
Abstract
Rationale
The Interventional Management of Stroke (IMS) I and II pilot trials demonstrated that the combined intravenous (i.v.) and intraarterial (i.a.) approach to recanalization may be more effective than standard i.v. rt-PA (Activase®) alone for moderate-to-large National Institutes of Health Stroke Scale (NIHSS ≥ 10) strokes, and with a similar safety profile.
Aims
The primary objective of this NIH-funded, Phase III, randomized, multicenter, open-label clinical trial is to determine whether a combined i.v./i.a. approach to recanalization is superior to standard i.v. rt-PA alone when initiated within 3 h of acute ischemic stroke onset. The IMS III trial will develop and maintain a network of interventional centers to test the safety, feasibility, and potential efficacy of new FDA-approved catheter devices as part of a combined i.v./i.a. approach to recanalization as the IMS III study progresses. A secondary objective of the IMS III trial is to determine the cost-effectiveness of the combined i.v./i.a. approach as compared with standard i.v. rt-PA. Trial enrollment began in July of 2006. Design A projected 900 subjects with moderate-to-large (NIHSS ≥ 10) ischemic strokes between ages 18 and 80 will be enrolled over the next 5 years at 40-plus centers in the United States and Canada. Patients must have i.v. treatment initiated within 3 h of stroke onset in both arms. Subjects will be randomized in a 2: 1 ratio with more subjects enrolled in the combined i.v./i.a. group. The i.v. rt-PA alone group will receive the standard full dose [0·9 mg/kg, 90 mg maximum (10% as bolus)] of rt-PA intravenously over an hour. The combined i.v./i.a. group will receive a lower dose of i.v. rt-PA (~0·6 mg/kg, 60 mg maximum) over 40 min, followed by immediate angiography. If a treatable thrombus is not demonstrated, no i.a. therapy will be administered. If an appropriate thrombus is identified, treatment will continue with either the Concentric Merci® thrombus-removal device, infusion of rt-PA and delivery of low-intensity ultrasound at the site of the occlusion via the EKOS® Micro-Infusion Catheter, or infusion of rt-PA via a standard microcatheter. If i.a. rt-Pa therapy is the chosen strategy, a maximum of 22 mg of i.a. rt-PA may be given. The choice of i.a. strategy will be made by the treating neurointerventionalist. The i.a. treatment must begin within 5 h and be completed within 7 h of stroke onset. Study outcomes The primary outcome measure is a favorable clinical outcome, defined as a modified Rankin Scale Score of 0–2 at 3 months. The primary safety measure is mortality at 3 months and symptomatic ICH within the 24 h of randomization.
Keywords
Introduction
Dating back to the National Institute of Neurological Disorders and Stroke (NINDS) pilot trials of intravenous (i.v.) rt-PA, acute stroke trial experience has shown that it is not enough to open occluded blood vessels; we must strive to open them quickly (1–4). However, even after ultra-early thrombolytic therapy, patients with moderate-to-large strokes National Institutes of Health Stroke Scale (NIHSS ≥ 10) frequently have persistent major arterial occlusions and poor outcomes (5,6). In the NINDS tPA study, only 17% of patients with NIHSS ≥ 20 treated with i.v. rt-PA had mild or no disability [modified Rankin Scale (mRS) Score 0–1] (7).
A combined i.v. and intraarterial (i.a.) approach to acute ischemic stroke therapy was designed to offer rapidly initiated i.v. therapy, followed by additional titrated local i.a. therapy, to patients with moderate-to-large strokes (NIHSS ≥ 10). The goal was to achieve higher rates of early, successful reperfusion in a widely accessible manner. This approach has been tested in clinical trials of over 200 patients, starting with the Emergency Management of Stroke (EMS) pilot trial from 1994 to 1995, followed by the Interventional Management of Stroke (IMS) I trial in 2001, the IMS II trial from 2003 to 2006, and several additional cohorts (8–13). In EMS and IMS I, subjects were given low-dose i.v. rt-PA (0·6 mg/kg), followed by additional rt-PA (up to 22 mg) intraarterially via a standard microcatheter. As shown in Tables 1 and 2, data suggested that the combined approach to recanalization may be more effective than standard i.v. rt-PA (Activase®) alone for moderate-to-large (NIHSS ≥ 10) strokes, while maintaining a similar safety profile.
Since the inception of the combined i.v./i.a. thrombolytic approach, two mechanical devices were cleared by the FDA for use during i.a. revascularization procedures based on nonrandomized studies. Based on two pivotal trials, the Concentric L5, X5, and X6 Merci® Retrievers received FDA clearance for thrombus removal in patients with acute ischemic stroke who are ineligible for rt-PA or fail thrombolysis (14,15). The EKOS® Micro-Infusion System (Bothell, WA, USA) was studied in the IMS II trial (12). This system (which includes low-energy ultrasound administration) was issued a substantial equivalence determination by the FDA for the administration of contrast into the neurovasculature. Both devices add to the armamentarium of acute stroke i.a. therapy and, therefore, were incorporated into the design of the IMS III trial.
As an interventional approach to cerebral revascularization becomes increasingly available and utilized throughout the world, it becomes critical to establish its benefit in the setting of large-scale Phase III clinical trials.
Methods
Design
The IMS III trial is a Phase III, NIH U01-funded, randomized, multicenter, open-label clinical trial coordinated by the University of Cincinnati and the Data Coordination Unit (DCU) at the Medical University of South Carolina in collaboration with the University of Calgary. The primary objective of the trial is to determine whether a combined i.v./i.a. approach (0·6 mg/kg i.v. rt-PA followed by i.a. therapy) to recanalization is superior to standard i.v. rt-PA alone when initiated within 3 h of acute ischemic stroke onset in subjects with moderate-to-severe strokes (NIHSS ≥ 10). The trial is designed to test hypothesis that there is an overall absolute difference of 10% in the favorable outcome (mild or no disability as measured by a mRS of 0–2) for subjects treated with the combined i.v./i.a. approach as compared with those treated with standard i.v. rt-PA.
Comparative efficacy in clinical trials of the i.v./i.a. approach
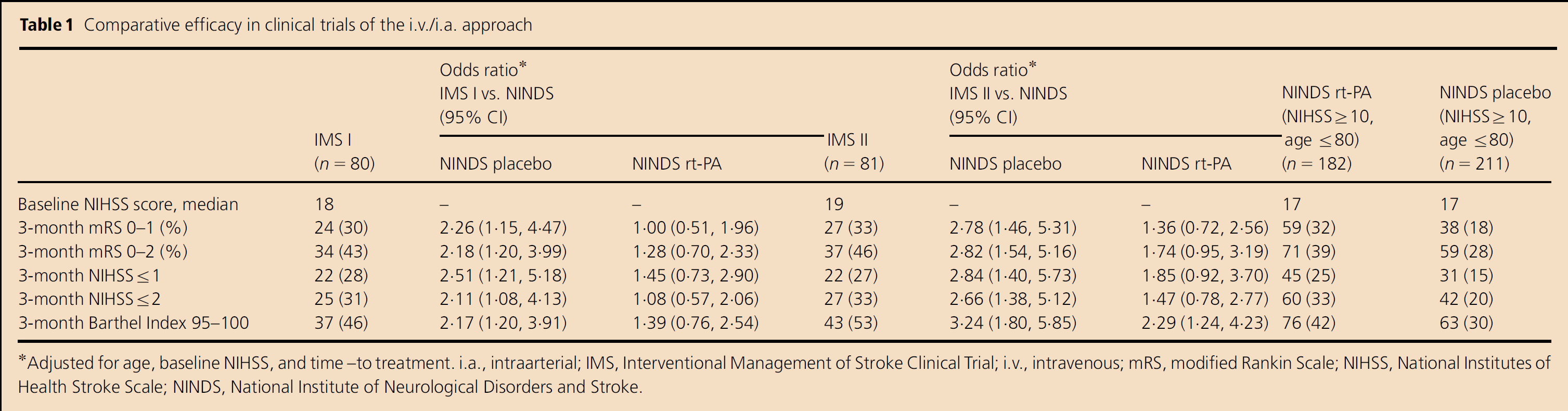
Adjusted forage, baseline NIHSS, and time–to treatment. i.a., intraarterial; IMS, Interventional Management of Stroke Clinical Trial; i.V., intravenous; mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; NINDS, National Institute of Neurological Disorders and Stroke.
Comparative safety in clinical trials of the i.v./i.a. approach
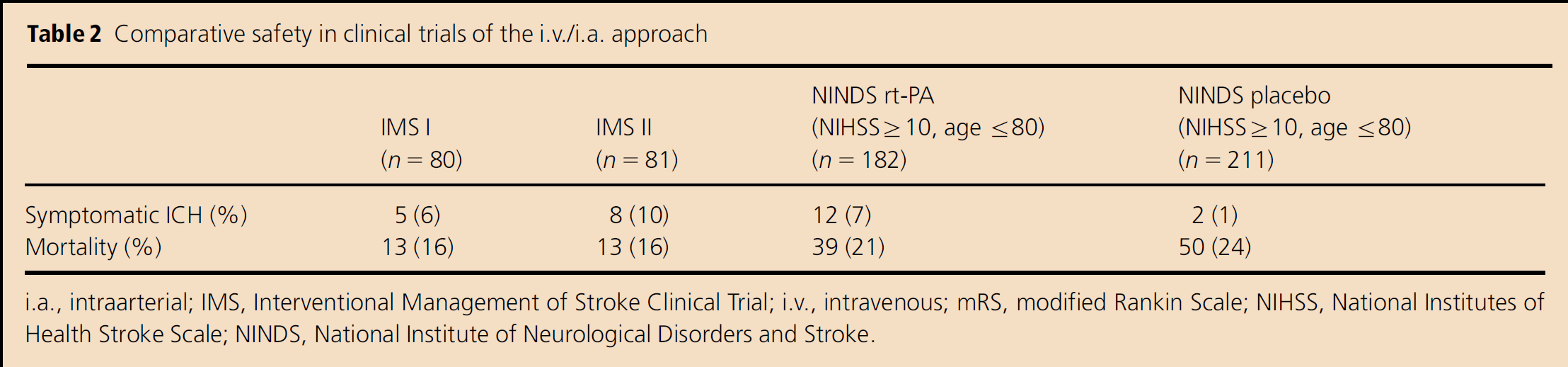
i.a., intraarterial; IMS, Interventional Management of Stroke Clinical Trial; i.v., intravenous; mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; NINDS, National Institute of Neurological Disorders and Stroke.
A secondary objective of the IMS III trial is to determine the cost-effectiveness of the combined i.v./i.a. approach as compared with standard i.v. rt-PA as measured by differences in utilization of resources and quality of life over 12 months after the stroke between the two treatment arms of the trial (as described further in the accompanying paper in this issue).
An additional secondary objective of this trial is to develop and maintain a network of interventional centers to test the safety, feasibility, and potential efficacy of new mechanical devices as part of a combined i.v./i.a. approach to recanalization as the IMS III study progresses.
Patient population
A maximum of 900 subjects with moderate-to-large (NIHSS ≥ 10) ischemic stroke between ages 18 and 80 will be enrolled from 42-plus centers in the United States and Canada. In order to be eligible for the trial, subjects must have had moderate-to-large strokes (baseline NIHSS ≥ 10) before initiation of i.v. rt-PA. In addition, i.v. rt-PA must be initiated within 3 h of symptoms onset (i.e., the time at which the subject was last seen to be well).
The protocol adheres to current medical practice regarding inclusion and exclusion criteria for revascularization therapies with only a few exceptions. Specifically, subjects older than 80 years have been excluded at this time due to possibly increased ICH rates based on prior i.v. and i.a. studies (8). However, this population may still benefit from combined i.v./i.a. therapy overall. Additional notable exclusions include seizure at stroke onset, which precludes knowledge of the baseline NIHSS score; ongoing hemodialysis or peritoneal dialysis, due to possibly increased ICH risk based on the IMS II trial; and hyperglycemia greater than 400 mg/dl (22 mmol/l), based on increased ICH risk.
Imaging criteria for exclusion are consistent with the NINDS tPA stroke trial. Specifically, patients with large regions of clear hypodensity (i.e., darker than white matter and brighter than CSF) on CT scan, such as greater than one-third of the middle cerebral artery vascular territory, are excluded. An Alberta Stroke Program Early CT Score (ASPECTS) of ≤ 4 can be used as a guideline when evaluating > 1/3 region of territory involvement. However, sulcal effacement and/or loss of gray–white matter differentiation alone are not contraindications for treatment. The complete list of inclusion and exclusion criteria is given in Table 3.
IMS III inclusion and exclusion criteria

ASPECTS, Alberta Stroke Program Early CT Score; i.a., intraarterial; IMS, Interventional Management of Stroke Clinical Trial; i.v., intravenous; mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; NINDS, National Institute of Neurological Disorders and Stroke.
Randomization
Subjects are randomized in a 2: 1 ratio, with more subjects assigned to the combined i.v./i.a. group. This randomization regimen was chosen based on pilot data suggesting a higher rate of favorable clinical outcome in the combined i.v./i.a. group in the IMS I and II trials as compared with comparable historical controls from the NINDS rt-PA stroke trial. In addition, this randomization scheme is likely to better maintain participation of neurointerventionalists in the IMS III trial.
The design and implementation of the randomization scheme is conducted by the DCU. The randomization scheme was designed to ensure a 2: 1 (i.v./i.a.: i.v. only) treatment group distribution within each baseline NIHSS score stratum (10–20 or ≥ 20), each performance site, and throughout the overall study.
Randomization is implemented using a combination of a web-based minimization + biased coin scheme and sealed randomization envelopes placed at each clinical site. A patient is randomized by opening a prespecified sealed envelope that contains the treatment assignment. The prespecification of the envelope is performed through the web within 8 h after the previous patient is randomized and his/her enrollment data are entered into the database via the study web site. This system was designed to allow for rapid randomization because it does not require a phone call, fax back system, or access to a web site before treatment.
Treatment
One of the guiding principles of the IMS III trial is rapid initiation of thrombolytic therapy to an eligible subject to provide maximal benefit. To minimize any delay in the administration of a proven effective therapy (i.e., i.v. rt-PA), the standard dose of open-label i.v. rt-PA (0·9 mg/kg; 90 mg maximum) can be initiated before enrollment and randomization in the trial if standard eligibility criteria are met. In addition, the i.v. rt-PA can be initiated in a patient who cannot provide informed consent for the study and for whom an authorized legal representative is not yet available (standard of care).
Randomization can be accomplished at any time before completion of 40 min of the i.v. rt-PA infusion (approximately two-thirds of the 0·9 mg/kg dose). This allows time to complete the determination of trial eligibility, confirm interventional staff availability, obtain consent/HIPAA authorization from the patient and/or legal representative, and identify the randomization assignment. If the patient and/or authorized legal representative cannot provide informed consent for participation or the subject cannot be successfully randomized into the trial before completion of 40 min of i.v. rt-PA infusion, the remainder of the 0·9 mg/kg standard dose of rt-PA can be completed within 1 h. This patient would not be included as a participant in the IMS III study and not followed as an intent-to-treat subject. A subject is considered to be in the IMS III trial only when the sealed randomization envelope is opened.
If the patient is entered in the trial and randomized to the i.v. rt-PA alone group, he/she will receive the full dose (0·9 mg/kg, 90 mg maximum) of rt-PA intravenously over an hour, as FDA-approved standard of care.
The combined i.v./i.a. group will undergo open-label rt-PA over 40 min only instead of 1 h (~ 0–6 mg/kg, 60 mg maximum) and then undergo immediate angiography. Ifa thrombus is not demonstrated, no additional treatment (i.a. rt-PA or mechanical devices) is provided. If an appropriate thrombus is identified, treatment continues with the concentric thrombus-removal device, infusion of rt-PA with low-energy ultrasound at the site of the thrombus via the EKDS® Microinfusion Catheter System, or infusion of rt-PA via a standard microcatheter. The choice of approach is at the discretion of the treating neurointerventionalist based upon the location and extent of lesion and vascular anatomy, documented prior experience and training with Concentric or EKOS® catheters, and documented instructions for use of the various catheters. However, use of both the Concentric and EKOS® catheters in the same patient is not permitted. The rt-PA used for i.a. treatment is provided as an investigational drug. No more than 22 mg of rt-PA will be administered intraarterially, and i.a. treatment must begin within 5 h of onset and be completed within 7 h (Fig. 1).
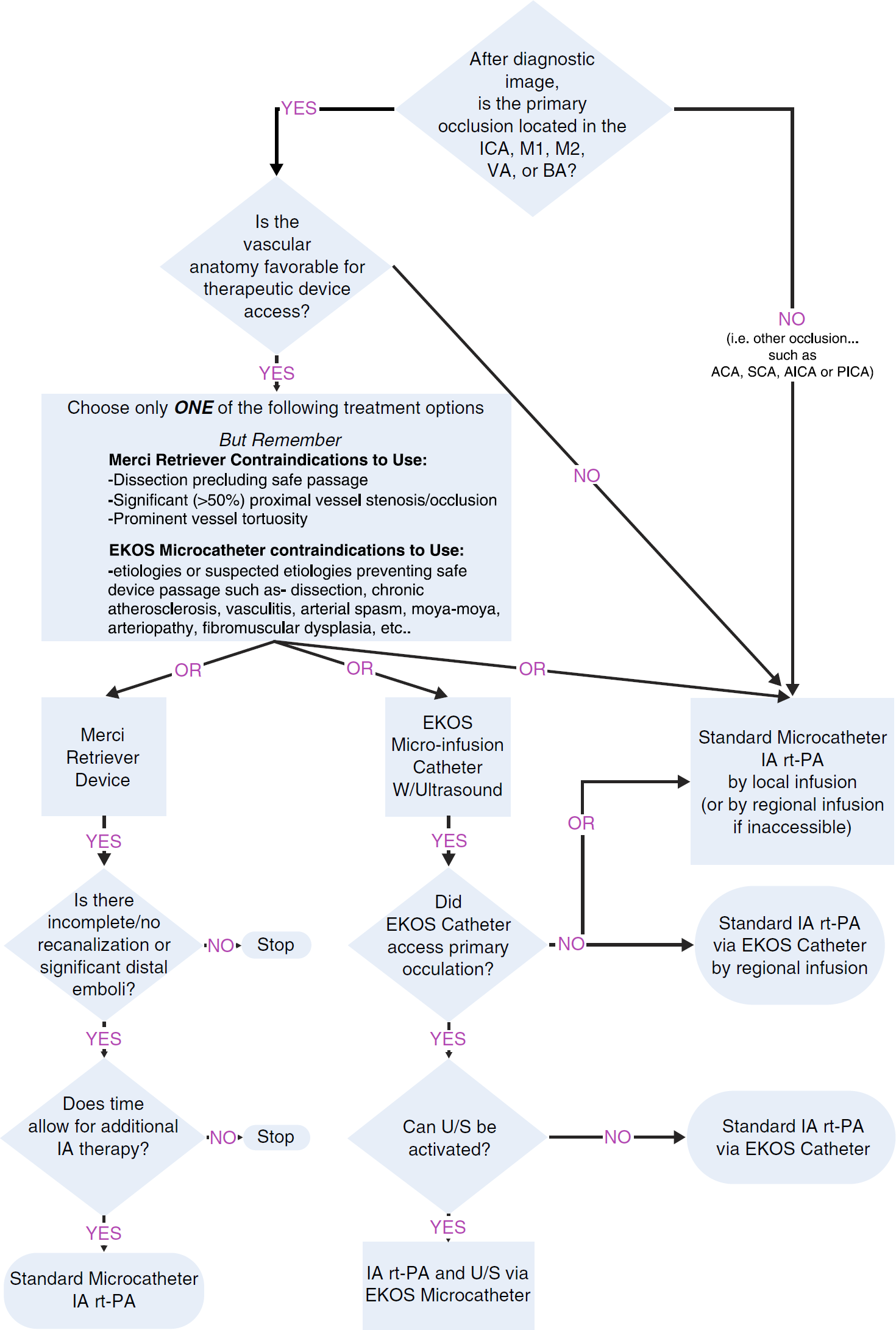
Intraarterial Treatment Protocol.
Primary outcomes
The efficacy of the i.v./i.a. approach, compared with i.v. rt-PA therapy, will be tested using the primary outcome measure of a mRS at 3 months from randomization. A score of 0–2 will be considered to be the favorable outcome. An mRS score of 0–2 was selected for assessing the outcome of moderate-to-severe strokes based on PROACT II, IMS I, and IMS II results (11,12,16). The measure of a favorable clinical outcome will also be analyzed in two prespecified subgroups based on stroke severity, consisting of those with a baseline NIHSS of 10–19 and those with a baseline NIHSS of ≥ 20.
The primary safety measures will be mortality at 3 months and symptomatic ICH within 30 h after stroke onset.
Secondary outcomes
The IMS III trial will also evaluate the effectiveness of the combined i.v./i.a. approach compared with the i.v. rt-PA therapy alone by a number of secondary outcome measures including:
the Barthel Index, Glasgow Outcome Scale, NIHSS, EuroQol EQ-5D, and Trail Making Test Parts A and B at 3 months;
an improved early response to treatment as determined by an NIHSS of 0–2 at 24 h;
a CT angiography assessment of intracranial vascular patency at 24 h (both treatment groups);
the volume of cerebral infarction as measured by a CT scan at 24 h from onset;
change in ASPECTS score from baseline to 24 h on CT scan; and
the rate of TICI Grade II or III perfusion flow and recanalization of the primary arterial occlusion at completion of angiography (combined i.v./i.a. group only).
Secondary safety measures will include:
the proportion of subjects with Type II parenchymal intracerebral hematomas within the first 30 h and
the incidence of any asymptomatic hemorrhage within the first 24 h (17).
Additionally, the following measures of safety will also be monitored:
incidence and severity of nonintracerebral bleeding complications;
incidence and severity of nonbleeding SAEs;
incidence of clinically significant abnormal laboratory values at 24 h;
changes in hemoglobin and hematocrit from baseline to 5 days or discharge;
incidence of blood and/or blood products transfusion of three or more units during hospitalization; and
incidence and severity of procedure and device-related complications (i.e., vessel perforation, clinically significant groin complications, etc.) in the i.v./i.a. rt-PA group.
Data and safety monitoring
An independent data safety and monitoring board, consisting of clinicians familiar with the treatment of stroke, biostatisticians, a neurointerventionalist, and other experts has been established by NINDS to monitor the progress of the trial. Additionally, two stroke neurologists have been appointed as internal and external safety monitors. The internal safety monitor will review safety data on an ongoing basis, including monitoring the trend in serious adverse outcome events and submitting FDA Medwatch reports. The external safety monitor, an independent and experienced neurologist, will review all serious life-threatening bleeding events during this study and will be the final adjudicator of intracranial hemorrhage endpoints (symptomatic or asymptomatic and the relationship with study intervention) when there is disagreement between the local site and the internal medical monitor. This is the same methodology used by the NINDS rt-PA stroke trial and IMS I and II pilot studies.
Regionally based, industry-experienced, independent contracted clinical research monitors will perform onsite data verification for the trial. The initial monitoring visit at a performance site will take place after the first i.v./i.a. treatment subject is enrolled at a site. Thereafter, each SAE will be monitored and each site will be visited no less than once each year during the trial.
All de-identified imaging data will be sent to the Clinical Coordinating Center at the University of Cincinnati for standardized CD archiving and data blinding (if needed). CT and CTA imaging will then be transferred to the Imaging Analysis Center in Calgary for central interpretation by a blinded three-member consensus panel. To expedite safety reporting, all acute CT data received from a clinical center will initially be reviewed by a blinded central reader at the University Cincinnati. All angiographic data will be reviewed by two independent readers, and a third reader will provide final adjudication as needed.
Sample size
The total sample size of 900 was determined based on an anticipated effect size of 10% (the absolute difference between the i.v. rt-PA and i.v./i.a. rt-PA arms in the proportion of subjects with favorable outcomes). Specifically, we assumed that the proportion of i.v. rt-PA proportion with favorable clinical outcome would be 40% (obtained from the NINDS rt-PA study), the 2:1 ratio of i.v./i.a. to i.v. only group assignment, and Type I and Type II error probabilities of 0·05 and 0·20, respectively, and three prespecified interim analyses. Because the intent-to-treat principle will be applied to the analysis, the sample size of 900 includes inflation by 1·03 to safeguard against dilution of the effect size by patients lost to follow-up and/or treatment cross-over in approximately 1–3% of the cases.
Statistical analyses
The set of statistical hypotheses for the overall sample is: H0: πi.v. = πi.v./i.a. vs. HA: πi.v. ≠πi.v./i.a., where πi.v. and πi.v./i.a. are the proportion of subjects with favorable outcome (mRS of 0– 2 at 3 months after randomization) for the i.v. and i.v./i.a. arms, respectively. The primary efficacy hypotheses will be tested with the Cochran–Mantel–Haenszel (CMH) test adjusting for the dichotomized baseline NIHSS score (< 20 or ≥ 20). The statistic will be tested at an α level of 0·05. Three interim analyses will be performed after approximately 225 (25% of 900), 450 (50%), and 675 (75%) subjects complete the 3-month mRS assessment and tested at appropriate nominal α levels that ensure an overall α level of 0·05. Ideally, all stratification variables should be accounted for in the analysis of primary efficacy outcome measure. For the IMS III trial, the stratification variables are the dichotomized baseline NIHSS (10–19 vs. ≥ 20) and Clinical Center Performance Site, both assumed to be fixed effects. Owing to the large number of participating Clinical Centers (approximately 42), a statistical analysis model with Clinical Center as a fixed effect would yield unstable estimates of the treatment effect. Therefore, the Clinical Center effect will be omitted in the primary analysis of the dichotomized mRS score at 3 months. However, as a secondary analysis, we will reanalyze the primary efficacy outcome variable with Clinical Center as a random effect and the treatment group and baseline NIHSS category as fixed effects in the mixed-effects model for the overall sample.
For other exploratory analyses of the primary outcome variable, the dichotomized mRS outcome will be assessed for treatment differences adjusting for a variety of covariates deemed clinically or prognostically important, such as age, time between symptom onset and randomization, and baseline NIHSS score. Each covariate will be assessed individually first with a model that includes interaction effect with the treatment (i.v. only or i.v./i.a.). If a significant interaction (P < 0·10) is observed, subgroup analyses will be considered. Quantitative and qualitative interactions between the treatment and each covariate will also be visually assessed by graphical methods.
An exploratory hypothesis of clinical interest for the primary outcome measure is the differential effect of various i.a. devices. Generalized linear regression analysis of the dichotomized mRS score at 3 months will be conducted with dummy variables representing different devices. Because of the 2:1 randomization ratio for the treatment assignment, we anticipate a sample size of approximately 600 for this analysis.
Analyses of other secondary clinical outcome, imaging, and safety measures will be conducted at the completion of the trial. Inferences made from these analyses will be supportive evidence (or lack thereof) of the effectiveness of i.v./i.a. therapy rather than primary evidence.
To compare the safety of the i.v./i.a. approach and i.v. rt-PA therapy, noninferiority hypothesis tests for the two primary safety parameters, mortality at 3 months and the incidence of symptomatic ICH within the first 30 h, will be conducted. The margin of noninferiority (δ) chosen will be tentatively set at absolute 5%. Furthermore, these safety outcomes will be compared between the treatment groups adjusting for possible confounders, such as baseline NIHSS and time from symptom onset to randomization, in a regression model.
At the end of the study, the cumulative incidences of selected adverse events will be compared between the two treatment groups using Fisher's exact test. Each of the secondary outcomes and exploratory analyses will be tested at the a level of 0·01 in order to control the Type I error probability inflation nominally.
Data management
An electronic copy of the CRF is made available to the Clinical Centers before initiation of the study to be used as worksheets to capture data for the trial. The study database, WebDCU™ System, has been developed in Microsoft SQL Server based on the approved CRFs. This system allows for a web-based data entry and management. The data are captured and entered (single keyed) at the Clinical Centers via the web interface. The data are managed (including data queries) by the DCU using a secured trial web site. During the design of the database, automated consistency checks and data validation rules were programmed to check for potential data errors, including missing required data, data out of the prespecified range, data conflicts, and disparities within and among the CRFs. The validation procedure is implemented and any changes made on the web site have a full audit trail. All data items that fail the programmed consistency checks are queried via the data clarification request (DCR) process initiated by the DCU. The DCRs are generated, communicated between the DCU and the Clinical Centers, and resolved on the secured study web site. In addition to the study database, the DCU provides the Clinical Center staff access (via password) to a standard set of web-enabled tools, including subject visit calendar, accrual status, CRF completion status, and outstanding DCR status pertaining to their respective centers. These tools allow the staff to receive regular updates on overall study status, new external information relevant to the trial, Committee meetings calendar, etc.
Study organization and funding
The participating units in this collaborative clinical trial will include at least 42 individual clinical centers, the Clinical Coordinating Center and the Angiographic Imaging Center at the University of Cincinnati (CCC), the IMS III DCU at the Medical University of South Carolina, the CT Imaging Center at the University of Calgary, Canadian Drug Distribution and Coordination Center at the University of Calgary, the Scientific Advisory Committee, the Data and Safety Monitoring Committee, and the NINDS Division of Stroke and Trauma.
The IMS III trial is funded as a collaborative U01 trial by the NIH/NINDS. Johnson and Johnson/Cordis Neurovascular, EKOS® Corporation, and Concentric Medical supply catheter devices used in the IMS III trial. Genentech Inc. supplies study medication for i.a. treatment in the IMS III study. As the trial proceeds, if new revascularization strategies become FDA approved, other companies could become involved in the conduct of the IMS III trial.
Summary
The primary objective of this NIH-funded, Phase III, randomized, multicenter, open-label clinical trial is to determine whether a combined i.v./i.a. approach to recanalization is superior to standard i.v. rt-PA alone when initiated within 3 h of acute ischemic stroke onset in subjects with moderate-to-severe strokes (NIHSS ≥ 10). The IMS III trial will develop and maintain a network of interventional centers to test the safety, feasibility, and potential efficacy of new FDA-approved catheter devices as part of a combined i.v./i.a. approach to recanalization as the IMS III study progresses. Trial enrollment began in July of 2006. As of June of 2007, 25 sites were actively screening and 58 patients were enrolled.
Footnotes
Acknowledgements
This study was funded by the National Institute of Neurological Diseases and Stroke (NINDS # U01 NS052220 and U01 NS054630). The trial receives rt-PA from Genentech, and devices from Concentric Medical, EKDS® Corporation, and Cordis Neurovascular (Johnson & Johnson Co.).
Conflicts of interest: Funding sources are as above; there are no additional conflicts of interest.