
Abstract
Large randomised studies have established that early intensive glycaemic control reduces the risk of diabetic complications, both microvascular and macrovascular. However, epidemiological and prospective data support a longterm influence of early metabolic control on clinical outcomes. This phenomenon has recently been defined as ‘metabolic memory'. Furthermore, evidence suggests that ‘glucose variability’ may also be an independent risk factor for cardiovascular complications in diabetes. Studies suggest that all these different situations of hyperglycaemia share a common pathogenetic mechanism, increased oxidative stress, producing an endothelial dysfunction. The therapeutic challenge derived from these evidences is a need not only for an early tight glycaemic control, but also for maintaining glycaemia within a strict normal narrow range. Eur J Cardiovasc Prev Rehabil 17 (Suppl 1):S15-S19 © 2010 The European Society of Cardiology
The hallmark of diabetes is hyperglycaemia and in both type 1 and type 2 diabetes, large prospective clinical studies have shown a strong relationship between time-averaged mean levels of glycaemia, as measured by haemoglobin A1c (HbA1c), and cardiovascular complications [1–2]. However, in recent years, several pieces of evidence have raised the possibility that several different aspects of hyperglycaemia may contribute, perhaps more than HbA1c levels, to the development of diabetic complications.
An early intensive glycaemic control reduces the risk of diabetic complications. However, data support a long-term influence of early metabolic control on clinical outcomes. This phenomenon has recently been defined as ‘metabolic memory’ [3]. Furthermore, glycaemic ‘instability,’ in terms of ‘postprandial hyperglycaemia’ [4] or ‘glucose variability’ [5] has recently been claimed as an independent risk factor, particularly for cardiovascular disease, in diabetes.
The endothelium is a major organ involved in the development of cardiovascular disease even in diabetes [6]. Therefore it is of great interest to understand how these different aspects of hyperglycaemia impact on the vessel wall and contribute to the global burden of cardiovascular risk in diabetes.
Different ‘hyperglycaemias’ share the activation of the same damaging pathways
Brownlee [7] has recently pointed to an excess of superoxide anion (•O− 2), a reactive species, in the mitochondria of endothelial cells in response to hyperglycaemia with the formation of diabetic complications. This new insight further links with four key pathways suggested to be involved in the development of these complications [increased polyol pathway flux, increased advanced glycation endproduct (AGE) formation, activation of protein kinase C and increased hexosamine pathway flux], into a unifying hypothesis regarding the effects of hyperglycaemia on the development of diabetic complications [7]. Oxidative stress underlines the effect of hyperglycaemia in producing endothelial dysfunction, which consistently contributes to the development of cardiovascular complications [8] (Fig. 1).
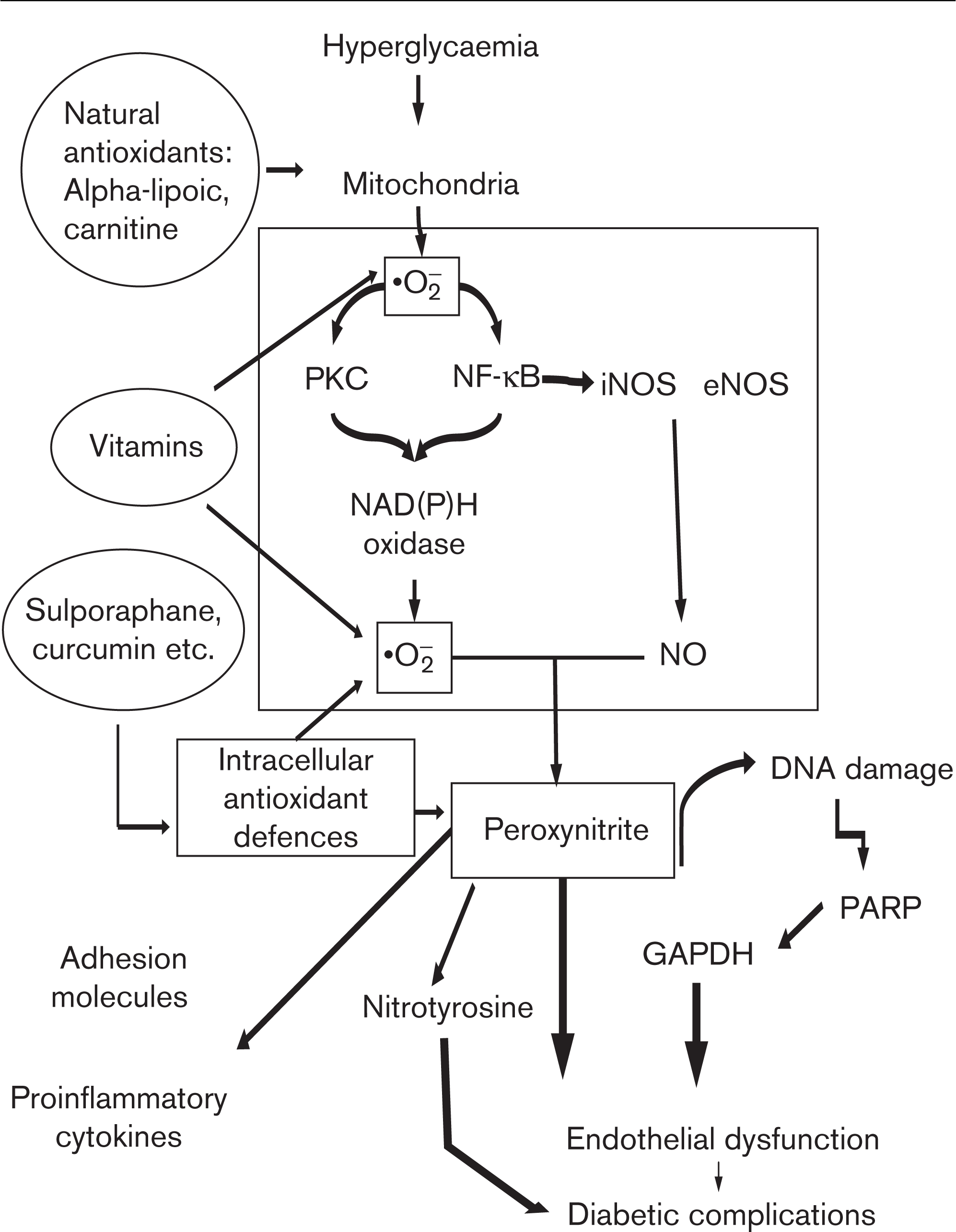
In the cells, hyperglycaemia induces an overproduction of superoxide (•O− 2) at the mitochondrial level, and nitric oxide (NO) overproduction through both inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS), whereas protein kinase C (PKC) and nuclear factor kappa B (NF-kB) are activated and they favour an overexpression of the enzyme NAD(P)H. Oxidase NAD(P)H generates a great amount of superoxide. Superoxide overproduction, accompanied by increased nitric oxide generation, favours the formation of the strong oxidant peroxynitrite, which, in turn, damages DNA. DNA damage is an obligatory stimulus for the activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP). PARP activation, in turn, reduces glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity. This process results in endothelial dysfunction, which, in turn, contributes to the development of diabetic complications. The new approach to a possible natural antioxidant therapy now takes into account three different possibilities: vitamins, substances which can balance free radical production at the mitochondrial level and substances able to induce their own antioxidant defenses in the cells. All these three different substance types are present in vegetables. Adapted from Ref. [8].
Evidence suggests that all the above-mentioned different ‘hyperglycaemia’ share oxidative stress as a common mediator. In the metabolic memory it has been shown that oxidative stress may induce persistent epigenetic changes [9]. These data are consistent with results obtained from human endothelial cells: after 14 days of culture in high glucose followed by 7 days of culture in normal glucose, both endothelial and retinal cells show an overproduction of free radicals, persistent after the normalisation of the glucose and accompanied by a prolongation of the induction of protein kinase C-β, NAD(P)H oxidase, apoptosis, collagen and fibronectin, in addition to 3-nitrotyrosine production [10]. The role of oxidative stress has been confirmed because the use of several intracellular antioxidant approaches, reducing intracellular production of free radicals particularly at mitochondrial level, has been able to switch off the metabolic memory [10]. Similar results have been reported in diabetic rats, in which a-lipoic acid was used as an antioxidant [10].
It is assumed that chronic hyperglycaemia alters mitochondrial function through glycation of mitochondrial proteins [11]. This is important in the light of a recent study, which for the first time described a direct relationship between the formation of intracellular AGEs on mitochondrial proteins and the decline in mitochondrial function and excess formation of reactive species [11]. Therefore, mitochondrial respiratory chain proteins which have undergone glycation are, independently of the level of hyperglycaemia, prone to produce more •O− 2. The glycation of mitochondrial proteins may be a contributing explanation for the phenomenon of the metabolic memory. Mitochondrial overproduction of free radicals independent of the actual glycaemia, can lead to a catastrophic cycle of mitochondrial DNA damage and functional decline, further oxygen radical generation and cellular injury [3], maintaining the activation of the pathways involved in the pathogenesis of diabetic complications. Furthermore, mitochondrial proteins become damaged or posttranslationally modified because of a major change in a cell's redox status [3]. This may also affect mitochondrially destined proteins, which are imported into the mitochondrial outer membrane, inner membrane or matrix space through specific import machinery transport components [3]. Finally, oxidative stress may alter mitochondrial protein expression and turnover, possibly leading to a perpetuation of the phenomenon [3].
Evidence suggests that AGEs and their receptor (RAGE) axis may be involved in the metabolic memory. There is a growing body of evidence that engagement of RAGE with AGEs elicits oxidative stress [12]. Binding of AGEs to RAGE results in the generation of intracellular reactive oxygen species and subsequent activation of the redoxsensitive transcription factor nuclear factor-kB in vascular wall cells, which promotes the expression of a variety of atherosclerosis-related genes and RAGE itself [12]. Therefore, self-maintaining conditions linked to the formation of AGEs would be a conceivable contributor to the metabolic memory.
On the basis of available evidence, it may be postulated that the cascade of the events in metabolic memory is the same as that proposed by Brownlee [7], that is, the source of •O− 2 is still the mitochondria. In addition, the subsequent production of reactive species becomes unrelated to the presence of hyperglycaemia, depending on the earlier production of AGEs, which will maintain RAGE overexpression in relation with the level of glycation of mitochondrial proteins and the amount of mitochondrial DNA produced, all conditions are able to induce an altered gene expression which is persistent even when glycaemia is normalized [3] (Fig. 2).
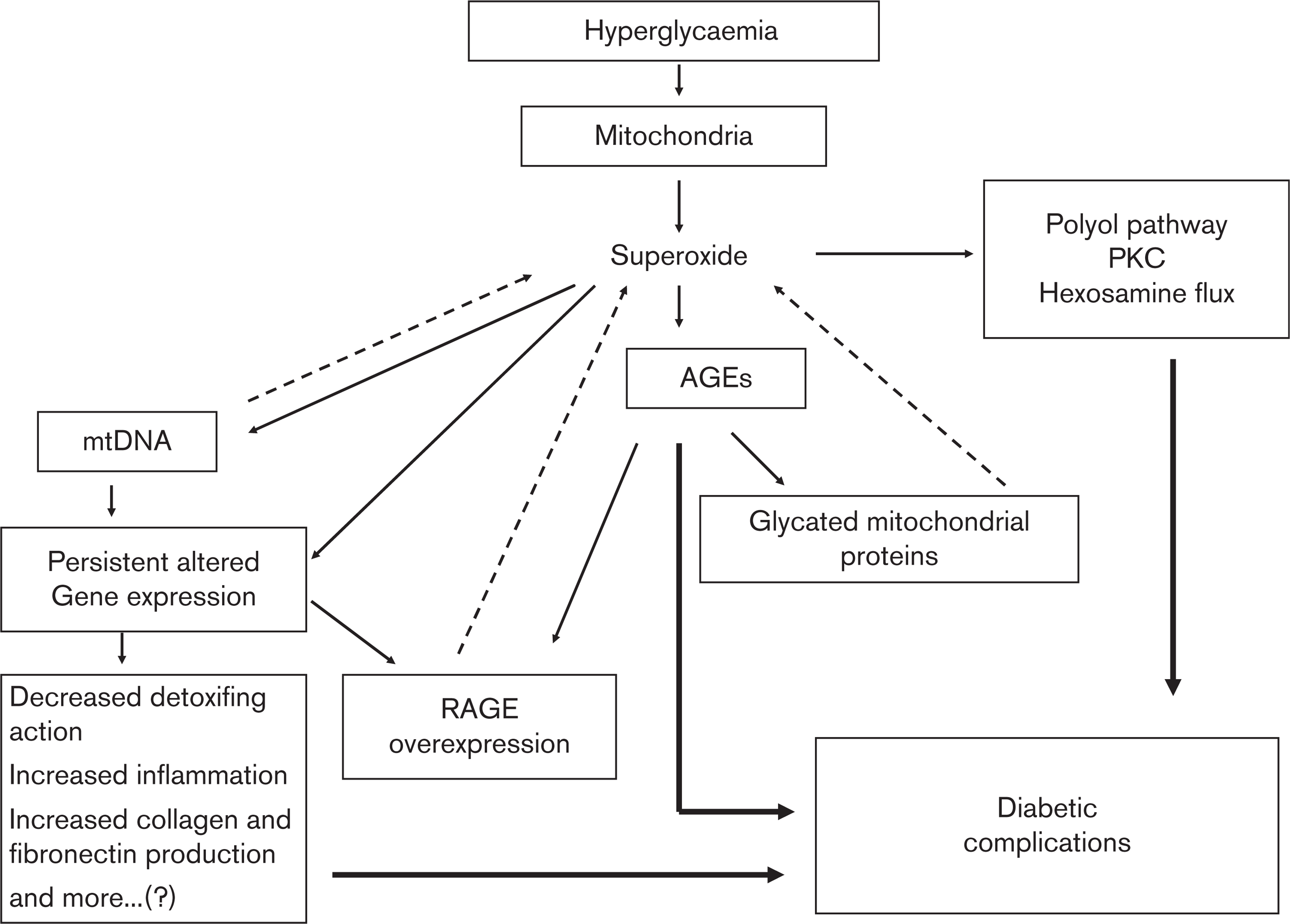
Intracellular hyperglycaemia induces overproduction of superoxide at the mitochondrial level. Overproduction of superoxide is the first and the key event in the activation of all other pathways involved in the pathogenesis of diabetic complications, such as polyol pathway flux, increased advanced glycation end-product (AGE) formation, activation of protein kinase C (PKC) and increased hexosamine pathway flux. Mitochondrial proteins are glycated in hyperglycaemia and this effect induces mitochondria to overproduce superoxide anion, a condition, which does not depend on the actual glycaemia. Binding of AGEs to receptor AGE (RAGE) results in the generation of intracellular reactive oxygen species generation, which promotes the expression of RAGE itself. Oxidative stress may alter gene expression, favouring the permanence of a hyperaction of free radicals. Mitochondrial DNA (mtDNA) may influence gene expression and at the same time may contribute to an overgeneration of free radicals at the mitochondrial level. These self-maintaining conditions, leading to a persistent oxidative stress generation independently from the actual levels of glycaemia, may contribute to the appearance of the metabolic memory.
Several laboratory studies have already approached the issue of the ‘glucose variability'
Increased apoptotic cell death has been observed in endothelial [13] cells in response to fluctuating high glucose levels as compared with continuous high glucose. Evidence suggests that the same phenomenon, increased superoxide production at the mitochondrial level, underlines the deleterious effect of oscillating glucose levels [14,15]. Experiments in animals support the hypothesis of a deleterious effect of fluctuating glucose levels. Recently, Azuma et al. [16] established a method, which permits the observation of the entire surface of the endothelium of a rat aorta to quantitate the number of attached monocytes, a marker of vascular inflammation [16]. Using this method, these investigators showed that repetitive fluctuation of hyperglycaemia resulted in significantly induced monocyte-endothelial adhesion as compared with sustained hyperglycaemia [17]. Furthermore, they assessed the role of glucose fluctuations on atherogenesis in atherogenic-prone mice, fed with maltose twice daily to model repetitive glucose spikes [18]. Fluctuations in blood glucose concentrations accelerated macrophage adhesion to endothelial cells and the formation of fibrotic arterosclerotic lesions. The role of oxidative stress in this phenomenon, in animals, has recently been confirmed [19].
The entire laboratory data presented are consistent with clinical data about metabolic memory. Therefore, several studies have shown that hyperglycaemia induces endothelial dysfunction in both diabetic and nondiabetic individuals [8]. A clear demonstration that controlling hyperglycaemia can restore or normalise endothelial function is, however, still lacking. For example, in patients with type 1 diabetes endothelial dysfunction has been reported to be present even when normoglycaemia was achieved [20–21]. Furthermore, several studies indicate that hyperglycaemia induces endothelial dysfunction through the generation of oxidative stress [4]. A recent study confirmed that endothelial dysfunction persists in patients with type 1 diabetes even after normalisation of hyperglycaemia [22]. Simultaneously, the possibility of improving endothelial function in type 1 diabetes using an antioxidant, particularly vitamin C, was confirmed [22]. This study was indeed the first to show that combining vitamin C and glucose normalisation almost normalised endothelial function [22]. The role of oxidative stress in this phenomenon appeared crucial: when endothelial function was still altered after 12 h of normalisation of glycaemia or 12 h of vitamin C treatment, nitrotyrosine, a good marker of peroxynitrite production and nitrosative stress, was still increased, whereas when endothelial function was normalised, combining glycaemia control and vitamin C, nitrotyrosine levels were also normalised. However, in all these studies the duration of the disease and long-term glycaemic control were not taken into consideration. In more recent studies these conditions were accounted for. Results indicate that endothelial dysfunction, inflammation and oxidative stress are almost normalised after normalising glycaemia or vitamin C administration into type 1 diabetic patients with less than 1 month since the diagnosis and in patients with a disease duration between 4.5 and 5.2 years and with HbA1c levels ≤7%, whereas a normalisation was not accomplished in patients with a disease duration between 4.5 and 5.2 years and a HbA1c greater than 7% since diagnosis. This suggests that long-lasting poor glycaemic control can lead to long-term endothelial dysfunction, which does not simply respond to plasma glucose normalisation or to antioxidants [23,24].
The role of glucose variability has also been widely studied in vivo. Repeated fluctuations of glucose produce increased the circulation levels of inflammatory cytokines, as compared with stable high glucose in normal individuals, and endothelial dysfunction in both normal individuals and patients with type 2 diabetes [25]. The role of oxidative stress also seems to be a key causative factor clinically, as the use of an antioxidant reduced the phenomenon [25]. Consistent with the hypothesis of an involvement of oxidative stress is the evidence that in type 2 diabetes, glucose fluctuations are strongly predictive of increased generation of oxidative stress [26]. In a recent study, looking at the effect of oscillating hyperglycaemia on endothelial function and oxidative stress, endothelial function and nitrotyrosine never returned to basal levels during periods with normal glycaemia in normal individuals in contrast to the normalisation noted in those with diabetes [27]. At the same time there was carry-over effect of oscillating glucose, however, only in normal individuals: after the second period of exposure to high glucose both endothelial dysfunction and nitrotyrosine were more compromised than after the first period of exposure to high glucose. Furthermore, vitamin C was able to normalise endothelial function and oxidative stress only in normal, but not in diabetic patients during the period of hyperglycaemia. When glycaemia was normalised in diabetic patients the simultaneous administration of vitamin C almost normalised both endothelial function and oxidative stress [27]. A hypothetical explanation of all these findings is that two pathways are simultaneously working: one because of the actual level of glycaemia and another one because of the long-lasting damage induced in the endothelial cells by chronic hyperglycaemia.
Even if the generation of oxidative stress seems to be the key player of all the phenomena reported above, the precise mechanism through which oscillating glucose levels may be more deleterious than constantly high glucose levels remains to be completely defined. Although further studies are necessary they would be difficult to accomplish in humans. A possible explanation is that in oscillating glucose conditions the cells are not able to sufficiently increase their own intracellular antioxidant defenses [28], a condition which has been suggested to favour the development of diabetic complications [29,30].
Conclusion
Consistent newly emerging evidence suggests that hyperglycaemia can leave an early imprint in cells of the vasculature, favouring the future development of vascular complications and that this effect is mediated by oxidative stress. In addition, evidence suggests that this ‘memory’ can appear even when a good control of glycaemia is achieved. This phenomenon has been named as metabolic memory [3]. Furthermore, it has been suggested that glucose swings could also be very deleterious for the vasculature [4,5]. These evidences raise many questions regarding the therapeutic management of patients with diabetes. In particular, the existence of the metabolic memory suggests that an early aggressive treatment of hyperglycaemia seems to be mandatory, and that an even more challenging therapeutic strategy would be necessary to maintain glycaemia within a very strict normal narrow range during long-term follow-up.
Footnotes
Acknowledgements
Antonio Ceriello reports no conflicts of interest.