice (Murakami et al., 1998). Although there is a difference of species and ischemia model, our results detecting cytosolic cytochrome c at 4 hours after ischemia may imply that an increase of cytosolic cytochrome c precedes the loss of mitochondrial membrane potential after ischemia. These observations, taken together, suggest that translocation of cytochrome c from the mitochondria to the cytosol occurred as early as 4 hours after transient cerebral ischemia.
Neuronal cell death after focal cerebral ischemia has previously been attributed to passive necrotic processes. However, increasing evidence suggests that an active process similar to programmed cell death or apoptosis may contribute to the death of neurons after focal ischemia (Linnik et al., 1993; Tominaga et al., 1993; Li et al., 1995a,b,d; Charriaut-Marlangue et al., 1996; Choi, 1996; Chopp and Li, 1996; Gillardon et al., 1996; Asahi et al., 1997). Fragmented DNA as shown by agarose gel electrophoresis was present in rat brains subjected to permanent MCA occlusion (Linnik et al., 1993; Tominaga et al., 1993). The DNA fragmentation is known to be associated with increased intranucleosomal endonuclease activity (Tominaga et al., 1993), or to be reduced by the protein synthesis inhibitor cycloheximide (Linnik et al., 1993; Du et al., 1996). Recent morphologic studies with TUNEL staining demonstrate that the inner boundary zone of the caudate putamen is vulnerable to DNA fragmentation after focal cerebral ischemia (Li et al., 1995b; Charriaut-Marlangue et al., 1996), and that the number of DNA-damaged neurons is maximized at 24 to 48 hours after ischemia (Li et al., 1995c). In the current study, we used a transient focal ischemia model in rats, which has been shown to present some evidence of neuronal apoptosis such as TUNEL-positive cells and internucleosomal DNA fragmentation (Li et al., 1995a). In fact, a significant number of TUNEL-positive cells were detected in the current study, which were maximized at 24 hours after ischemia. More DNA-damaged cells were found in the inner boundary of the caudate putamen than in the core of caudate putamen, MCA territory cortex, and cortical penumbra. These data are consistent with previous reports, as shown above, and indicate that DNA-damaged neuronal death also contributes to the expansion of the lesion in our ischemia/reperfusion injury model.
Cytochrome c has drawn particular attention because of a recent cell-free study suggesting its critical role in apoptosis (Liu et al., 1996). This recent study showed that mitochondria are involved in apoptosis by releasing cytochrome c to the cytoplasm where it activates CPP32, a molecule of the ICE family that has been shown to trigger apoptosis when activated (Liu et al., 1996). However, the involvement of cytochrome c in apoptosis has not been demonstrated in vivo. In cerebral ischemia, apoptosis is thought to accompany neuronal necrosis (Li et al., 1995c), and even in vitro a wide variety of apoptotic and necrotic stimuli are reported to induce progressive mitochondrial swelling and outer membrane rupture (Vander Heiden et al., 1997). Therefore, it is difficult to reveal a direct relationship between cytochrome c release and apoptosis in vivo. In the current study, we show that cytochrome c is released from mitochondria to the cytoplasm, which precedes the peak of DNA fragmentation, after transient focal ischemia in rats. From our observations, it is conceivable that cytochrome c release takes place in the induction of DNA fragmentation after ischemia. Further studies are warranted for elucidating the direct correlation between cytochrome c release and DNA-damaged neuronal cell death after ischemia.
Recent evidence shows that the expression of Bcl-2 process (Kluck et al., 1997; Yang et al., 1997) or Bcl-XL (Kharbanda et al., 1997; Kim et al., 1997) prevents cells from undergoing apoptosis and acts to inhibit cytochrome c translocation to cytoplasm, thereby blocking CPP32 activation and the apoptotic process. Martinou et al. (1994) have reported that overexpression of Bcl-2 in transgenic mice protects neurons from ischemic insult, and our previous report demonstrates that Bcl-2 protein is induced in neurons in the ischemic hemisphere that survive transient focal ischemia in rats (Chen et al., 1995). Although the relationship between Bcl-2 expression and ischemic apoptosis in these studies is not clear, it is conceivable that Bcl-2 also inhibits the neuronal apoptosis of cerebral ischemia by preventing cytochrome c translocation and subsequent activation of CPP32. A cytochrome c study using Bcl-2 transgenic or knockout animals will provide evidence necessary to establish the relationship between cytochrome c translocation and ischemic apoptosis.
In conclusion, our studies have demonstrated that cytochrome c was released from the mitochondria to the cytosol, which precedes the peak of the induction of DNA-fragmented cells, after transient focal ischemia in rats. These results imply that the cytochrome c release might play a role in DNA-damaged neuronal cell death after ischemia/reperfusion injury.
Footnotes
Acknowledgments
The authors thank L. Reola, B. Calagui, and S. F. Chen for their technical assistance, and C. Christensen for editorial assistance.