
Abstract
Background
We designed a Phase 3 clinical trial to determine the safety and efficacy of adding transcranial ultrasound using an operator-independent headframe to recombinant tissue-plasminogen-activator for the treatment of acute ischemic stroke.
Methods
Combined lysis of thrombus with ultrasound and systemic tissue-plasminogen-activator for emergent revascularization in acute ischemic stroke is a randomized, double-blind, placebo-controlled clinical trial that will enroll subjects with the following main inclusion criteria: less than 4·5 hours from symptom onset (three-hours in US and Canada), age 18–80 years, baseline National Institutes of Health Stroke Scale score ≥ 10, and premorbid modified-Rankin-score of 0–1, eligibility for full dose recombinant tissue-plasminogen-activator. Subjects will receive two-hours of 2-MHz pulsed wave transcranial ultrasound (target group) or sham ultrasound (control group). The projected sample size is approximately 824 subjects.
Results
The primary endpoint, based on intention-to-treat criteria of patients enrolled within three-hours of symptom onset is the comparison between target and control groups of modified-Rankin-score scores at day 90 poststroke assessed using the proportional odds method. The study will have two planned interim analyses after approximately one-third and two-thirds of subjects have reached the 90-day modified-Rankin-score evaluation. Safety outcomes are symptomatic intracranial hemorrhage within 24 h and an overall analysis of adverse events.
Conclusions
Since intravenous recombinant tissue-plasminogen-activator remains the only medical therapy to reverse ischemic stroke applicable in the emergency department, our trial will determine if the additional use of transcranial ultrasound improves functional outcomes in patients with severe acute ischemic stroke (NCT#01098981).
Background
Intravenous thrombolysis (IVT) with recombinant tissue plasminogen activator (rt-PA) remains the fastest way to initiate treatment for ischemic stroke, yet one-half of treated patients remain disabled or die despite receiving therapy (1, 2). Major reasons for incomplete recovery include the initial severity of ischemic insult, proximity of the intracranial occlusion, as well as slow and incomplete thrombolysis (3–5).
Over the past 30 years, experimental studies have provided evidence that IVT can be enhanced with ultrasound (6, 7). Although all the mechanisms are still not fully determined, it is believed that ultrasound accelerates enzymatic fibrinolysis primarily through nonthermal mechanisms by increasing transport of drug molecules into the clot (8, 9). Moreover, mechanical effects of ultrasound radiation forces have the ability to influence drug transport and promote the motion of fluid through and around the thrombus, an effect called acoustic streaming (10). Finally, sonothrombolysis may promote nitric oxide release and improve microcirculation, and this may be associated with reduction in infract size (11).
A phase II controlled trial of patient exposure to a 2-MHz diagnostic ultrasound [transcranial Doppler (TCD)] and a meta-analysis of other similar studies showed that ultrasound aimed at residual flow/thrombus interface can at least double the chance of early recanalization (12–14). The group of acute ischemic stroke patients that appeared to benefit the greatest from sonothrombolysis included patients with National Institutes of Health Stroke Scale (NIHSS) scores ≥10 and proximal intracranial occlusions (15).
A major obstacle for emergency physicians, neurologists, and health professionals to use diagnostic ultrasound in acute stroke is its operator dependency. To address this issue, we developed a novel therapeutic device that requires no operator targeting of the intracranial vessels and no sampling of returned intracranial echoes. The safety of this approach to enhance the activity of rt-PA was determined in phase II studies (16, 17). A detailed description of the operator-independent device (Sonolysis Headframe™, Cerevast Therapeutics, Inc., Seattle, WA, USA), ultrasound parameters, vessel targeting, and safety testing was previously published (16).
In brief, the headframe used in the CLOTBUST-ER study (Fig. 1) employs 16 transducers fixed into three different arrays (two temporal, one suboccipital conventional windows fro TCD ultrasound) to transmit sequentially transcranial ultrasound energy to the principal regions in which vessel occlusions associated with severe strokes are generally known to occur (5). The emitted ultrasound exposure is in accordance with the parameters mandated by the FDA for currently marketed TCD diagnostic ultrasound devices.
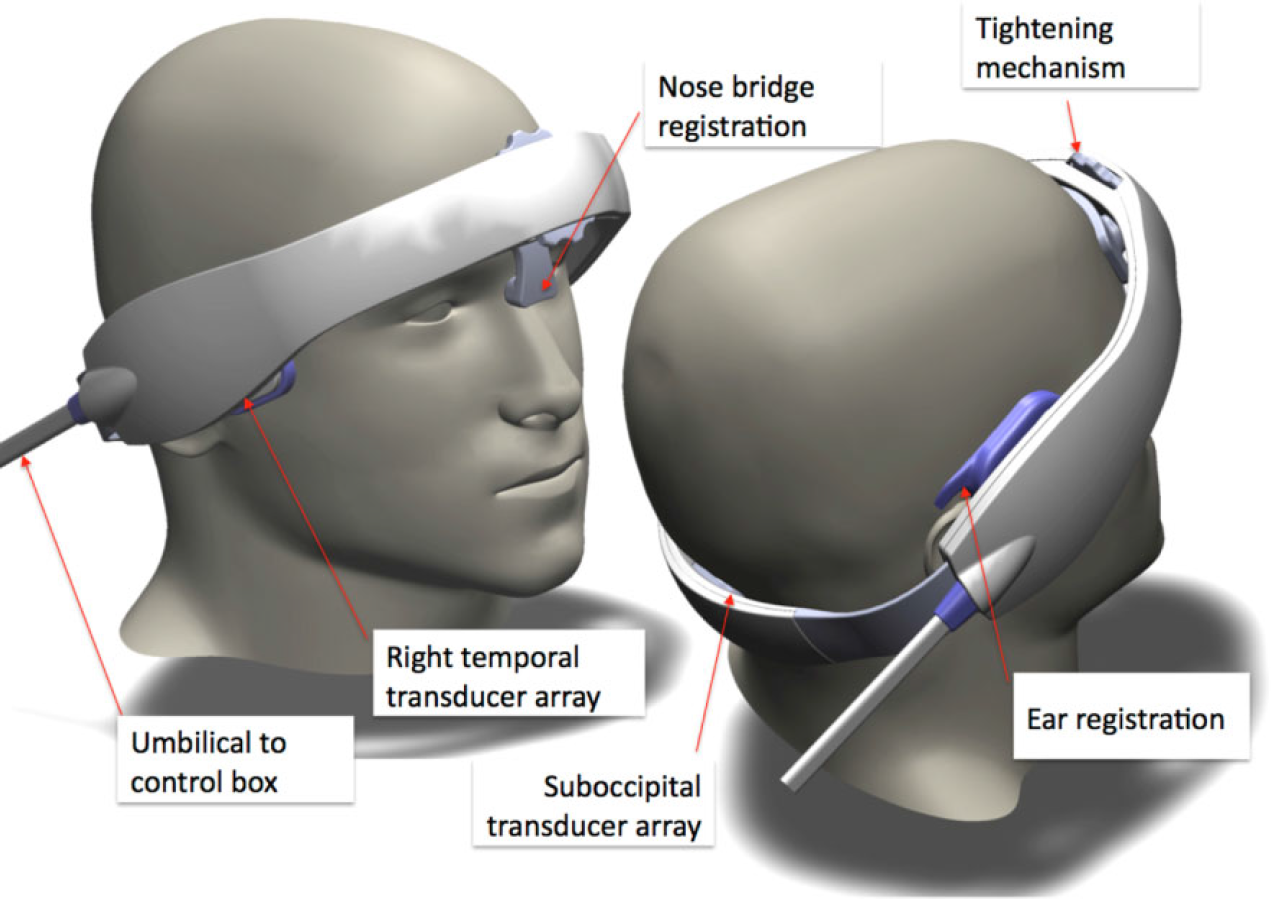
Sonolysis headframe.
Our objective is to confirm the safety and determine the efficacy of adding transcranial ultrasound to rt-PA for the treatment of acute ischemic stroke in the setting of a phase III randomized controlled trial.
Methods
Study design
CLOTBUST-ER is a prospective, multicenter, randomized, double-blind, placebo-controlled clinical trial. The study is designed to add the ultrasound to standard of care treatment of acute ischemic stroke (IVT). Subjects will be randomized 1 : 1 to the target group (active US + IVT) or the control group (sham US + IVT) using web-based central randomization. The patient can be randomized either prior or subsequent to administration of the rt-PA bolus. The headframe is identically activated in the target and control groups, with the control group given a sham ultrasound treatment in a manner that is blinded. All patients with the headframe activated will be included in the intention-to-treat analysis, but headframe activation must occur within 30 mins of rt-PA bolus for the subject to be included in the per-protocol analysis. A schematic of the study design is shown in Fig. 2.
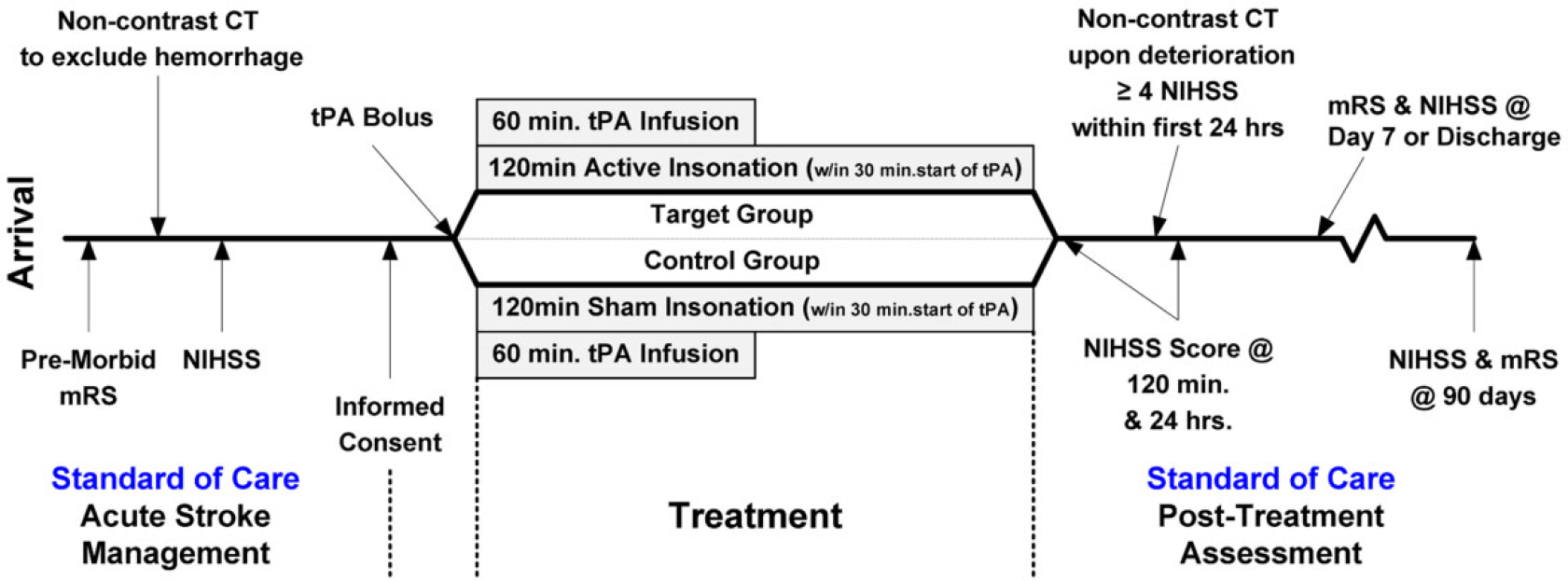
Schematic of study design.
Patient population
The inclusion and exclusion criteria are listed in Table 1. In brief, adult patients aged 18–80 years, with baseline NIHSS ≥ 10 ischemic stroke within a 4·5-hour treatment window (three-hour treatment window in the US and Canada as per label), who were independently functioning in the community immediately prior to their stroke, are eligible for study participation. Subjects in the optional arterial recanalization substudy must have an occlusion located in the intracranial carotid terminal, M1 thru mid M2 or proximal A2, or intracranial vertebrobasilar or P1/proximal P2 segments or tandem lesions as determined within 15 mins of rt-PA bolus by computed tomographic angiography (CTA) or magnetic resonance angiography (MRA), and must have a glomerular filtration rate ≥60 ml/min. Follow-up imaging is mandated two-hours after end of study treatment using the same imaging modality as in the baseline assessment.
Inclusion and exclusion criteria
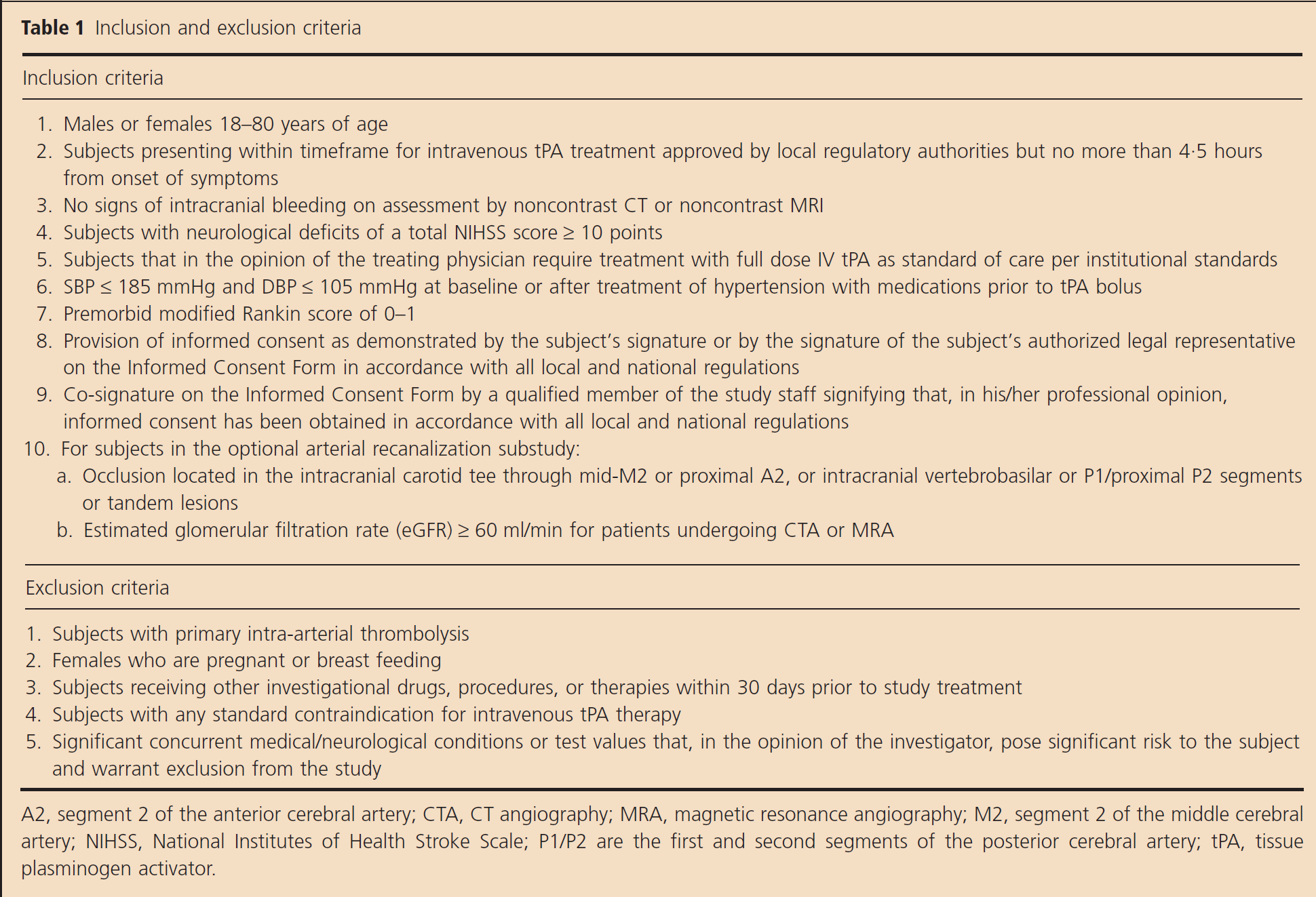
A2, segment 2 of the anterior cerebral artery; CTA, CT angiography; MRA, magnetic resonance angiography; M2, segment 2 of the middle cerebral artery; NIHSS, National Institutes of Health Stroke Scale; P1/P2 are the first and second segments of the posterior cerebral artery; tPA, tissue plasminogen activator.
Baseline measures
The timing of all study procedures is listed in Table 2. All patients will have standard of care assessments for demographics, medical history, prestroke function, prior medications, baseline laboratory tests, and stroke severity (NIHSS). All subjects will undergo a noncontrast head CT or noncontrast MRI scan to rule out intracranial hemorrhage.
Schedule of enrollment, interventions, and assessments
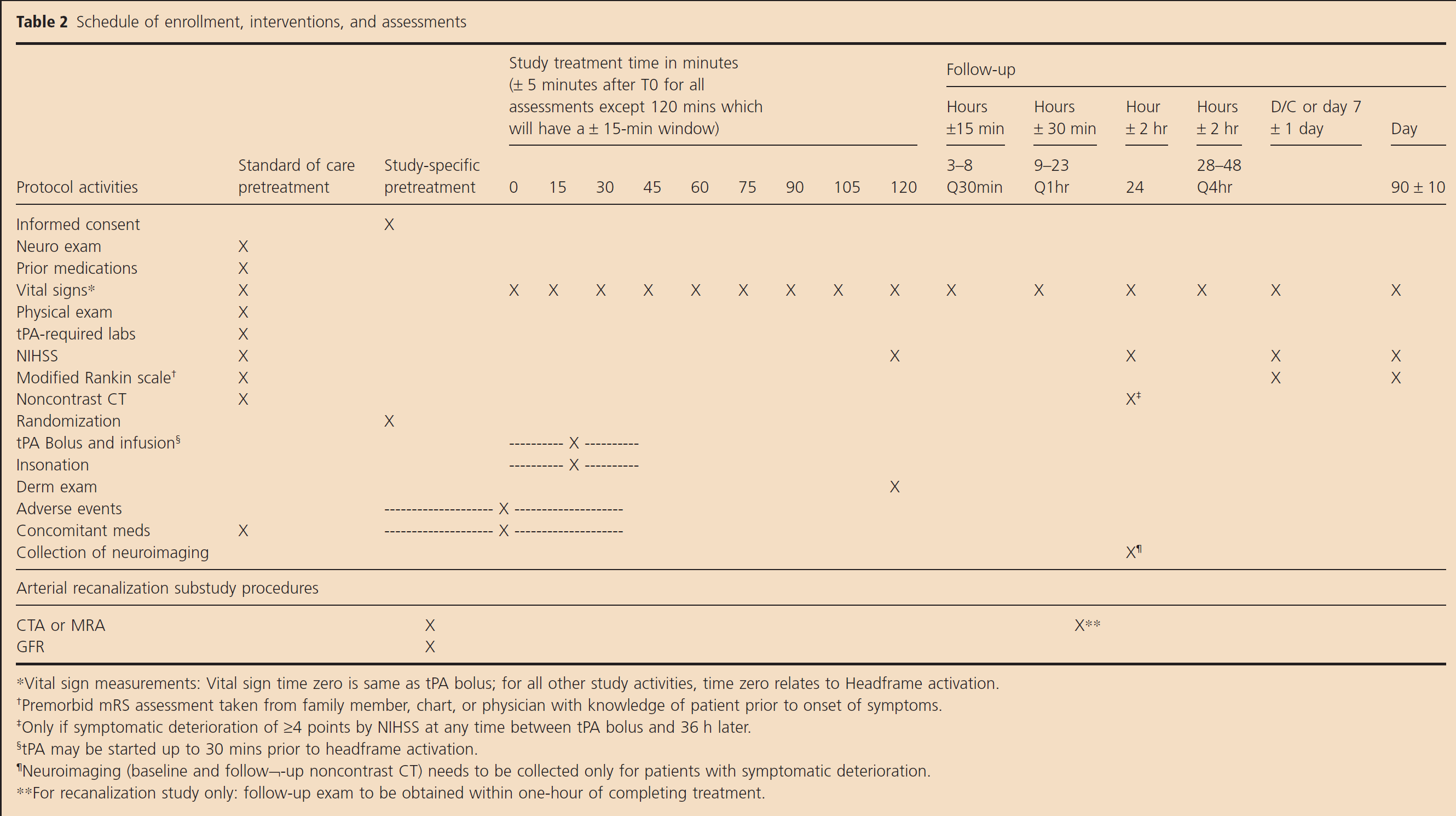
Randomization
Following signed informed consent, subjects are randomized 1 : 1 to the target group (active US + IVT) or the control group (sham US + IVT) via web-based central randomization using a permuted block randomization by site (block sizes of 2, 4, or 6). The randomization code (treatment A or B) is entered into the headframe control box which results in the delivery of either active ultrasound or a sham (no ultrasound) treatment. All subjects will be wearing a headframe whether they are receiving active ultrasound or sham treatment. There are no observable differences when the headframe is operating in active vs. sham mode; thus, double blinding of the study is maintained.
Treatment
All eligible subjects will receive standard of care treatment with full-dose rt-PA (0·9 mg/kg; 90 mg maximum; 10% bolus followed by 90% IV infusion over 60 mins) within the timeframe after onset of stroke symptoms approved by local regulatory authorities (three-hours USA and Canada, 4·5 hours all other participating countries globally). The headframe will be placed on all subjects prior to or as shortly after rt-PA bolus as possible. The headframe must be activated within 30 mins of rt-PA bolus for the subject to be in the per protocol analysis group. The target group (US + IVT group) will receive standard of care rt-PA bolus/infusion over 60 mins and transcranial ultrasound at 2 MHz for 120 mins. The control group will receive standard of care rt-PA bolus/infusion over 60 mins and sham (inactive) ultrasound for 120 mins. Standard of care evaluations include NIHSS evaluation pretreatment, at two-hours, 24 h, day 7 or discharge (whichever occurs earlier), and at day 90, and mRS evaluations at day 7 or discharge (whichever occurs earlier) and at day 90. Significant neurological worsening, defined as a total NIHSS score increase by ≥4 points from the best score at any time during first 24 h post rt-PA bolus, will require a noncontrast CT to rule out intracranial hemorrhage. Routine poststroke imaging is not required for this clinical trial but is allowed at facilities where this is standard of care.
The goal for blood pressure prior to rt-PA infusion and for 24 h thereafter is a systolic blood pressure ≤ 185/180 mmHg and diastolic blood pressure ≤ 110/105 mmHg respectively using site-specific blood pressure lowering algorithms. Blood pressure is measured every 15 mins for the first two-hours after rt-PA bolus, every 30 mins from two to eight-hours, and hourly thereafter up to 24 h per standard of care.
A brief dermatological examination is performed before and after placement of the headframe to evaluate for clinically significant swelling and/or erythema at the site of headframe contact.
Primary outcome
The primary outcome is assessed on the modified Rankin Scale (mRS) at 90 ± 10 days from randomization. The primary analysis will assess the mRS using a cumulative ordinal logistic regression [proportional odds method (POM)] for those subjects enrolled within three-hours of stroke symptom onset. The primary outcome assessments at 90 days will be conducted by trained and certified personnel. Sites are encouraged to have the same person administer all NIHSS and mRS assessments for a given subject.
Secondary and safety outcomes
Secondary end points include day 90 mRS assessment in subjects enrolled within 0–4·5 and 3–4·5 hours of symptom onset (POM), and for subjects in both the 0–3- and 0–4·5-hour postsymptom onset timeframes: day 90 dichotomous mRS 0–1 and mRS 0–2; rates of clinical recovery; and length of hospital stay. An optional arterial recanalization study using pre/post-treatment CTA or MRA will evaluate rates of two-hour recanalization.
Clinical recovery will be defined as the following: dramatic clinical recovery assessed at 120 ± 15 mins after headframe activation includes a reduction of 10 or more points on NIHSS compared with pretreatment, or a total NIHSS score of 3 or less. Clinical recovery assessed at 24 ± 2 h after headframe activation includes a reduction of 10 or more points on NIHSS compared with pretreatment, or a total NIHSS score of 3 or less. Neurological improvement assessed at 24 ± 2 h after headframe activation requires a reduction of 5 or more points on NIHSS compared with the pretreatment score. Neurological worsening assessed at 24 ± 2 h after headframe activation requires an increase of 4 or more points on NIHSS compared with the pretreatment score. Independent functional outcome adjusting for pretreatment NIHSS assessed at 90 ± 10 days post-treatment includes an mRS score of 0–1 for subjects with pretreatment NIHSS 10–14, or an mRS score of 0–2 for subjects with pretreatment NIHSS > 14.
In addition, a comparison will be conducted between target and control groups for NIHSS at two-hours, 24 h, day 7 or discharge (whichever is earlier) and day 90, and mRS at day 7 or discharge (whichever is earlier) and day 90.
The duration of hospital stay until discharge will be captured as an indicator of cost of treatment. ‘Discharge’ is interpreted to mean either the ‘date of discharge’ or ‘date of transfer/transition to rehabilitation’ – in other words, the end of acute care and start of subacute care.
Subjects who participate in the optional arterial recanalization substudy will have images interpreted by a central imaging core lab at the University of Calgary. Recanalization rates for CTA will use the following arterial occlusive lesion (CTA AOL) scoring adapted from the angiographic AOL score: AOL score 0: no recanalization; AOL score 1: incomplete or partial recanalization with no contrast perfusion beyond thrombus; AOL score 2: incomplete or partial recanalization with distal contrast perfusion; and AOL score 3: complete recanalization with distal contrast perfusion despite the possible presence of distal emboli (18). Recanalization rates for MRA will use a simple dichotomous scale of no or any recanalization. No recanalization was defined as no signal beyond the original site of occlusion. Any recanalization was defined as signal now present beyond the original site of occlusion despite the possible presence of distal emboli (19).
Safety outcomes include the proportion of subjects in the target vs. control group experiencing symptomatic intracranial hemorrhage (sICH) within 24 h of rt-PA bolus and an overall analysis of adverse events. sICH is defined as neurological deterioration (≥4 points worsening on the NIHSS compared with the best prior examination) within 24 h after rt-PA bolus with documented parenchymal hemorrhage type 2 or type 2 remote (PH2/PH2r) where PH2 is defined as ICH volume at least one-third of the infarct volume, or death due to hemorrhage within 24 h after rt-PA bolus. All intracranial bleeds within 24 h which are associated with neurological deterioration as defined above are sent to a central imaging core lab at the University of Calgary for ECASS scoring. All cases with ECASS PH2/PH2r are sent for independent adjudication by one neuroradiologist and one stroke neurologist chosen at random from a panel of neuroradiologists and stroke neurologists. Should the two evaluations result in contradictory conclusions, the case is sent to a third, randomly chosen, adjudicator to evaluate; the two matching outcomes will determine the result. Additionally, to allow comparison of sICH results in this study with sICH results from prior stroke trials (2, 20), the above sICH definition and adjudication process will be applied to 36 h post rt-PA bolus.
All patients who have received any therapy with the headframe (defined as having the headframe placed, the control box turned on and placed into A or B mode) will be included in the overall analysis of safety. All adverse events will be coded and tabulated by MedDRA System Organ Class (SOC), individual events within each SOC and presented in descending frequency. Adverse events will also be tabulated by severity and relationship to the investigational device. Serious adverse events will be summarized separately. Death from any cause within 90 days of treatment and the proportion of subjects discontinuing the study due to adverse events will also be summarized by treatment group.
DSMB/Safety monitoring
Unblinded safety data will be reviewed by the DSMB at regular intervals during the study. If the incidence and severity of adverse events, including sICH, are within the expected range as defined prospectively by the DSMB committee members and there are no other safety concerns, enrollment will progress to the planned sample size.
Statistical analysis plan and sample size
The primary endpoint analysis will use cumulative ordinal logistic regression (POM) of 90-day mRS (21–26) in order to demonstrate that the subjects treated with the headframe and systemic rt-PA (target group) compared with systemic rt-PA alone (control group) will be a significant factor influencing better outcomes in the target group. The trial is powered to detect a statistically significant shift in the distribution of scores on the mRS scale at 90 days between the target and the control arm. The Mann–Whitney U test was used to estimate the sample size required, as it approximates the POM with respect to power, significance, and required sample size (27). Relative proportions of 90-day mRS outcomes were estimated according to the outcomes observed in the CLOTBUST study6, for subjects with baseline NIHSS ≥ 10. Assuming a power of 90%, a two-sided α = 0·05, a target mean (SD) of 2·97 (1·15), a control mean (SD) of 3·25 (1·12), and three equally spaced analyses (two interim and one final), it was determined that a total of 700 subjects (350 per arm) were required. Because the primary endpoint analysis is conducted only in those subjects who receive rt-PA within three-hours of symptom onset, and approximately 15 percent of subjects are expected to be in the 3- to 4·5-hour symptom onset timeframe, the total sample size has been set at 824 subjects.
For those subjects missing the 90-day mRS data, multiple imputation methods will be used to estimate the probability of the outcome at 90 days. This multiple imputation will incorporate information from participants who withdrew prior to the final visit and to implement the full intention-to-treat principle. Additional sensitivity analyses will be conducted using last observation carried forward, missing equals nonresponse, and mixed-model/GEE approaches to assess if the method of analysis has an impact on the interpretation of the study. Additionally, a sensitivity analysis of the primary endpoint with missing values imputed as worst case (i.e. mRS of 6) will also be presented. This approach will allow all patients randomized to be included in the ITT analysis.
Interim analyses assessing the primary outcome between treatment groups will be performed after approximately one-third and two-thirds of all of the 90-day mRS results are available. Differences between treatment groups will be assessed using imputation approaches identical to those employed for the final analyses. Using O'Brien-Fleming boundaries for the group sequential design with 90% power and testing at approximately one-third and two-thirds of the subjects imply critical values of P = 0·0003525 and P = 0·0120085 at the first and second interim analyses, respectively, and P = 0·0462386 at the final analysis. In addition, a conditional power futility analysis will be performed at each of the interim analysis point, where the study will be stopped should the conditional power fall below 15%.
For the arterial recanalization substudy, the analyses will include a comparison of the rates of complete recanalization in the target vs. control groups as determined by vascular imaging, and a comparison of the rates of ‘any recanalization’ where ‘any recanalization’ includes complete or partial recanalization on CTA and any recanalization on MRA. The observations of complete recanalization and any recanalization will be correlated with occurrence of dramatic clinical recovery.
Discussion
Our multicenter randomized clinical trial will determine the safety and efficacy of adding ultrasound to IVT with rt-PA for treatment of acute ischemic stroke. IVT remains the fastest way to initiate treatment of an acute ischemic stroke, and it was given as part of standard medical therapy in all recent trials of mechanical thrombectomy (25, 26). In these trials, systemic thrombolytic therapy was administered hours prior to initiation of thrombectomy procedures. Time delays to endovascular intervention are unavoidable in clinical practice since the majority of stroke patients present to facilities lacking neuro-endovascular specialists (i.e. stroke ready or primary stroke centers), and there is always a delay for preparation of endovascular therapy and the angiosuite. Even in the fastest trials, the door to thrombectomy time is in a median range of 4–5 hours from stroke symptom onset. Therefore, the ability to augment the efficacy of rt-PA remains critical to the initial treatment of stroke patients, particularly in the setting of patient transfer to comprehensive centers while rt-PA is being administered en route. For patients presenting directly at comprehensive stroke centers, time from symptom onset to rt-PA therapy is about 120 mins, leaving ample time to administer rt-PA with ultrasound before thrombectomy.
We foresee that the addition of ultrasound to IVT will fit into the current stroke treatment paradigm and systems of care since our novel device allows noninvasive bedside initiation of treatment that can be continued during patient transfer. A recent comprehensive benefit–risk analysis of available acute reperfusion therapies in acute cerebral ischemia in comparison with standard IVT has shown that sonothrombolysis appears to have a high benefit-to-risk ratio adjusted for baseline stroke severity (29). Our device requires no prior experience with diagnostic ultrasound, is compact, and is battery operated and can be rapidly applied at the bedside without delaying rt-PA administration.
Our trial has limitations since it is focused on severe strokes as measured by the NIHSS and, as such, may not be generalizable to all ischemic stroke patients. We focused on this patient group since patients with milder strokes tend to respond well to intravenous rt-PA, and inclusion of these patients would have required a larger sample size. Moreover, ultrasound-enhanced thrombolysis appeared to be more effective in terms of functional outcome in patients with moderate-to-severe stroke according to a subgroup analysis of our pivotal phase II trial (15).
Another potential limitation is a change in the protocol while the trial was underway, and approximately 300 patients had been recruited. In order to have interpretable results for stroke patients in both the United States and Canada (where rt-PA is approved for use in the 0- to 3-hour window) and Europe and Australasia (where patients in the 3- to 4·5-hour window may also be treated), we designed the trial in accordance with the FDA to enroll patients in the 0- to 4·5-hour timeframe, reflecting global practice. After trial enrollment had commenced, the Sponsor was notified that according to US regulatory requirements, only those patients in the 0- to 3-hour window would be eligible for inclusion in the primary analysis. In order to preserve the original inclusion criteria/sample size, we elected to convert the primary analysis methodology from a dichotomous to an ordinal analysis, to provide the requisite statistical power for the reduced size of the primary analysis data set comprising only those patients in the 0- to 3-hour window. This revision was implemented prior to the first interim analysis and while all data remained blinded. Considering the fact that the 0- to 4·5-hour time window is recommended for IV thrombolysis with rt-PA in national and international guidelines, and this timeframe currently is the clinical practice in the United States and Canada (off label but on guideline), we felt these steps to be necessary in order to maintain the integrity of our trial.
In conclusion, intravenous rt-PA remains the only medical therapy to reverse ischemic stroke applicable in the emergency department, and the amplification of its efficacy to dissolve intracranial thrombi and reverse ischemic stroke damage is highly desirable (28). Our controlled trial will determine if the addition of transcranial ultrasound to rt-PA improves functional outcomes in patients currently believed to benefit least from systemic thrombolysis.
Footnotes
Acknowledgements
Please see Appendix S1 for the full list.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: