
Abstract
Background
Both angiotensin-converting enzyme genotype and plasminogen activator inhibitor type 1 genotype have an effect on fibrinolytic components and hence, may increase risk or advance occurrence of coronary heart disease. We examined the association of the angiotensin-converting enzyme and the plasminogen activator inhibitor type 1 genotypes, and their combinations, with early onset of coronary heart disease in a cohort of 907 patients with coronary heart disease. Design and methods All patients with a coronary heart disease (International Classification of Diseases, 9th Rev. pos. 410–414), aged 30–70 years and participating in an inpatient rehabilitation program between January 1999 and May 2000 in two clinical centres in Germany were enrolled. The plasminogen activator inhibitor type 1 and the angiotensin-converting enzyme genotypes were determined by polymerase chain reaction, and the distribution was compared between patients with early (>55 years) and late (>55 years) onset of coronary heart disease. A multivariate analysis was employed to adjust for potentially confounding factors.
Results
Of the 907 included patients, 408 (45.0%) developed coronary heart disease before the age of 55 years. For the 4G/4G genotype of plasminogen activator inhibitor type 1 the odds ratio for early onset of coronary heart disease was 1.68 [95% confidence interval (CI) 1.01–2.57] and for the D/D genotype of angiotensin-converting enzyme the odds ratio was 1.22 (95% CI 0.84–1.76) after adjustment for covariates. In multivariate analysis an odds ratio of 3.10 (95% CI 1.51–6.36) was found for the association between the combined homozygosity for both polymorphisms (plasminogen activator inhibitor type 1 genotype 4G/4G and angiotensin-converting enzyme genotype D/D) and onset of coronary heart disease before the age of 55 years after controlling for sex, age, smoking, diabetes, hypertension, hyperlipidemia and school education.
Conclusion
The co-existence of the 4G/5G polymorphism of the plasminogen activator inhibitor type 1 gene and the I/D polymorphism of the angiotensin-converting enzyme gene increases the risk for early onset of coronary heart disease in this population. Eur J Cardiovasc Prev Rehabil 13:449–456 © 2006 The European Society of Cardiology
Keywords
Introduction
Coronary heart disease (CHD) is the leading cause of death in Western industrialized countries [1]. A large number of risk factors such as hypertension, diabetes, smoking, body mass index, hyperlipidemia, physical inactivity and others have been established as determinants for the development of CHD. Beyond these environmental and behavioral determinants, genetic factors have been implicated in the development of CHD and it has been assumed that the disease process itself might be highly polygenic [2]. Especially polymorphisms in genes involved in blood coagulation, in the regulation of blood pressure, and in the metabolism of lipoproteins have been suggested as possible genetic determinants of CHD.
Experimental studies and clinical trials have shown that the renin-angiotensin system (RAS) plays an important role in the pathogenesis of CHD and prognosis of myocardial infarction [3]. The three major components of the RAS are the angiotensin-I-converting enzyme (ACE), angiotensinogen, and the angiotensin-II type 1 receptor (AGT1R). A homozygous deletion in the ACE gene (DD-genotype) is associated with increased serum concentration of ACE [4] which enhances conversion of angiotensin-I to angiotensin-II. The pathophysiological role of ACE [5] and the established benefits of ACE-inhibitor therapy [6] suggested the ACE gene as an important susceptibility factor for CHD. An association between the DD-genotype of the ACE gene and increased risk of CHD has been described [7]. Many other studies have subsequently investigated the association between the ACE I/D polymorphism and CHD, but results are still inconclusive [8–12].
The plasminogen activator inhibitor type-1 (PAI-1) is the primary physiologic inhibitor of plasminogen activation in blood and has strong prothrombotic properties [13]. Several polymorphisms have been described within the PAI-1 gene including a single base-pair polymorphism in the promotor region [14]. Subjects who are homozygous for the 4G allele have plasma PAI-1 concentrations that are approximately 25% higher than those who are homozygous for the 5G allele [15]. Elevated PAI-plasma levels are associated with an increased incidence of acute coronary syndromes including myocardial infarction [16]. Yet, the clinical role of the PAI-1 4G/5G polymorphism and the subsequent risk of CHD is discussed controversially. Both, positive risk associations [15, 17, 18], as well as absence of such associations [19–21] have been reported.
As ACE may play an important role in regulating plasma levels of angiotensin-II and hence, PAI-1 plasma levels, a gene-gene interaction of ACE with the PAI-1 genotype seems biologically plausible and has recently been suggested [22, 23]. The DD genotype of the ACE gene has been shown to be associated with enhanced conversion of angiotensin-I to angiotensin-II [24]. Recently, in-vivo and in-vitro studies demonstrated that angiotensin-II increases PAI-1 mRNA and PAI-1 levels [25–28]. In addition, persons with the DD-genotype of ACE showed elevated plasma levels of PAI-1 [29]. So far no corresponding genotype combination regarding the risk of CHD or the risk of early onset of CHD has been investigated in an observational study, although both polymorphisms may have a functional relevance for CHD. We therefore examined the association of combined homozygosity for the D allele of ACE and the 4G allele of PAI-1 with early onset of CHD after carefully adjusting for other established risk factors in a large group of patients with coronary heart disease.
Methods
Study design and sampling
Patients aged 30–70 years participating in an inpatient rehabilitation program due to CHD (International Classification of Diseases, 9th Revision. pos. 410–414) between January 1999 and May 2000 in one of two participating rehabilitation clinics in Germany (Isny and Bad Nauheim) were enrolled in the study (initial response 58%). All patients included in the program had suffered an acute myocardial infarction or had undergone surgical treatment of CHD (percutaneous transluminal coronary angioplasty, bypass surgery or stent).
Germany has a very comprehensive inpatient rehabilitation program and patients who were hospitalized due to acute manifestation or surgical treatment of CHD (mostly percutaneous coronary intervention or coronary artery bypass graft) are offered the chance to participate in an inpatient rehabilitation program after discharge from the acute care clinic. The aims of this 3-week program are the reduction of cardiovascular disease risk factors, improvement of health-related quality of life, and the preservation of the ability to work (the latter only if a participant was at work at onset of disease). This inpatient rehabilitation program usually takes place approximately 3 weeks after the acute event or cardiac surgery but this time interval may be larger in some cases. In this study only patients who were admitted within 3 months from the acute event or cardiac surgery were included.
Patients included in the study filled out a standardized questionnaire at the beginning and at the end of the rehabilitation program and underwent a medical examination. This analysis is based on information collected at the beginning of the rehabilitation program. The questionnaire included a detailed medical history regarding prior diagnoses of various cardiovascular diseases, as well as physician-diagnosed diseases related to CHD, such as diabetes mellitus. In addition, we asked for the age at which the diagnoses were first made, and a lifetime history of various risk factors was obtained.
Overall, 1222 persons were recruited in the study. This retrospective analysis is restricted to 907 persons for whom exact information on age at onset (first diagnosis by physician) of CHD was available and ACE and PAI genotypes could be determined.
The study was approved by the Ethics Boards at the Universities of Ulm and Heidelberg and of the Medical Chambers of Baden-Wuerttemberg and Hessen.
Laboratory methods
Genomic DNA was prepared from EDTA anticoagulated peripheral blood by using a common salting-out procedure [30].
The PAI-polymorphism was determined as described by Nauck and colleagues [31]. To confirm genotypes, allele-specific restriction enzyme analysis was performed as described. This protocol employs a modified upstream primer producing a Bsl I cutting site only in the presence of the 5G but not of the 4G allele. A 99 or 98 base pair fragment, depending on the promotor allele present, was amplified from genomic DNA using the primers described by Margaglione et al. [32].
Amplifications were performed in volumes of 50μl containing 100 ng of genomic DNA, 0.8 μmol/l of each primer, 200 μmol/l of each deoxyribonucleotide, 10 mmol/l Tris-HCl (pH 8.3), 1.5 mmol/l MgCl2, 50 mmol/l KCl, 0.1 g/l gelatin and one unit thermus aquaticus DNA polymerase (Roche Molecular Biochemicals, Germany). The protocol included an initial denaturation step at 94°C for 2 min, followed by 40 cycles with each cycle consisting of 20 s of denaturation at 94°C, 20 s of annealing at 60°C, and 20 s of elongation at 72°C.
Thermocycling was carried out on a conventional programmable thermocycler (UNO II; Biometra, Goettingen, Germany), followed by a final elongation step of 72°C for 5 min. Aliquots of 10 μl of the polymerase chain reaction (PCR) mixture were then digested for 4 h at 55°C with 5 U of Bsl I using the restriction buffer recommended by the manufacturer (New England Biolabs, Beverly, Massachusetts, USA). The resulting fragments (77 and 22 base pairs for the 5G allele; 98 base pairs for the 4G allele) were electrophoresed on 3.5% GTG agarose (FMC Bioproducts, Rockland, Maine, USA), visualized with 0.5 μg/ml ethidium bromide and examined under ultraviolet illumination.
The I/D ACE-polymorphism was determined as described by Winkelmann and colleagues [33]. This protocol employs three different primers to amplify three fragments of different length.
Amplifications were performed in volumes of 25 μl containing 100ng of genomic DNA, 25pmol respectively 12.5 pmol of each primer, 200 μmol/l of each deoxyribonucleotide. The protocol included an initial denaturation step at 96°C for 3 min., followed by 40 cycles with each cycle consisting of 20 s of denaturation at 95°C, 60 s of annealing at 56°C, and 30 s of elongation at 72°C.
Thermocycling was carried out on a conventional programmable thermocycler (UNO II; Biometra), followed by a final elongation step of 72°C for 7 min. The resulting fragments (371 and 65 base pairs for the inserted allele; 84 base pairs for the deleted allele) were electrophoresed on 3-GTG agarose (FMC Bioproducts), visualized with 0.5 μg/ml ethidium bromide and examined under ultraviolet illumination.
Statistical methods
Demographic characteristics as well as prevalence of known risk factors for CHD (hypertension, diabetes, hyperlipidemia, smoking cigarettes) of subjects with early (>55 years) and late (>55 years) onset of CHD were compared in a descriptive way. For persons with early onset of CHD the status of the risk factors hypertension, diabetes, hyperlipidemia and smoking cigarettes at age of onset of CHD given in the questionnaire was determined. For subjects with late onset of CHD, the status of these risk factors was determined at the age of 47 years, which is the mean age of disease onset among patients with early onset of CHD. With this approach, we simulated the data situation given in a case-control study in which controls would ideally be matched by age and in which the prevalence of risk factors would therefore be ascertained at disease onset among cases (here among patients with early onset who had a mean age of disease onset of 47 years) and among a control group of subjects without the disease (here no early onset of CHD) at a comparable age (at age 47 years).
The association of homozygosity for one polymorphism as well as the association of combined homozygosity with early onset of CHD and differences in the distribution of alleles and genotype combinations between individuals with early (>55 years) and late (>55 years) onset of CHD were assessed calculating X 2 statistics. Furthermore we used unconditional logistic regression to assess the risk of early onset of CHD (>55 years) associated with homozygosity for the two polymorphisms controlling for sex, age, diabetes, hypertension, hyperlipidemia, smoking and school education. Power estimations found the study to have a power of 80% to detect an odds ratio (OR) of 2.25 at α = 0.05 for early onset of CHD for persons with combined homozygosity. All procedures were carried out with the SAS-software package (version 8.0; SAS Institute, Cary, North Carolina, USA).
Results
Nine hundred and seven persons were included in the analysis (Table 1). The majority of the patients (84.2%) were male. The mean age of patients was 59 years. An early onset of the CHD (at or before the age of 55 years) had occurred in 45.0% of the CHD patients. A body mass index over 25kg/m2 was found in 71.4% of the patients, a history of hypertension, diabetes, hyperlipidemia or cigarette smoking was found in 69.7, 20.6, 74.4, and 70.3% of patients, respectively.
Main characteristics of the study population
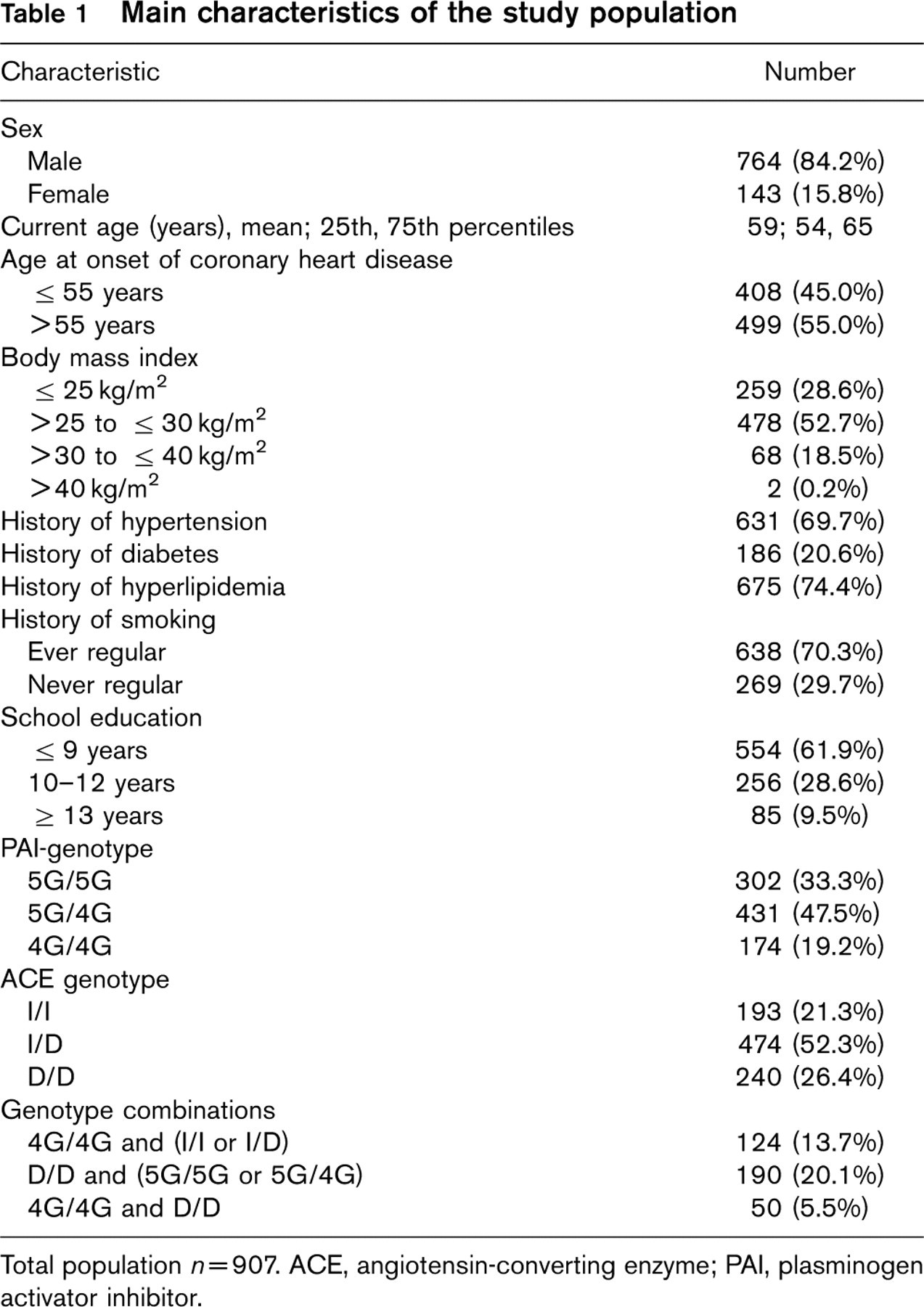
Total population n = 907. ACE, angiotensin-converting enzyme; PAI, plasminogen activator inhibitor.
With respect to the PAI-1 genotype 302 (33.3%) of the CHD patients were homozygous for the wild-type allele 5G, 431 (47.5%) of the patients were heterozygous, and 174 (19.2%) of patients were homozygous for the 4G allele. The PAI-1 genotype distribution in the study population was compatible with Hardy-Weinberg expectations (P = 0.57; X 2 = 0.84).
With respect to ACE genotype 193 (21.3%) of the CHD patients were homozygous for insertion allele (wild-type allele), 474 (52.3%) of the patients were heterozygous, and 240 (26.4%) of patients were homozygous for the deletion. The ACE genotype distribution in the study population was compatible with Hardy-Weinberg expectations (P = 0.47; X 2 = 2.14).
Characteristics of the study population with respect to age at onset of coronary heart disease (CHD)
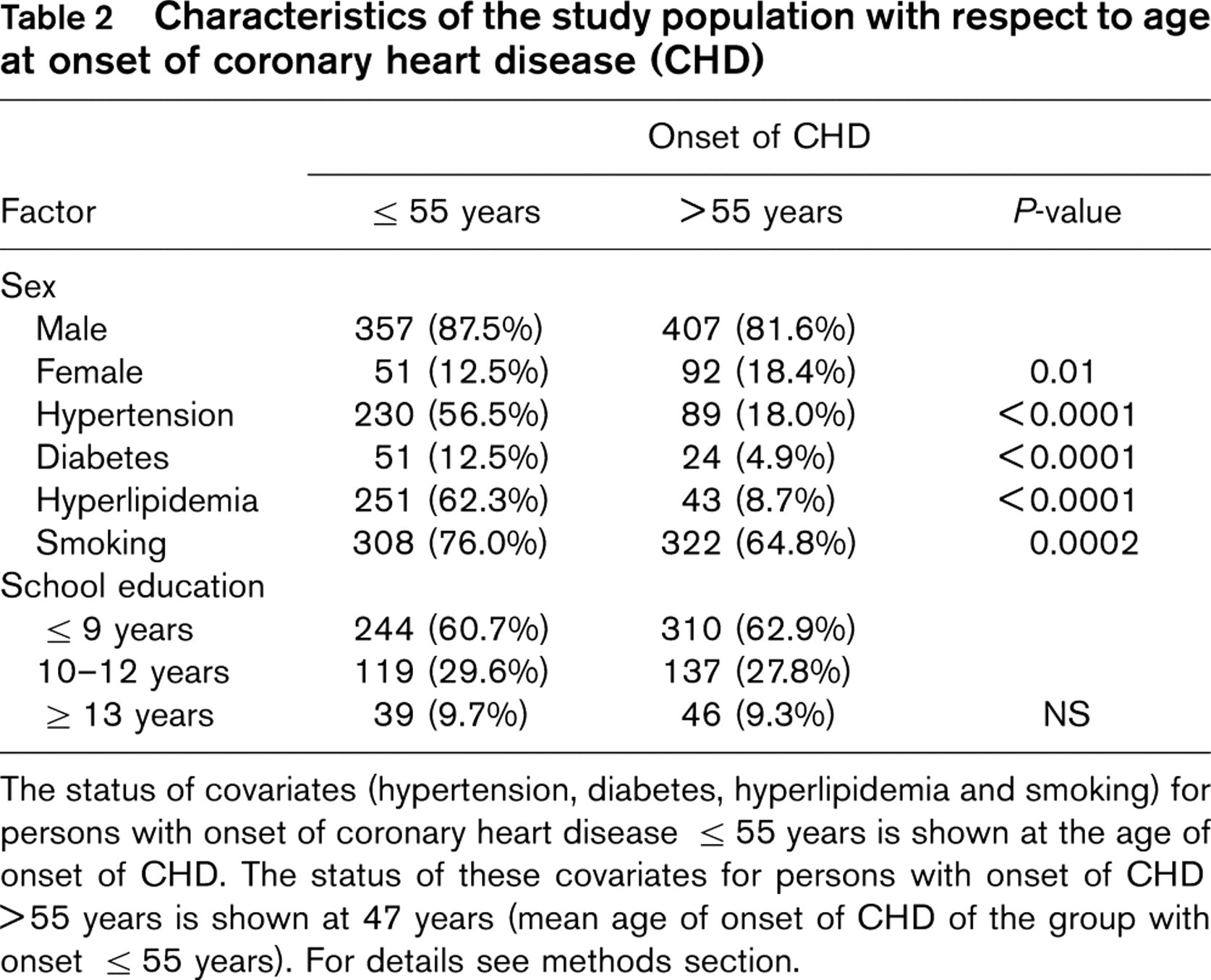
The status of covariates (hypertension, diabetes, hyperlipidemia and smoking) for persons with onset of coronary heart disease >55 years is shown at the age of onset of CHD. The status of these covariates for persons with onset of CHD >55 years is shown at 47 years (mean age of onset of CHD of the group with onset >55 years). For details see methods section.
Plasminogen activator inhibitor type-1 and angiotensin-converting enzyme genotype distribution and onset of coronary heart disease (CHD)
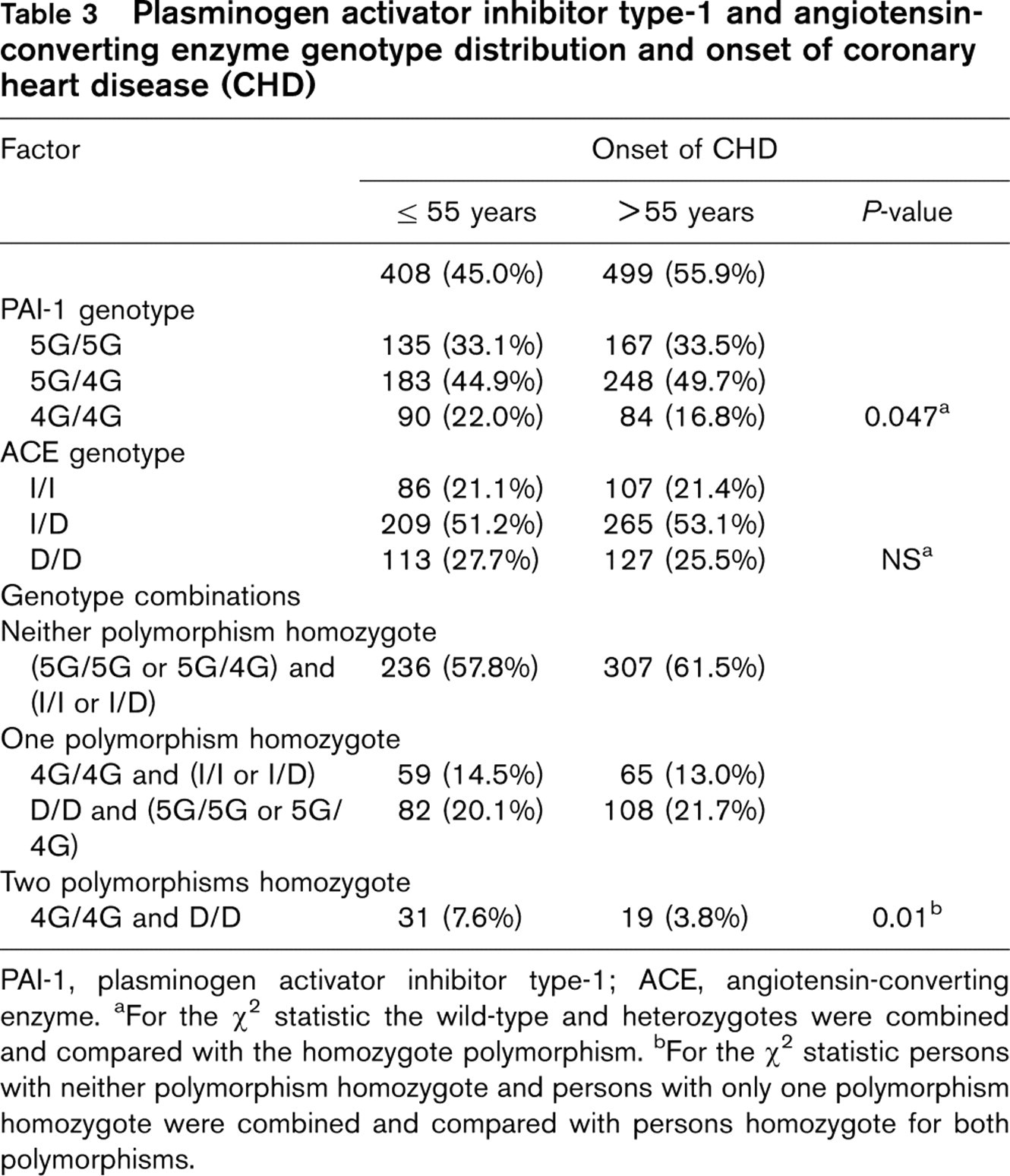
PAI-1, plasminogen activator inhibitor type-1; ACE, angiotensin-converting enzyme. aFor the X 2 statistic the wild-type and heterozygotes were combined and compared with the homozygote polymorphism. bFor the X 2 statistic persons with neither polymorphism homozygote and persons with only one polymorphism homozygote were combined and compared with persons homozygote for both polymorphisms.
If both genotypes were combined, 50 (5.5%) CHD patients were homozygous for the PAI-1 and for the ACE polymorphism showing the 4G/4G genotype of PAI-1 and the D/D genotype of ACE.
Table 2 shows the distribution of various characteristics according to onset of disease. Among individuals with an onset of CHD at or before the age of 55 years the proportion of males was significantly higher (87.5%) than in the group with later onset of disease (81.6%) (P = 0.01). The proportion of persons with classical risk factors (hypertension, diabetes, hyperlipidemia, smoking) was statistically significantly higher among persons with an early onset of CHD than in the group with later onset of disease. There was no statistically significant difference among the two groups with respect to duration of school education.
Crude and adjusted odds ratios for early onset of coronary heart disease (CHD) (>55 years)
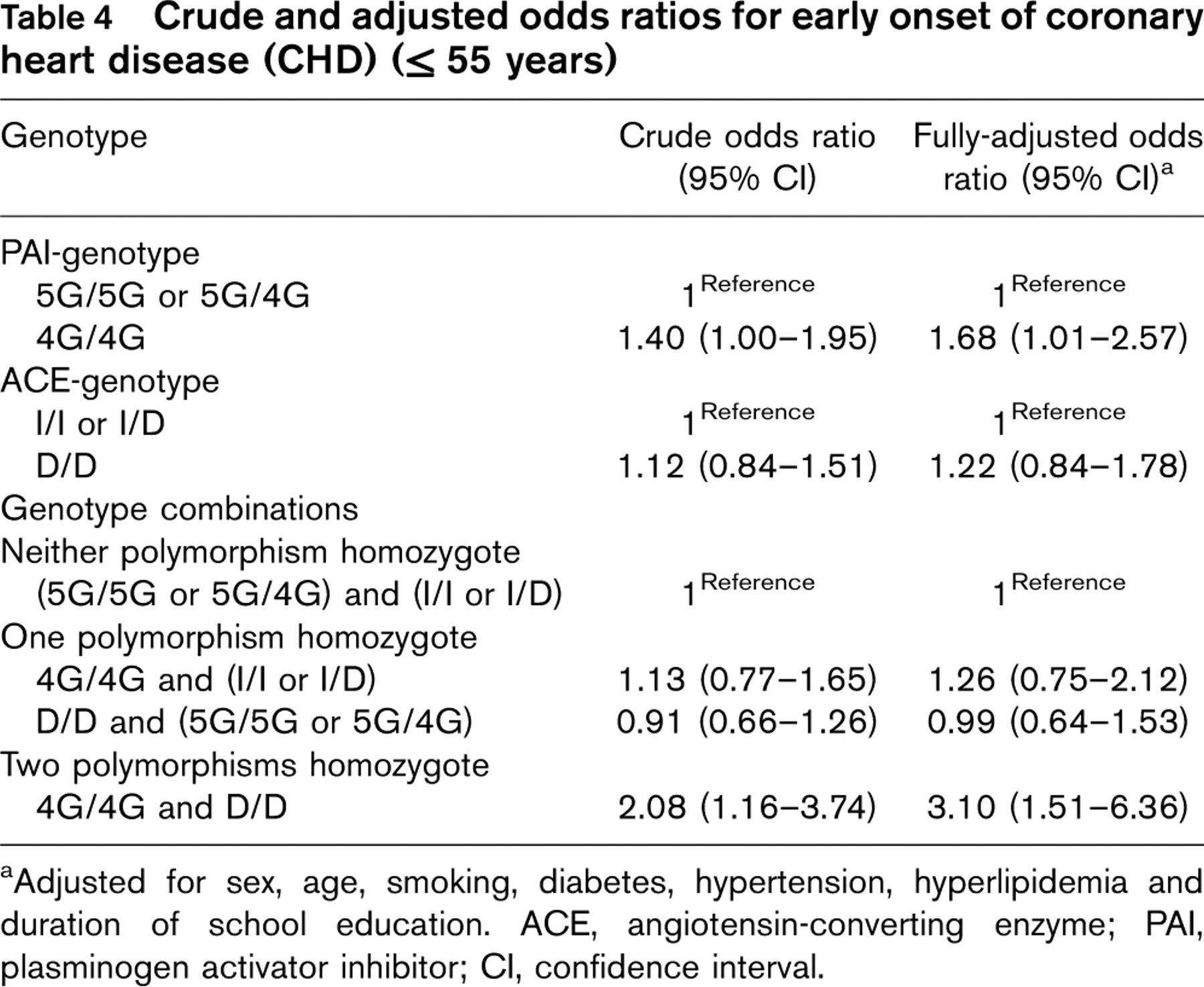
aAdjusted for sex, age, smoking, diabetes, hypertension, hyperlipidemia and duration of school education. ACE, angiotensin-converting enzyme; PAI, plasminogen activator inhibitor; CI, confidence interval.
Table 3 shows the distribution of the PAI-1 and ACE-genotypes alone and in combinations according to onset of disease. The proportion of individuals who were homozygous for both polymorphisms (i.e. had the 4G/4G genotype of PAI-1 and the D/D genotype of ACE) was statistically significantly higher in the group with early onset of CHD (7.6 versus 3.8%; P = 0.01).
Table 4 shows the estimated OR for early onset of CHD (at or before an age of 55 years) for each genotype alone and in combination. For the PAI-1 genotype the crude OR was 1.40 [95% confidence interval (CI) 1.00–1.95] when the 4G/4G genotype was compared to the 5G/4G or 5G/5G genotype. Adjustment for sex, age, smoking, diabetes, hypertension, hyperlipidemia and school education resulted in an OR of 1.68 (95% CI 1.01–2.57; P = 0.02). For the D/D genotype of ACE the crude OR for early onset of CHD was 1.12 (95% CI 0.84–1.51) and it increased to 1.22 (95% CI 0.84–1.76) after full adjustment for covariates.
When each of the genotypes was considered alone, no risk elevation was observed. By contrast the crude OR for early onset of CHD was 2.08 (95% CI 1.16–3.74) when the combined 4G/4G and D/D genotype was compared to the reference group of subjects with neither of the polymorphisms. After adjustment for sex, age, smoking, diabetes, hypertension, hyperlipidemia and school education, the OR for early onset of CHD increased to 3.10 (95% CI 1.51–6.36; P = 0.002).
Discussion
In this large study of a homogenous group of patients with coronary heart disease, we found a statistically significant association between the genotype combination 4G/4G of PAI-1 with D/D of ACE and early onset of CHD (at or before the age of 55 years) which seems biologically plausible. The genotype combination was associated with a threefold increased risk for early onset of CHD after adjustment for established risk factors compared with all other genotype combinations. This findings suggests that, besides the well established causal role of environmental and behavioral risk factors, this genotype combination may play an additional role in the etiology of CHD and may allow the identification of persons with increased risk for early onset of CHD.
First indications of a significant genetic contribution to the overall risk of CHD came from cases with a positive family history of CHD [34] and from twin studies [35]. So far many polymorphisms of genes possibly involved in CHD have been investigated. Yet many of the positive associations initially reported have not been replicated in subsequent studies. The ethnic diversity of the populations studied, findings by chance, the over-interpretation of subgroup analyses or the inadequate selection of cases or of controls may explain part of the inconsistencies in the literature [36]. The selection of controls is a critical point. In most studies, patients with diseases other than CHD were used as controls. Whether these controls adequately reflect the true prevalence of the polymorphisms in the population base which gave rise to the cases is often, however, questionable.
The clinical role of the PAI-1 4G/5G polymorphism remains controversial as both, positive [15, 17, 18] and negative associations have been reported [19–21]. One reason for this discrepancy may be the use of different endpoints in some studies because the underlying pathophysiology may be different. Small studies, apart from the limitations already discussed, may lack power to demonstrate a statistically significant difference. In a meta-analysis of nine studies Iacoviello and colleagues [37] found a slight increase in the risk of myocardial infarction in 4G homozygotes.
A recent study [38] in which 112 polymorphisms of 71 candidate genes were investigated in 2819 Japanese CHD patients and 2242 controls in a two-step analysis process gave support to a possible contribution of the PAI-1 polymorphism to CHD risk [38]. In that study, the 4G/5G polymorphism of the PAI-1 gene was among very few other gene polymorphisms associated with an increased risk for myocardial infarction. The result, however, was only statistically significant in women. In the current study, we did not find indication of a sex difference, but the power of our study to detect such a difference was limited due to the limited number of women included in the sample.
The pathophysiological role of ACE [5] and the established benefits of ACE-inhibitor therapy [6] made the ACE gene a candidate for susceptibility to CHD; an association between the DD-genotype and increased risk of CHD has been described [7]. Many other studies have subsequently investigated the association of the ACE I/D polymorphism with CHD. Results, however, are still inconclusive [8–11]. Recently a large case-control study with more than 10 000 participants showed no difference in frequency of DD-genotype between cases with myocardial infarction and controls [12].
One way to choose a control group representing the adequate study base is the use of an internal comparison group as it was performed in the current study. As atherosclerosis is considered to be a multi-factorial disease that results from a complex interplay of environmental, behavioral and possibly, genetic factors, an early onset of disease (respectively, first clinical manifestation) associated with a specific genetic factor may point to a possible role of the factor under investigation. This is even more the case if the increased risk, as in the current study, is independent of other established risk factors.
Currently, there are only few studies describing the role of genetic factors for the onset of CHD. Nora et al. [34] found the highest risk for CHD before the age of 55 years in cases with a positive family history of disease. In their twin study, Marenberg and colleagues [35] assumed genetic factors to be important for the onset of CHD at younger ages. Some studies have found polymorphisms in the apolipoprotein E gene to be independent risk factors for early onset of CHD [39–41]. Petrovic et al. [3] described an association between polymorphisms of the renin-angiotensin system and onset of CHD before the age of 55 years, whereas others did not find such an association [42, 43]. Furthermore polymorphisms in the thrombospondin genes were associated with early onset of CHD [44] and Nasser et al. described a spurious association between butyrylcholinesterase K and early onset of CHD [45]. Recently an Italian study group found no association between nine polymorphisms, including 4G/5G PAI-I polymorphism, and myocardial infarction at a young age in a case-control study, involving 1210 patients with myocardial infarction and 1210 healthy controls matched for age, sex and geographical origin. In their study, however, only myocardial infarction was used as endpoint and an age of 45 years was used as cut point for infarction at a young age [46].
An additional way to deal with the problem of many involved genes with small impact of CHD risk is considering pathophysiologically based genotype combinations [2, 47]. An interaction between the DD genotype of ACE and the 4G/5G polymorphism of PAI-1 regarding PAI-1 plasma level was recently described [23].
Our study was the first to describe an association between combined homozygosity for the 4G/5G polymorphism of PAI-1 and the I/D polymorphism of ACE with early onset of CHD. The described OR of 3.1 suggests that it may be possible to identify high-risk persons by combined investigation of PAI and ACE polymorphisms to intervene at a very early stage of disease development.
Our study has the following limitations. The recruitment of cases from a rehabilitation program may lead to some bias as only survivors of acute coronary events will get into such a program. Furthermore, because participation was limited to patients under 70 years, women may be under-represented.
We determined the age at onset of CHD retrospectively from the patient questionnaire. Therefore, there is some room for recall bias. As a potential reporting error (with respect to age of onset of CHD), however, would most likely be independent of the exposure (i.e. genotype) a potential misclassification bias would most likely be towards null: the true association might even be higher than the described one. Our study population comprised solely subjects with CHD and so we could not compare subjects with early onset of CHD with subjects without CHD. This limitation is at the same time an advantage, also: we had a homogeneous study population for internal comparison with respect to the age at onset of CHD and were not confronted with the various problems possibly related to other sources of controls [36].
We further used a novel statistical approach to simulate the prevalence of risk factors in an ideally matched case-control study to adequately estimate the prevalence of these risk factors in the comparison population. Finally, we assessed in our analysis two important polymorphisms and their combination based on an a priori hypothesis raised by pathogenetic considerations. Some authors suggest considering many polymorphisms simultaneously, respectively, genotype combinations [2, 47]. Testing multiple polymorphisms, however, would lead to chance findings unless statistical tools are used to adjust for multiple testing [36], an issue less important in our study approach.
In summary our results suggest an association between the combined homozygosity for the 4G-allele of the PAI-1 gene and the D-allele of the ACE gene and early onset of CHD. Clearly these results require confirmation by subsequent studies. If confirmed, our findings might have important implications regarding identification of subjects at increased risk of early onset of CHD who might benefit most from possible prevention measures.