
Abstract
Background
Elevated plasma (Lp(a)) levels may represent an independent risk factor for atherothrombotic complications but the relation between Lp(a) levels and the extent of coronary artery disease (CHD) has been discussed controversially. Little is known about potential atherothrombogenic mechanisms of Lp(a).
Design
Case—control study.
Methods
We assessed the relationship between plasma Lp(a) and angiographically defined CHD, evaluating the severity of coronary atherosclerosis by three different scores. A total of 312 patients with stable angina aged 40-68 years with at least one coronary stenosis > 50% were studied. A group of 479 voluntary blood donors matched for age and sex served as controls. A complete lipid profile and a large number of markers of coagulation, fibrinolysis and inflammation were measured.
Results
Plasma levels of Lp(a) were significantly higher in patients (14.8 mg/dl; 5.4-47.1 mg/dl; median/interquartile range) than in controls (9.7 mg/dl; 3.5-25.3) (P>0.0001). In a logistic regression model, the fully adjusted Odds Ratio for CHD was 3.3 (95% confidence interval (CI) 1.8-5.6, P >0.0001) for patients in the upper quartile of the Lp(a) distribution compared to the bottom quartile. There was no appreciable association between Lp(a) and apolipoproteins, markers of haemostasis, fibrinolysis and inflammation and the severity of CHD.
Conclusions
These results indicate that elevated plasma Lp(a) levels may be an independent risk factor for CHD but unrelated to the severity and extension of CHD. Furthermore, there is no good evidence that the presumed link between Lp(a) and CHD is mediated by increased levels of markers of inflammation, or interference with markers of fibrinolysis or coagulation.
Introduction
There is increasing evidence that Lp(a) represents an independent risk factor for coronary heart disease (CHD). Recently, a formal meta-analysis of 27 prospective studies clearly demonstrated an association between Lp(a) levels and CHD with a combined risk ratio of 1.6 (95% CI 1.4-1.8) for incident CHD if the top third of Lp(a) was compared with the bottom third [1]. However, some studies have failed to confirm this relationship [2–5]. In contrast to the evidence of an association between Lp(a) and CHD itself, the relationship between Lp(a) levels and the severity and extension of angiographically defined CHD is discussed controversially [6–13]. Furthermore, Lp(a) excess has been found to be associated with a history of previous myocardial infarction [6, 14]. It has therefore been suggested that besides its potential role in the initiation and progression of CHD, Lp(a) levels might be important for plaque destabilization and thrombotic coronary occlusion [6, 15].
Mechanistically, an inhibitory effect of Lp(a) on fibrinolysis through structural similarity of apolipoprotein (a) with plasminogen might represent the patho-physiological link between Lp(a) and CHD. Apolipoprotein(a) competes with plasminogen for binding sites on molecules, mononuclear and endothelial cells and thus may accentuate thrombosis [16, 17]. It binds to fibrin and attenuates the fibrin-dependent enhancement of tissue-type plasminogen activator mediated activation of plasminogen [18]. Low levels of plasminogen seem to promote the deleterious effect of Lp(a) [19].
We conducted a large case-control study to assess the significance of Lp(a) as a risk factor for angiographically determined CHD in patients with stable disease and, more specifically, to investigate the relation between plasma Lp(a) levels and the severity and extension of CHD defined by three different scores. Additionally, we sought to assess potential patho-physiological links between Lp(a) and CHD by correlating its plasma concentrations with other lipid variables, markers of inflammation, coagulation and fibrinolysis.
Methods
Patients and controls
Between October 1996 and November 1997 a total of 312 patients of German nationality aged 40-68 years who underwent elective coronary angiography at the Department of Cardiology at the University of Ulm Medical Center were recruited for the study. All of them showed a > 50% diameter stenosis of at least one major coronary artery, and diagnosis of CHD had been established within the previous 2 years. Patients with acute ischaemic syndromes within the previous 4 weeks, with acute or chronic infectious diseases, malignancies, severe liver or renal disease, and patients on anticoagulants were excluded.
The control group consisted of 479 occasional blood donors from the local Red Cross centre serving the University hospitals of Ulm and was frequency matched for age and sex. Controls with a history of definite or suspected CHD according to the Rose angina questionnaire were excluded, as were subjects reporting infectious diseases or surgery within the previous 4 weeks.
All participants were interviewed in a standardized manner by the same trained personnel. They were asked about medical history particularly including questions related to history of CHD, cardiovascular risk factors, current medication, socioeconomic information and lifestyle habits like smoking, physical activity and alcohol consumption.
Participation was voluntary and written informed consent was obtained from each subject. The study was approved by the ethics committee of the University of Ulm. Participation rate was 78% in eligible patients and 84% in eligible controls.
Laboratory methods
Venous blood was drawn in the morning in a standardized manner and a complete blood cell count was conducted in a Coulter STKS chamber (Coulter, Krefeld, Germany). Within 30 min the remaining blood was centrifuged at 3000 g for 10 min, immediately aliquotted and frozen at −70°C until analysis.
Total cholesterol (CHOD-PAP) and triglycerides (GPO-PAP) were measured enzymatically using reagents from WAKO Chemicals (Neuss, Germany). HDL cholesterol was measured in the supernatant after precipitation of apolipoprotein B containing lipoproteins with phosphatungstate acid and MgCl2 obtained from Roche Diagnostics (Mannheim, Germany). Apolipoproteins AI, B, CIII, E and Lp(a) were determined by immunoturbidimetry with antisera from Greiner Biochemicals (Flacht, Germany). Apolipoproteins AII and CII were measured by immunoturbidimetry with antisera from Roche Diagnostics (Mannheim, Germany) and Kamiya Biomedial (Seattle, USA), respectively. The apolipoprotein assays were calibrated using the following reference sera: N apolipoprotein standard (Behring, Marburg, Germany) for apo AI, AII, B, and E; reference standard from Immuno (Heidelberg, Germany) for Lp(a), and multi-calibrator set from Kamiya (Seattle, USA) for apo CII and apo CIII. All analyses were performed on a WAKO R-30 automated analyser in a laboratory setting certified according to ISO 9001.
C-reactive protein (CRP) measurements were carried out by an immunoradiometric assay (range 0.05-10 mg/l) calibrated with the WHO reference standard 85/506 [20]. Albumin, serum amyloid A (SAA) and fibrinogen were determined by immunonephelometry (Behring, Marburg, Germany). The following parameters were determined by ELISA: plasminogen-activator-inhibitor-1 (PAI-1) activity (Immuno, Heidelberg, Germany), von Willebrand factor (vWF) (Haemochrom, Essen, Germany), D-Dimer (Agen Biomedical, Acacie Ridge, Australia), ICAM-1 (Diaclone Research, Besancon, France), IL-6 and TNF-α (Quantikine HS, R&D Systems, Wiesbaden, Germany). Plasminogen activity was determined by extinction method (Chromogenix, Milano, Italy). Measurements of plasma viscosity were taken in a Coulter Harkness capillary viscometer (Coulter Electronics Co, Luton, UK). Intra-assay coefficient of variation was >6% for Lp(a). All laboratory analyses were carried out in a blinded fashion.
Angiographic evaluation
Coronary angiography was performed by the Judkins method. The severity of CHD was assessed by means of three different scores. Besides the clinical 1-3 vessel disease score, the quantitative AHA extension score evaluating 1-15 segments of major coronary arteries was applied. Additionally, a qualitative and quantitative evaluation was performed using the Gensini score. All coronary angiograms were evaluated by a single observer blinded to clinical and laboratory data. The kappa-coefficient for repeated analyses of 30 randomly selected angiograms after a mean of 3 months was 1.0 for the 1-3 vessel disease score, 0.79 for tertiles of the extension score, and 0.85 for tertiles of the Gensini score, respectively.
Statistical analysis
Demographic and clinical characteristics in patients and controls were compared in a descriptive way. Because of their skewed distribution, Lp(a) levels and various other variables are expressed as medians together with their interquartile ranges. Levels of further blood, plasma, or serum parameters are reported as means and their standard deviations (SD). Comparison of Lp(a) levels between patients and controls was performed by means of the Kruskal—Wallis test.
Multivariable logistic regression analyses were performed to assess the independent association between Lp(a) and angiographic CHD, while controlling for age and sex, TC (mmol/l), HDL-cholesterol (mmol/l), body mass index (BMI, kg/m2), smoked pack-years, history of hypertension and diabetes, alcohol consumption, fibrinogen and years of formal school education. The results of the multivariate logistic regression analyses are expressed as Odds Ratios (OR) together with their 95% CI for the comparison of risk for CHD between each of the upper three quartiles and the bottom quartile of the Lp(a) distribution within controls. Additionally, the adjusted OR of subjects with levels of Lp(a) in the upper quartile combined with levels of plasminogen in the lowest quartile were calculated in the same way. We then assessed the association between Lp(a) and the different coronary scores using a linear regression model adjusting for age and sex.
Spearman rank correlation analyses were performed between Lp(a) and other lipid variables, markers of coagulation, fibrinolysis and inflammation. Additionally, each of these variables was then included in the fully adjusted model to assess its potential role as mediator of the effect of Lp(a). A P-value >0.05 was considered statistically significant. All computations were carried out with the help of SAS for Windows, Release 6.12.
Results
Clinical and biochemical characteristics
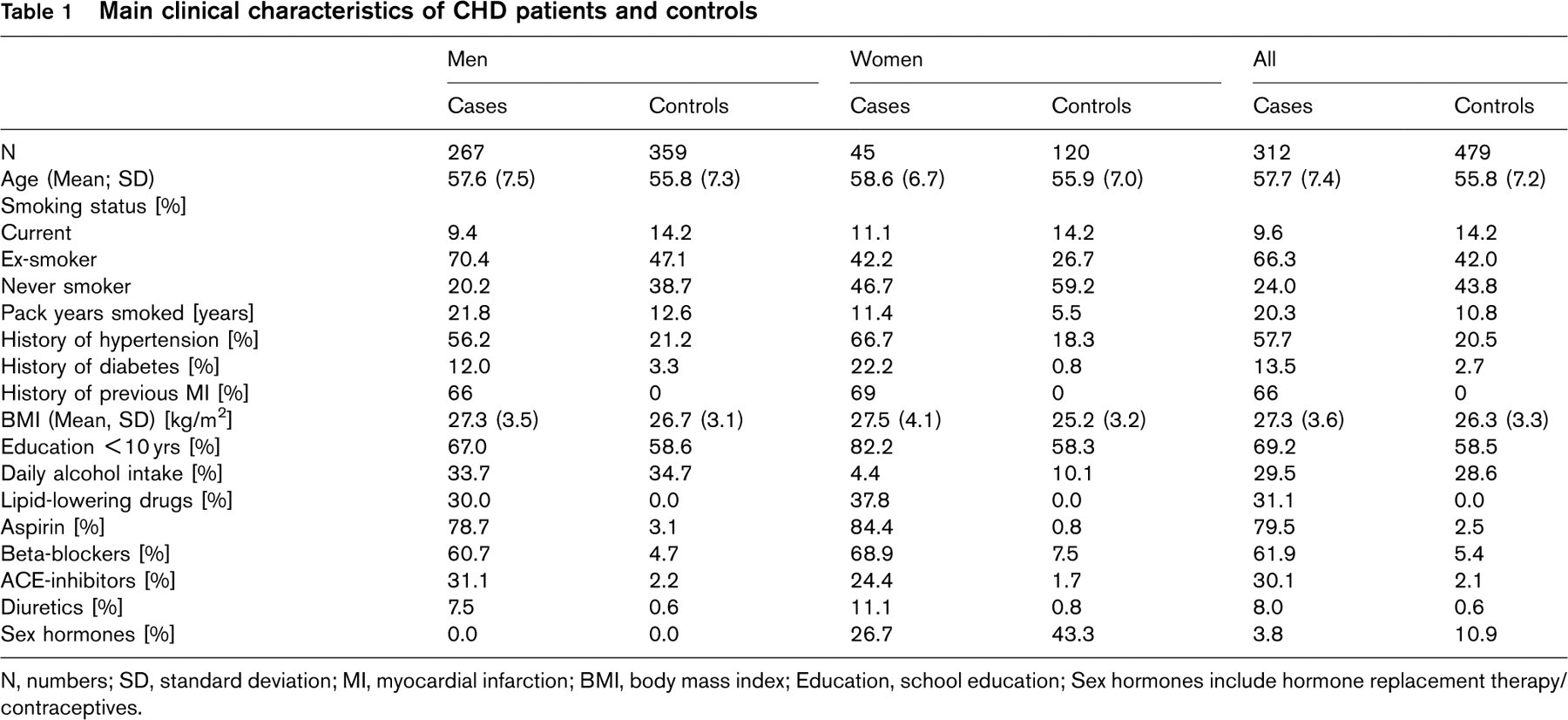
Demographic, lifestyle and clinical characteristics as well as other laboratory markers of patients and controls are summarized in Table 1 and 2, respectively. Fewer patients were current smokers whereas more of the controls had never smoked (Table 1). Controls showed a somewhat lower mean BMI, and reported less frequently a history of hypertension or diabetes. About one third of patients took lipid-lowering drugs and ACE-inhibitors, about 80% were on aspirin, two-thirds took beta-blockers, one fourth of the female cases received sex hormones, and approximately two-thirds had suffered a myocardial infarction (MI) within the previous 2 years. Among controls less than 3% took aspirin, ACE-inhibitors or diuretics, 5.4% took beta-blockers, and ∼43% of females used oral contraceptives or received hormone replacement therapy.
Total cholesterol, as well as HDL cholesterol, was considerably higher in controls than in patients (Table 2), as were apolipoproteins ApoA1 and ApoA2. Conversely, ApoB, ApoC2, ApoC3, ApoE levels and levels of all measured markers of coagulation, fibrinolysis and inflammation, with the exception of plasminogen and albumin, were higher in patients than in controls.
Main clinical characteristics of CHD patients and controls
N, numbers;
SD, standard deviation;
MI, myocardial infarction;
BMI, body mass index;
Education, school education;
Sex hormones include hormone replacement therapy/contraceptives.
Distribution of lipid variables, markers of coagulation, fibrinolysis and inflammation∗
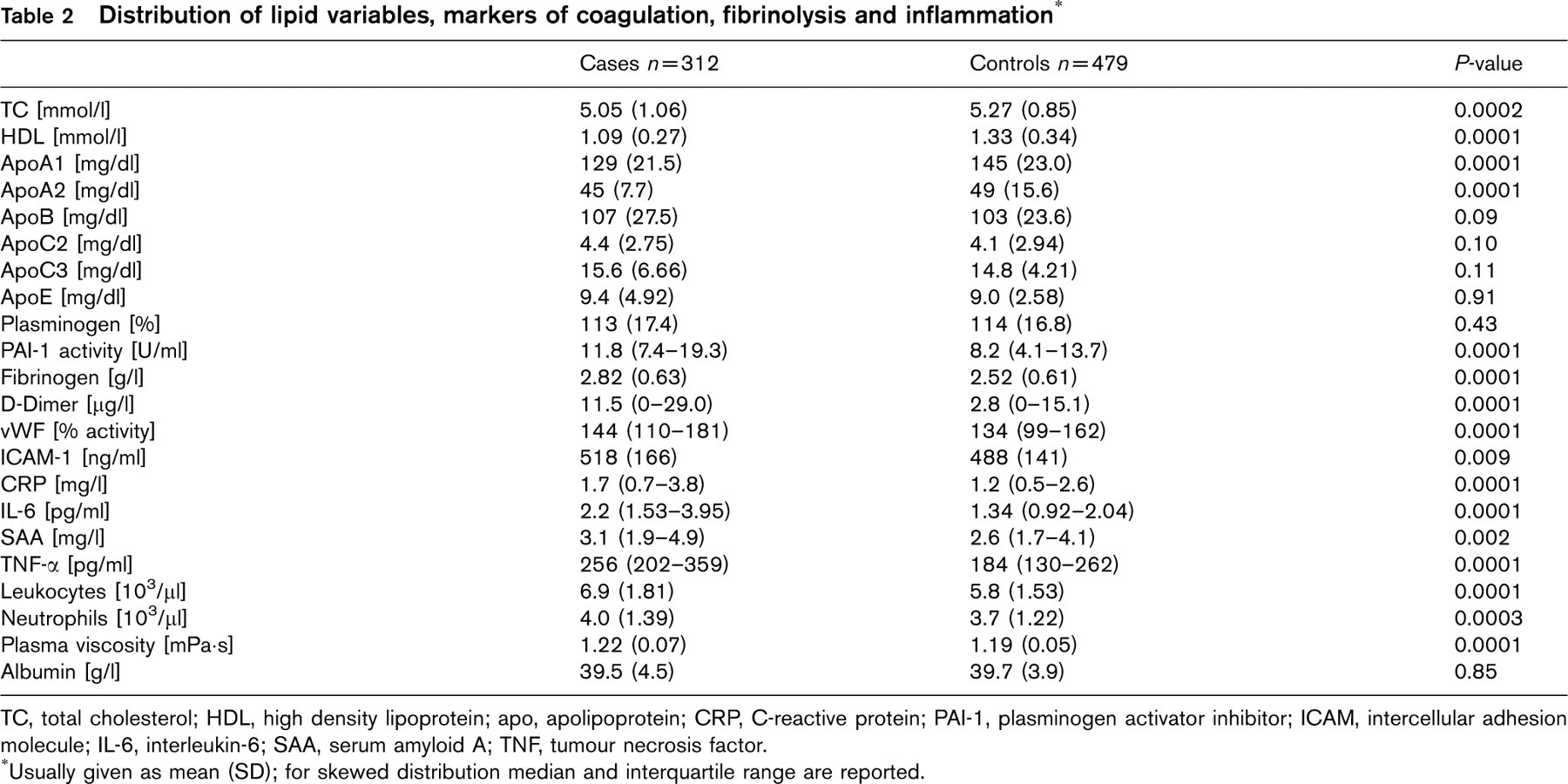
TC, total cholesterol;
HDL, high density lipoprotein;
apo, apolipoprotein;
CRP, C-reactive protein;
PAI-1, plasminogen activator inhibitor;
ICAM, intercellular adhesion molecule;
IL-6, interleukin-6;
SAA, serum amyloid A;
TNF, tumour necrosis factor.
∗Usually given as mean (SD);
for skewed distribution median and interquartile range are reported.
Lp(a) distribution
The distribution of Lp(a) was highly skewed in patients and in controls. Plasma levels of Lp(a) were significantly higher in patients (14.8 mg/dl; 5.4-47.1 mg/dl; given as median and interquartile range) than in controls (9.7 mg/dl; 3.5-25.3 mg/dl) (P >0.0001). The difference between median Lp(a) levels of patients and controls was mainly attributable to the high Lp(a) levels of cases with a history of MI (15.7 mg/dl; 6.6-55.0) whereas the median of Lp(a) among other cases did not differ from that of controls (9.1 mg/dl; 3.9-43.6).
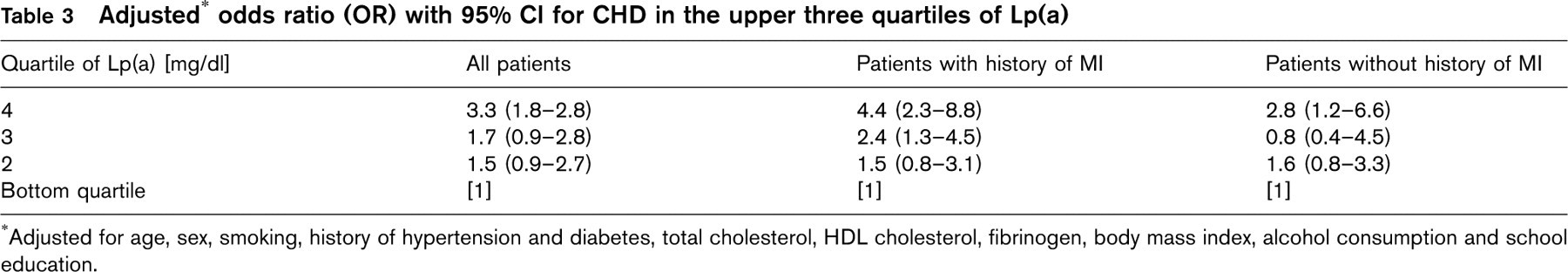
Adjusted∗ odds ratio (OR) with 95% CI for CHD in the upper three quartiles of Lp(a)
∗Adjusted for age, sex, smoking, history of hypertension and diabetes, total cholesterol, HDL cholesterol, fibrinogen, body mass index, alcohol consumption and school education.
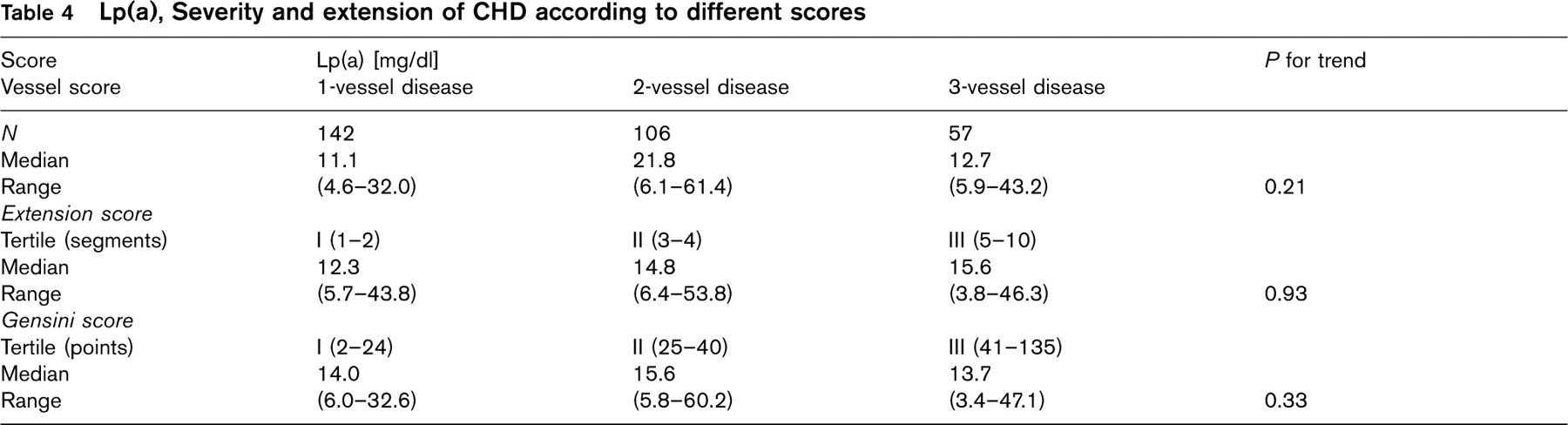
Lp(a), Severity and extension of CHD according to different scores
Lp(a) and risk of CHD
Table 3 presents the results of multivariate logistic regression analysis adjusted for age and sex as well as for TC, HDL, smoking, BMI, history of hypertension and diabetes, alcohol consumption, fibrinogen and educational status. In the fully adjusted model the OR for CHD in each of the upper three quartiles of the Lp(a) distribution within controls in comparison to the bottom quartile were 1.5 (95% CI, 0.9-2.7) for the second, 1.7 (95% CI, 0.9-2.8) for the third and 3.3 (95% CI 1.8-5.8) for the top quartile, respectively.
Exclusion of cases with lipid-lowering medication did not substantially alter these results (data not shown). Although the difference in Lp(a) levels between cases and controls was mainly due to high Lp(a) levels in patients with a history of previous MI, the OR for patients without previous MI remained substantial and statistically significant for the upper quartile 2.8 (95% CI 1.2-6.6). This reflects the different distribution of Lp(a) among cases without history of previous MI. Most of them showed comparable levels as controls did, whereas some of them had the highest levels among all study participants.
Lp(a) and extension and severity of CHD
There was no relationship between Lp(a) and the 1-3 vessel score, the extension of CHD according to the AHA extension score, or tertiles of the Gensini score (Table 4).
Lp(a) and markers of coagulation, fibrinolysis and inflammation
Table 5 shows Spearman rank correlation coefficients between Lp(a) levels and other lipid variables, markers of coagulation, fibrinolysis and inflammation. There was no appreciable association between either of these variables and Lp(a). The OR of patients with Lp(a) levels in the upper quartile combined with plasminogen levels in the lowest quartile (n = 60) did not substantially differ from those who only showed high Lp(a) concentrations. Neither markers of inflammation, nor markers of coagulation and fibrinolysis attenuated the relationship between Lp(a) and CHD in the logistic regression analysis (data not shown).
Spearman Rank Correlation Coefficients between Lp(a) and other lipid variables, markers of coagulation, fibrinolysis and inflammation
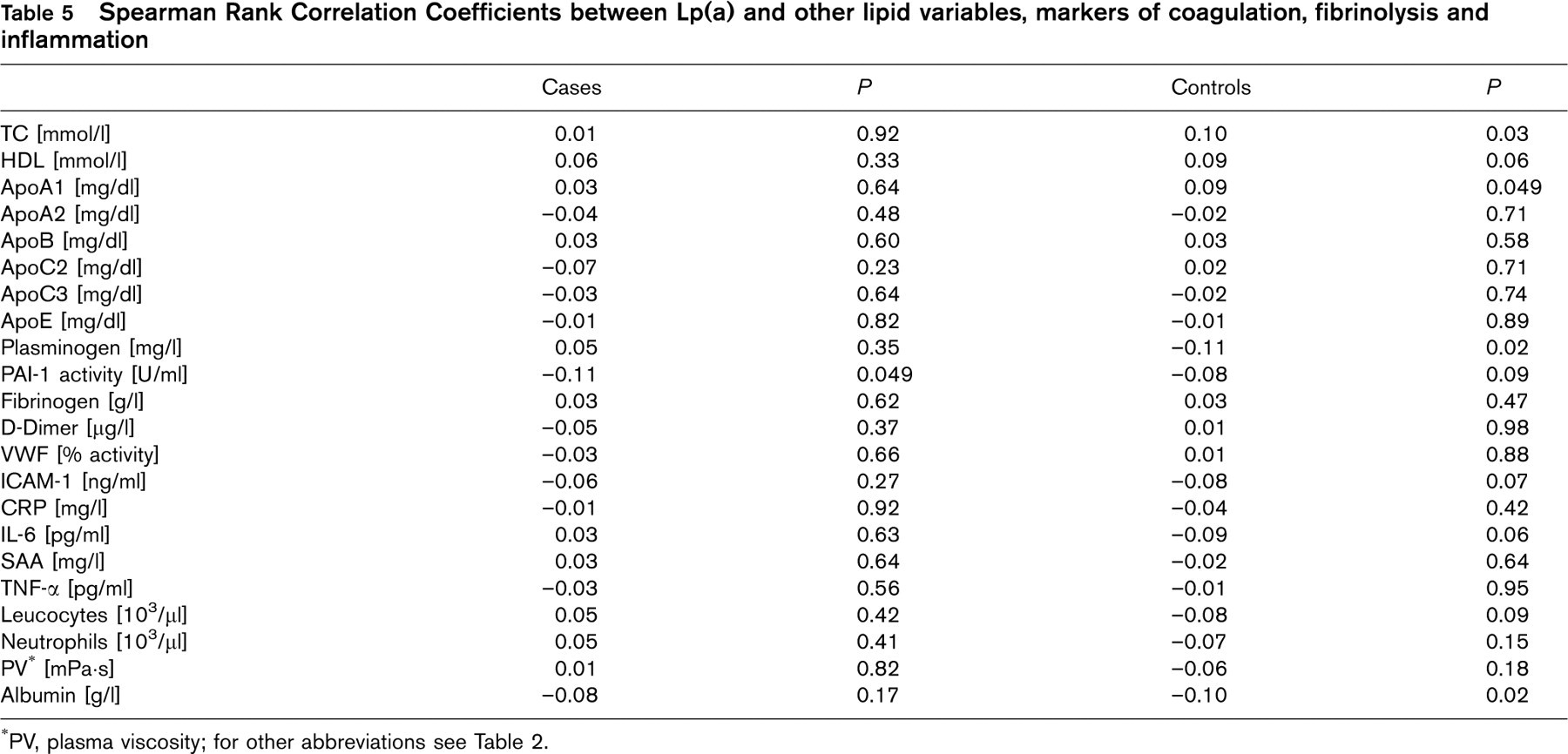
∗PV, plasma viscosity;
for other abbreviations see Table 2.
Discussion
This large case—control study provides further evidence for an association between elevated Lp(a) plasma levels and angiographically diagnosed CHD even after controlling for a variety of known risk factors. However, we could not demonstrate a significant relationship between Lp(a) levels and extension of CHD estimated by three different established scores.
Furthermore, there was no association with various markers of coagulation, fibrinolysis and inflammation.
Lp(a) and CHD
Results from several large, prospective studies indicate a strong, positive association between Lp(a) and atherothrombotic complications [21–32]. Several studies, however, did not confirm such a relationship [2–5, 33, 34], which was mainly attributed to storage problems of plasma samples, inadequate statistical power, or concomitant medication [2, 5, 33–35]. In the present study these possible limitations were carefully taken into account.
Severity and extension of CHD, previous MI and Lp(a) levels
The lack of association between Lp(a) levels and various CHD scores is in agreement with several other studies [6, 9, 10]. However, in some studies in which patients without significant CHD in the angiogram were used as reference group a statistically significant trend towards higher levels of Lp(a) could be demonstrated in unselected patients as well as in patients with CHD and a history of previous MI [6, 7, 12, 13, 36, 37]. In our study, patients with a history of MI within the previous 2 years had significantly higher Lp(a) levels than cases without. In patients with unstable angina, Lp(a) was the only baseline variable among a variety of risk factors including various lipoproteins, which identified those with an adverse prognosis [38]. In another study, Lp(a) was detected in larger amounts in lesions from patients with unstable angina than it was in lesions from patients with stable angina [39]. These findings strengthen the hypothesis that high Lp(a) levels may predispose to plaque rupture and thrombosis as suggested by Schwartzman et al. [6]. Thus, Lp(a) may have particular implications for atherogenesis as well as for clinical complications.
Lp(a) and other lipoproteins, markers of coagulation, fibrinolysis and inflammation
We could not demonstrate an appreciable association between levels of Lp(a) and plasma concentrations of other lipid variables, markers of coagulation, fibrinolysis and inflammation. These findings are in agreement with other studies [2, 40], and thus may underline the independent impact of Lp(a) on CHD.
Impaired fibrinolysis through competition of apolipoprotein(a) with plasminogen for binding sites on molecules, mononuclear and endothelial cells which will possibly enhance thrombosis [16, 17] represents the suggested interference between Lp(a) and atherosclerosis. In one study of 61 children with idiopathic nephrotic syndrome during a flare-up of the disease, binding of Lp(a) to fibrin and cell surfaces was significantly inversely correlated with plasminogen levels [19]. These data support the hypothesis that competitive binding of Lp(a) at plasminogen binding sites may represent an important pathophysiological link between Lp(a) and CHD. However, in our study, the OR for individuals with high Lp(a) were independent of plasminogen levels. One might speculate that in individuals with high Lp(a) levels and thus a high competitive potential of Lp(a) for plasminogen binding sites, an up-regulation of plasminogen synthesis would occur, for example, individuals with high Lp(a) levels would be expected to have high levels of plasminogen. If this up-regulation fails to occur plasminogen levels in normal ranges may already promote the deleterious effect of Lp(a).
Stimulation of human endothelial cell PAI-1 synthesis and activity has been suggested as another mechanism by which Lp(a) might inhibit fibrinolysis [41, 42]. In two studies elevated PAI-1 levels were detectable in patients with high Lp(a) [42]. However, our results are in agreement with several other studies in which no relationship between Lp(a) levels and concentration of PAI-1 or other fibrinolytic markers (for review see [43]) was seen.
Apolipoprotein(a) co-localizes with fibrin at intracellular and extracellular sites [44]. It binds to fibrin and attenuates the fibrin-dependent enhancement of tissue-type plasminogen activator mediated activation of plasminogen [18]. In addition, it was suggested that Lp(a) may play a role in the modulation of vascular function during inflammation [45], since it acts as an acute phase reactant [40].
Experimental data indicate that Lp(a) may represent an attractant for macrophages in the atheromatous plaque [46, 47], and significantly co-localizes with plaque macrophages in atherectomy specimen [39], possibly representing another link between Lp(a) and inflammation.
The present study has several limitations. First, case—control studies are unsuitable to draw causal conclusions. Secondly, controls did not undergo coronary angiography or a treadmill test, so that asymptomatic CHD within these subjects cannot be completely ruled out. However, prevalence of CHD in asymptomatic middle-aged individuals appears to be low [48], and selecting controls among patients who underwent coronary angiography because of suspected CHD without presenting significant stenosis may have caused an even more severe bias. Thirdly, blood donors may tend to be healthier than population-based controls. However, we tried to minimize this potential bias by carefully adjusting for various risk factors. Finally, since patients without significant CHD in the angiogram were not included our study may not have the power to detect a relatively smaller independent association between Lp(a) concentrations and extension and severity of CHD.
In conclusion, the present study provides further strong evidence for an association between elevated plasma Lp(a) levels and angiographically diagnosed CHD. However, our results do not indicate that inflammation, impaired fibrinolysis or enhanced coagulation present the presumed link between Lp(a) and CHD. Further research is needed to elucidate the molecular mechanisms involved in the effects of Lp(a) in atherogenesis and its complications.