
Abstract
Human immunodeficiency virus (HIV) is a virus that causes acquired immunodeficiency syndrome, a chronic and incurable disease of the human immune system. As the standard of care for the patients with HIV-1, current highly active antiretroviral treatment has been therapeutically effective in most patients; however, it is not curative, and highly active antiretroviral treatment is intolerable because of severe adverse effects. Therefore, nucleic acid-based therapeutics, such as antisense oligonucleotide, ribozyme, messenger RNA, RNA interference (RNAi)-based therapeutics, aptamer, and so on, have been actively developed as alternative or adjuvant agents for those chemical antiviral drugs to surmount those drawbacks. The combinatorial use of various antiviral nucleic acids could be more efficacious in blocking viral replication and preventing the emergence of resistant variants. In this regard, RNAi can function as a gene-specific therapeutic option for controlling HIV-1 replication. Another type of therapeutic nucleic acid—aptamers—shows promise as a new and potent class of anti-HIV agent and can additionally function as a cell-type–specific delivery vehicle for targeted RNAi. The combined use of small interfering RNA (siRNAs) and aptamers could effectively block viral replication and prevent the emergence of resistant variants. The present review offers a brief overview of the use of cell-type–specific aptamer and aptamer-siRNA conjugates’ development in our group for the treatment of HIV-1. Their potentials for targeted delivering RNAi therapeutics (eg, siRNA) and suppressing HIV-1 replication in vitro and in humanized animal model will be highlighted here.
Therapeutic strategies designed to treat human immunodeficiency virus (HIV) infection with combinations of antiviral drugs (eg, highly active antiretroviral treatment [HAART]) have proven to be the standard approach for delaying the progression to acquired immunodeficiency syndrome and reducing the risk for death and disease complication. 1 However, dosing regimens for the individuals are difficult, and drugs must be taken daily. Drug resistance and serious adverse effects remain a concern with some patients not responding to HAART.2,3 Nucleic acid-based therapeutics, such as ribozyme, antisense oligonucleotide (ASO), small interfering RNA (siRNA), aptamer/decoy, messenger RNA (mRNA), might represent a potential alternative or an adjuvant to the chemical antiviral agents.4,5
The early development of nucleic acid-based anti–HIV-1 agents mainly included ASOs and RNA decoys.6–8 For instance, ASOs specific to transactivating response region (TAR), Rev response element, HIV group-specific antigen and HIV envelop, and RNA decoys against TAR and Rev binding element have been reported for inhibiting HIV-1 replication.8–10 Later on, ribozyme and aptamer have rapidly emerged as important HIV-1 suppressors.11–14 Currently, a number of ribozymes or highly specific aptamers targeting various HIV-1 genomes and protein, or HIV-1 dependency factors (HDFs), including HIV-1 group-specific antigen, TAR, viral infectivity factor, long terminal repeat, reverse transcriptase, integrase, nucleocapsid, envelop (gp120 protein), transactivator of transcription (tat), regulator of expression of virion protein (rev), ribonuclease H cluster of differentiation 4 (CD4), and C-C chemokine receptor type 5 (CCR5), have been generated and indicated an effective inhibition to viral replication.15–19 Most recently, more efficacious RNAi-based therapeutics, such as siRNAs and small hairpin RNAs (shRNAs), have shown unique promise in controlling HIV-1 infection.20–22 The entire HIV genome can be specifically targeted by RNAi agents, whereas some cellular receptors that are essential for viral entry (eg, CD4, CCR5, C-X-C chemokine receptor type 4) or other HDFs (eg, transportin-3) and KH domain containing, RNA binding, signal transduction associated 1 also represent potential targets for siRNA-mediated gene silencing in vitro and in vivo models.23–25
To date, the most efficacious drug therapy for HIV uses a combination of 2 or 3 drugs targeting HIV reverse transcriptase and protease.1,26 It is very likely that combinations can prolong and probably prevent resistant viral variants.27–29 Therefore, it should be attractive to develop combinatorial nucleic acid-based therapies, which would be more potential in suppressing viral replication and minimizing the viral mutation escape in comparison with single nucleic acid therapeutic agent. Analogous to HAART, 4 shRNAs were incorporated in a single lentiviral vector, which prevented HIV-1 escape in an in vitro cell culture system. 30 In addition, a triple combination of an anti–HIV-1 tat/rev shRNA with a nuclear-localizing TAR RNA decoy and an anti-CCR5 hammerhead ribozyme has been constructed (lentiviral vector rHIV7-shI-TAR-CCR5RZ) to effectively combat HIV-1 replication in primary hematopoietic stem cells. 31 Importantly, a prolonged viral suppression for 6 weeks was observed, thus providing more effectiveness than an anti–HIV-1 tat/rev shRNA alone or double combinations of the shRNA with ribozyme or shRNA with decoy. Recently, the triple combinatorial construct has been evaluated in a human clinical trial (ClinicalTrials.gov identifier NCT00569985). 32
The technical term aptamer came from the Latin word aptus “to fit” and the Greek word meros for “part.” Nucleic acid aptamers are in vitro evolved single-stranded DNAs or RNAs that can fold into a specific 3-dimensional conformation to fit together with their targets. With the development for the past 24 years, numerous nucleic acid aptamers have been evolved against thousands of various targets, including small metal ions, organic dye molecules, short peptides, large proteins, viruses, and even whole cells or organisms. Many research and review articles have described their selection and application in the different fields. Because of their nature of nucleic acid, the DNA or RNA aptamers can form specific diverse structures, such as stem, internal loop, hairpin, bulge, pseudoknot, G-quadruplex motif, or kissing complex, which are essential for the target recognition. Because of the exquisite specificity and binding affinity, nucleic acid aptamer itself can stand alone as a potential therapeutics.33,34 In 2005, the first therapeutic aptamer, called pegaptanib (Macugen) offered by OSI Pharmaceuticals, a polyethylene glycol-modified RNA molecule, has been approved by the U.S. Food and Drug Administration for the treatment of age-related macular degeneration. In addition, at least 10 aptamer-based therapeutic agents are currently being evaluated in the clinic phases I and II.
On the other hand, cell-type–specific aptamers, as a building block, can be assembled with other therapeutics or delivery system, thus resulting in the specific recognition and further improving cellular internalization of the delivered cargoes.35,36 Various molecules or delivery vehicles, such as anticancer drugs, toxin, enzymes, radionuclides, virus, ASOs, RNAi triggers (siRNAs, shRNAs, microRNAs), and nanomaterials, have been functionalized by cell-type–specific aptamers for targeted delivery purpose.35,37 Recently, several groups have successfully adapted HIV-1 gp120-specific aptamers38,39 and host cellular receptor CD4 aptamers40,41 for the targeted delivery of siRNA molecules into cultured cells and humanized mice model. Our laboratory is interested in developing and testing complementary highly innovative strategies to target HIV-1 infected cells and specifically regulate both gene silencing of HIV-1 and inhibition of the CD4 receptor-HIV-1 envelope glycoprotein (gp120) interaction required for HIV-1 to enter cells. Through “purified protein-based SELEX,” we successfully identified 2’-fluoropyrimidine modified RNA aptamers from an RNA library that bind to the HIV-1 gp120 with nanometer affinity. 39 The selected anti-HIV gp120-specific aptamers have been successfully combined with anti-HIV siRNAs to achieve a dual-inhibitory drug that is able to deliver siRNAs selectively to HIV-infected cells for target mRNA knockdown as well as inhibiting viral entry via blocking of the envelope interaction with the CD4 receptor. Our efforts demonstrate that the nucleic acid drugs can be successfully used in combinations resulting in efficacious treatments for HIV infection. The importance of such a strategy for the treatment of HIV-1 cannot be overstated as viral variants resistant to current therapies are on the rise in developed nations and portend a new pandemic of resistant virus. In the following section, we will mainly focus on our own efforts in the development of cell-type–specific aptamer and combinatorial use of aptamer and siRNA for HIV-1 therapy. We will highlight representative examples and also discuss the challenges of advancing the combination of antiviral aptamer and siRNA therapeutics.
Development of Hiv-1 Gp120 Aptamers as Hiv-1 Inhibitor
Systematic evolution of ligands by exponential enrichment (SELEX) that is an evolutionary strategy to generate high-affinity nucleic acid aptamers was described primarily in 1990.42–44 The SELEX condition can be further refined and modified to drive the selection to aptamers with desired features. Currently, 2 typical SELEX strategies have been used to select and identify cell-type–specific, nucleic acid aptamers. One is to use traditional purified membrane protein as target, 45 whereas another is to use whole live cells as selection target.46,47 Recently, some variants also have been developed. 48 For example, alternate use of these 2 procedures has been reported to improve the specificity to the target protein. Apart from whole cells, live animal/organ has also been used as a target for SELEX, which is the so-called “in vivo SELEX”.49,50
The HIV external envelope gp120 glycoprotein that is exposed on the surface of viral particles and the plasma membrane of HIV-infected cells represents a biomarker to distinguish HIV-1 infected cells from noninfected cells.51,52 The interaction between the HIV gp120 and the human cellular CD4 receptor is a crucial trigger in the entry of HIV into T cells, subsequently resulting in fusion of the viral membrane with the host cell membrane.53,54 Therefore, generating some blockers directed to the virus surface gp120 protein should be a direct and rational antiviral strategy to prevent the viral infection. 55 Several success examples in the development of nucleic acid aptamers against HIV-1 gp120 that are capable of blocking the interaction of gp120 and CD4 have been reported in recent years.56–60 For example, William James and co-workers have described the selection and characterization of several anti-HIV gp120 RNA aptamers using a BIAcore biosensor system. They demonstrated that the selected aptamer specifically bound to the conserved co-receptor interacting region of an R5 HIV-1 stain and suppressed HIV-1 infectivity in human peripheral blood mononuclear cells (PBMCs). 57
Our own efforts have also added several novel, RNase-resistant RNA members in the family of anti-HIV gp120 aptamer. 61 We used a standard in vitro SELEX procedure, in which a purified HIV-1 BaL gp120 envelop protein (R5 stain) as target and an RNA library containing 50 random nucleotides were used for the selection. The RNA/protein complexes were isolated from the unbound RNA species through a nitrocellulose filter-based partition method. Subsequently, the bound RNAs were recovered from the nitrocellulose membrane for the complementary DNA synthesis and reamplification. New transcripts of the DNA products were applied for the next round of selection. After 12 rounds of selection, the individual aptamer sequences were identified via cloning and sequencing analysis. A-1, one of the best aptamer candidates, displayed good binding kinetics to the target gp120 protein with low nanomolar dissociation constant (52 nM of apparent Kd value) calculated from a native gel mobility shift assay. Our results demonstrated that A-1 aptamer was specifically bound and then was rapidly internalized into the cells expressing HIV-1 gp120 protein. Importantly, A-1 aptamer significantly neutralized the replication of various HIV-1 stains (R5- and X4-tropic laboratory and clinical HIV isolates) in cultured CEM T-lymphocytes and in human PBMCs, suggesting the anti–HIV-1 gp120 aptamers offer potential and lead anti–HIV-1 inhibitors for the treatment of HIV-1.
The Strategies of Aptamer-Sirna Targeting in Hiv Therapy
Because the selective binding of anti–HIV-1 gp120 aptamers to the cells expressing HIV-1 gp120 protein can trigger rapid cellular internalization, the gp120 aptamers probably had the potential to be a cell-type–specific, internalizing targeting molecule for selective drug delivery to HIV-1 infected cells. We therefore took advantage of this property to explore aptamer-targeted RNAi strategies. It is expected that the combinatorial use of 2 nucleic acid therapeutics (gp120 aptamer and siRNA) would offer a “dual-inhibitory function” bonus for combating HIV-1. Our laboratory has developed 2 different types of anti-gp120 aptamer-siRNA conjugates (Fig. 1). The design and activity will be discussed in the following paragraphs.
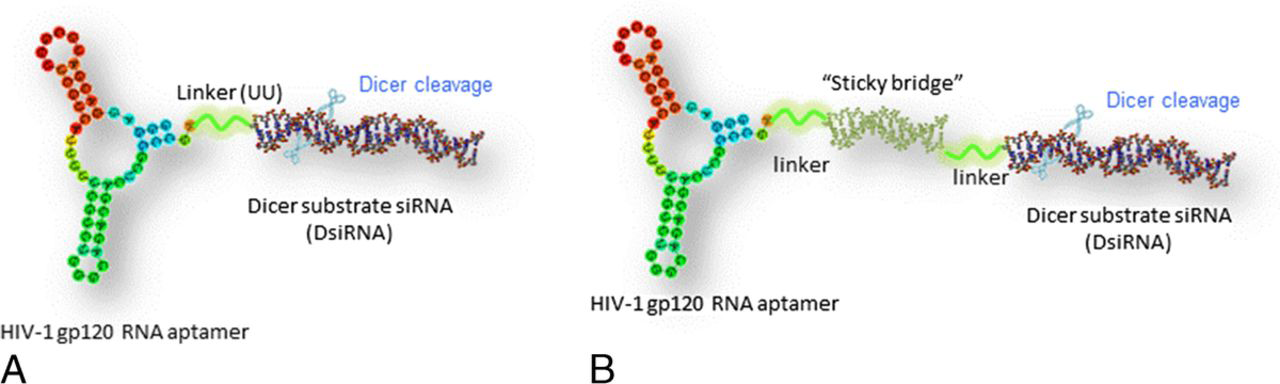
HIV-1 gp120 aptamer-mediated siRNA delivery system. A, Schematic of the covalent aptamer-siRNA chimera. The chimera was synthesized as 2 pieces followed by an annealing step to make the chimera. A 2-nucleotide (UU) overhang (green) was added between the aptamer and sense strand of siRNA. B, Schematic of the aptamer-stick-siRNA conjugate. The aptamer and siRNA were appended to complementary 17-nt 2’ OMe/2’ Fl GC-rich bridge sequences and were annealed by simple mixing that allowed Watson-Crick base pairing.
Covalent Aptamer-siRNA Chimeras
By adopting a similar strategy as previously described by Giangrande et al., 62 we covalently fused an anti-HIV gp120 aptamer (A-1) to a Dicer substrate siRNA (DsiRNA) targeting HIV-1 tat/rev common exon.38,61 In this design (Fig. 1A), the 2’-fluoropyrimidine modified gp120 aptamer A-1 covalently joined to the sense strand of tat/rev DsiRNA portion was generated via in vitro transcription and subsequently was annealed with an unmodified antisense strand to make the chimeric RNA molecule (A-1 tat/rev siRNA chimera). To improve the molecular flexibility and maintain the correct folding of aptamer, a 2-nucleotide spacer (UU) was inserted between the aptamer and the DsiRNA. Flow cytometric analysis and confocal microscopy study revealed that the chimera maintained the selective binding affinity and internalization efficiency as the aptamer alone. Moreover, anti-HIV efficacy of the resultant chimeric RNA was evaluated in the HIV-1 infected CEM T cells and PBMCs as described previously. We demonstrated that the gp120 aptamer-siRNA chimera was specifically taken up by HIV-1 infected cells, and the siRNA portion processed by Dicer was functioning along with the aptamer. The HIV-1 p24 enzyme-linked immunosorbent assay indicated that both of the aptamer and chimera neutralized the production of HIV-1 p24 antigen. However, the chimera treatment indeed was the most efficient. Furthermore, significant silencing of the target HIV-1 tat/rev gene was only observed in the infected cells treated with the chimera, but not the aptamer alone or the irrelative siRNA delivered by the same aptamer. These results supported that the aptamer intracellularly delivered siRNA, which subsequently triggered RNAi activity.
Encouraged by these cell culture results with aptamer-siRNA chimeras, we moved forward with the next step to evaluate the anti-HIV efficacy of the chimeras in an HIV-1 infected humanized Rag2−/− γc−/− mouse model (Rag-hu). 63 It has been demonstrated previously that HIV-1 infected Rag-hu mice can mirror the main features (eg, HIV-1 replication and CD4+ T cell depletion) of human HIV-1 infected patients, therefore providing an excellent model for the evaluation of anti–HIV-1 agents.64–67 In our study, approximately 3 weeks after HIV-1 infection with NL4-3 virus, the Rag-hu mice established infection became viremic. These animals were given 5 weekly injections of 0.25 nmol of gp120 aptamer A-1 or A-1-tat/rev siRNA chimera. The treatment efficacy was evaluated by the plasma viral loads and CD4+ cells counts. Our results indicated that the aptamer A-1 or chimeras strongly inhibited HIV-1 infectivity by 3 logs of magnitude and protected the viral-induced CD4+ T cell depletion. In contrast, naked tat/rev siRNAs or mutant gp120 aptamer-siRNA chimers did not prevent viral replication and CD4+ T cell decline. We also confirmed the specific delivery and RNAi efficacy of siRNA in PBMCs of these mice with Taqman polymerase chain reaction and quantitative real time-polymerase chain reaction. Our results showed that the tat/rev siRNA was detectable in the mice treated by the gp120 aptamer-siRNA chimeras, and 75% to 90% of knockdown in the levels of tat/rev gene was observed at both the first and third weeks of treatment. When compared with the gp120 aptamer alone, the combination of the aptamer and siRNA offered more significant suppression and prolonged inhibitory effect by several weeks beyond the last injected dose. Taken the results from 5’-rapid amplification of complementary DNA ends together, all of these data clearly indicated that the siRNA was specifically delivered by gp120 aptamer and triggered sequence-specific degradation of the target mRNA in vivo animal model.
Noncovalent Aptamer-siRNA Conjugation via a “Sticky” Bridge
Ideally, a combinatorial use of various antiviral nucleic acids (eg, aptamer and siRNA) would be capable of easily combining different aptamers or siRNAs to minimize the viral escape mutants, which often abrogate RNAi activity. To further improve the facility of conjugation, continued efforts have also been made by our group. Different from the aforementioned covalent conjugation in aptamer-siRNA chimera, a “sticky bridge” strategy has been designed to noncovalently assemble aptamer with different siRNAs. 39 In this strategy (Fig. 1B), the 3’-end of aptamer and 1 strand of siRNAs were chemically linked to 1 pair of 16-nt sticky bridge sequences, respectively. Because they are highly guanine-cytosine rich and complementary, a simple incubation at 37°C for 15 minutes allows the formation of a stable base-paired conjugate. A flexible 3-carbon atom hinge as a linker was added between the aptamer and sticky sequence to make molecular flexible, thus allowing the correct structure of aptamers and the siRNA processing by Dicer. Currently, commercial solid phase using phosphoramidite generally makes RNA sequences of less than 60 nt. It is a big challenge to chemically synthesize longer RNA (more than 80 nt) with various modifications, such as 2’-fluror, 2’-methyl, 2’-amino, and so on, via efficient, cost-effect manufacturing. In the sticky bridge strategy, all the RNA components as individual building blocks can be chemically synthesized with desired modifications. In comparison with the longer aptamer-siRNA chimera (>100 nt), it offers 2 notable advantages: (1) large-scale chemical production with a relative lower cost and (2) facile assembly of various nucleic acid components, which may mitigate viral resistance to the aptamer or siRNA therapeutics.
In our study, this strategy facilitated the ready exchange of 3 different 27-mer DsiRNAs, which targeted to HIV-1 tat/rev gene and HDFs (CD4 and transportin-3), associated with 1 single gp120 aptamer (A-1). The resultant gp120 aptamer A-1-stick-cocktailed siRNA conjugates showed selective binding and cellular uptake to gp120 positive cells and effectively inhibited viral replication in HIV-1 infected cells. Furthermore, using an HIV-1 infected humanized Rag-hu animal model, we validated the functional delivery of multiplexed siRNAs in vivo mediated by the gp120 aptamer via the sticky bridge. 68 As described previously in the case of aptamer-siRNA chimera, the plasma viral loads were assessed to determine whether the established viral loads could be suppressed by the treatment of aptamer-stick-cocktailed DsiRNA conjugates on a weekly basis (5 weekly injections of 0.25 nmol per injection). Compared with the control groups, a mean of 3 logs of suppression was observed with a statistical significance. When the treatment was terminated, HIV-1 level was rebounded in most of the treated animals, which facilitated a follow-up injection with the aptamer-stick-cocktailed siRNA conjugates. This follow-up treatment provided complete suppression of HIV-1 levels, which persisted for 2 weeks beyond the period of follow-up treatment. In the mice treated with the conjugates, the levels of CD4+ T cells remained near the levels of the uninfected mice. Moreover, substantial reduction of the 3 targeted mRNAs was observed in the PBMCs from the treated mice. Collectively, the sticky bridge strategy suggested a facile, combinatorial use of antiviral nucleic acids for targeted HIV-1 therapy.
Conclusions and Challenges
Nucleic acid-based therapeutics has captured the extensive attention of researchers for a long time. The combinatorial use of 2 or 3 antiviral nucleic acids has been proven to be more efficacious than single nucleic acid component in the cultured cells and in humanized mice model, even in HIV-1 patients. In our laboratory, we have successfully capitalized on the exquisite specificity of HIV-1 cell-specific aptamers against gp120 to deliver anti-HIV siRNAs in vitro and in vivo, which resulted in additive antiviral activity by the combined action of the aptamer and siRNA. The gp120 aptamers can function both as HIV-1 entry inhibitor and as targeted delivery agents for siRNA.
Despite these successful examples, several key challenges hinder the rapid translation of aptamer-mediated therapeutics to the clinic. Long-term and repeated administration of nucleic acid therapeutic may cause some potential toxicities, including nonspecific immune response, polyanionic effects, tissue accumulation, off-target effect, and so on. In addition, the pharmacokinetics of nucleic acid therapeutics should be taken into account.69,70 Because of the relatively smaller size and molecular weight (40–60 nt, 15–20 kDa), nucleic acid aptamers or siRNAs are vulnerable to nuclease degradation and are subjected to rapid renal filtration and short circulation half-time in the blood organisms. Therefore, rational modifications should be considered to improve their pharmacokinetics profile for clinical development.33,71 For example, we incorporate 2’-fluoropyrimidine protective group in the gp120 RNA aptamers, thus significantly enhancing their nuclease resistance in mouse serum and in animal model.
Moreover, effective endosomal release of the aptamer-mediated siRNA delivery system still remains an important concern. A higher concentration of aptamer-siRNA conjugates was required to obtain effective gene silencing. Although efficient cellular internalization of large amounts of conjugates was observed, siRNA-mediated knockdown was marginal. Most siRNA may be trapped in the endosome and be inaccessible by Dicer and RNA-induced silencing complex in the cytoplasm. Therefore, further efforts should be necessary. For example, formulating multiple aptamers and siRNAs into nanocarriers with endosome-rupturing capability may increase the targeting capability of aptamers and the loading capacity of siRNA. Advances in chemical synthesis and nanotechnology would hopefully solve these concerns in the near future.